
1
Fifth stage
Pediatric
Lec-4
.د
أ
ثل
1/1/2016
Celiac disease (Gluten Sensitive Enteropathy )
is an immunemediated enteropathy caused by permanent sensitivity to gluten in
genetically susceptible individuals
PATHOGENESIS
Celiac disease develops only after dietary exposure to the protein gluten, which is found in
wheat, rye, and barley. The activity of gluten resides in the gliadin fraction, that
lead to sensitization of lamina propria lymphocytes.
• The inflammatory response results in villus atrophy, crypt hyperplasia, and damage to the
surface epithelium in the small bowel. The injury is greatest in the proximal small bowel
and extends distally for a variable distance.
• Celiac disease results in a decrease in the absorptive and digestive capacity of the small
intestinal surface area and a relative increase in immature epithelial cells.
Genetic predisposition
• Concordance in monozygotic twins approaching 100%. Two to 5% of first-degree relatives
have symptomatic gluten-sensitive enteropathy, and as many as 10% of first-degree
relatives have asymptomatic damage to small bowel mucosa consistent with this disorder.
• Celiac disease is associated with
– (HLA) types (DQ8 and DQ2)
– Down syndrome
– Type 1 diabetes
– Viruses
CLINICAL PRESENTATION
• The typical presentation of celiac disease (diarrhea,abdominal distention, & FTT) appear
in toddler (6-24 mo.) after introduction of gliadin containing diet.
• The stools are characteristically pale, loose, and offensive.
• Neurologic symptoms do occur; many children are emotionally withdrawn, irritable, and
fretful.
• Other Manifestations: smooth tongue , oral ulcers, excessive bruising, finger clubbing,
peripheral edema
Non-Intestinal Manifestations and
Association of Celiac Disease
• Osteopenia/osteoporosis
• Short stature & delayed puberty
• IDA not responding to oral iron therapy
2
• Hepatitis
• Arthritis
• Epilepsy
• Ataxia
• Autoimmunity: DM1, thyroiditis, 1 ry biliary cirrhosis, addison disease & dermatitis
herpetiformis
• Syndromes: Turner, Down
• Malignancy
DIFFERENTIAL DIAGNOSIS
GIARDIASIS
MALNUTRITION
COWS MILK PROTEIN ALLERGY
laboratory invest
Anaemia usually IDA but dimorphic anaemia
may occur.
Hypoproteinaemia, hypoprothrombinaemia,
↑fecal fat estimation
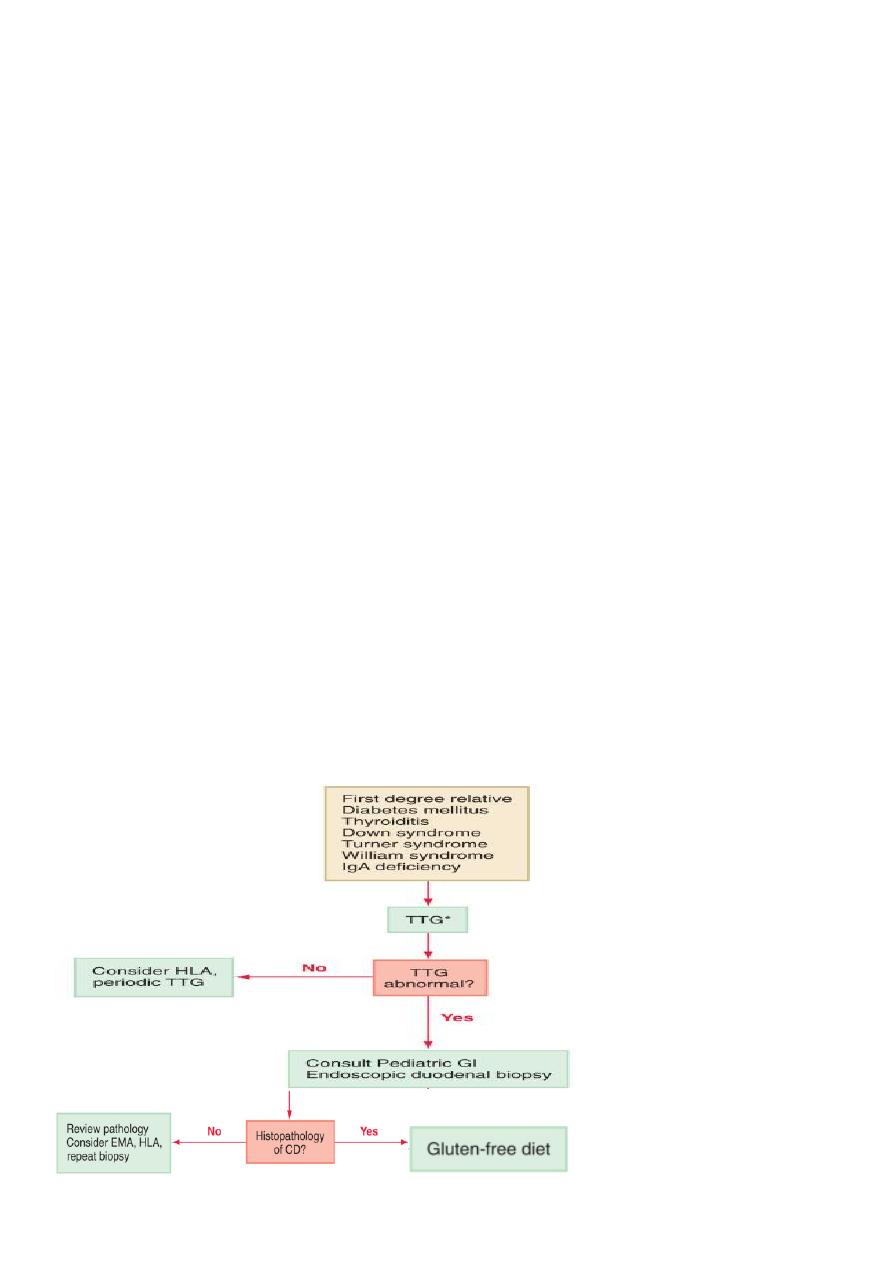
SCREENING AND DIAGNOSIS
• Screening for celiac disease has been recommended for specific risk factors.
• Serologic tests such as anti-endomysium IgA antibody test (EMA), anti-tissue
transglutaminase IgA antibody test (TTG), and HLA DQ2 or DQ8 genotype testing are useful
for evaluation of asymptomatic subjects with diabetes mellitus, thyroiditis, Down
syndrome, Turner syndrome, William syndrome, or IgA deficiency who have a higher
incidence of celiac disease, and first-degree relatives of patients with celiac disease (CD).

3
Serologic tests
The anti-endomysium IgA antibody and
anti-tissue transglutaminase IgA antibody
tests are highly sensitive and specific in identifying individuals with celiac disease.
Some 10% of patients whose disease is diagnosed earlier than 2 yr of age show
absence of IgA anti-TG2. For them, the measurement of serum antigliadin antibodies is
generally advised. Antibodies against gliadinderived deamidated peptides (D-AGA) have
been assessed. Compared with conventional AGA, the peptide antibodies (IgG and IgA)
have a greater sensitivity and specificity. A problem with serology is represented by the
association of celiac disease with IgA deficiency (10-fold increase compared to the general
population). Serum IgA should always be checked, and in the case of IgA deficiency,
D-AGA, IgG anti-endomysium, or TG2 should be sought. Negative serology should not
preclude a biopsy examination when the clinical suspicion is strong
Genetic tests
have an increasing role in the diagnosis. Less than 2% of celiac patients lack both HLA
specificities; at the same time, approximately one third of the “normal” population has one
or the other marker; that means that the measurement of HLA DQ2 and/or DQ8 has a
strong negative predictive value but a very weak positive predictive value for the diagnosis
of celiac disease.
Small Intestinal Biopsy
• Definitive diagnosis of celiac disease requires small intestinal biopsy, as none of the
available serologic tests are 100% reliable .
• The characteristic histologic changes include partial or total villous atrophy, crypt
elongation and decreased villous/crypt ratio, increased number of intraepithelial
lymphocytes .
• The mucosal involvement can be patchy, so multiple biopsies must be obtained.
ESPGHAN current criteria,
The 2 requirements mandatory for the diagnosis of celiac disease are:
– characteristic histologic features
+
– full clinical remission after withdrawal of
gluten from the diet
Reversal of positive serologic tests after gluten withdrawal is considered supportive
evidence .
• In children <2 yr of age, milk protein–sensitive enteropathy can produce changes similar
to celiac disease; confirmation of diagnosis after a gluten challenge is sometimes required.
This necessitates three biopsies: an initial biopsy at presentation, the 2nd to document
healing with gluten withdrawal, and the 3rd to show recurrent damage with reintroduction

4
of gluten.
• Gluten challenge is not considered mandatory except in situations where there is doubt
about the initial diagnosis
TREATMENT
• The only treatment for celiac disease is lifelong exclusion of gluten. This requires a wheat-
,barley-, and rye-free diet.
• After gluten withdrawal, there is rapid remission of symptoms, improved bone
mineralization, and reversal of growth failure and nutritional deficiencies.
• It is recommended that children with celiac disease be monitored with periodic visits for
assessment of symptoms, growth, physical examination, and adherence to the gluten-free
diet.
PROGNOSIS
• The clinical response to a gluten-free diet usually results in improvement of mood,
appetite, and lessening of the diarrhea within a week. Reduced bone mineral density also
improves with gluten exclusion. No long-term complications from a gluten-free diet have
been recognized.
• Celiac disease is associated with intestinal lymphoma and other forms of cancer,
especially adenocarcinoma of the small intestine, of the pharynx, and of the esophagus.
• Several follow-up studies suggest that a gluten-free diet protects from cancer
development, especially if started in the 1st years of life. Therefore, early diagnosis and
strict dietary restrictions appear to be the only possibility of preventing risk for rare but
very aggressive forms of cancer associated with celiac disease.
CYSTIC FIBROSIS
• A multisystem disease
• Autosomal recessive inheritance
• Commonest lethal genetic condition affecting Caucasian people.
• DEFECT IN CHLORIDE TRANSPORT IN EPITHELIAL TISSUE . This result in relative
dehydration of airway secretions.
GENETICS
• CF is inherited as an autosomal recessive trait.
Let C= normal CFTR
Let c= mutant CFTR
If mom and dad are both carriers then:
C c

5
With mom and dad
carriers, then:
50% chance of having child who is a carrier
25% chance of child being affected
25% of child with no mutant copies of CFTR
GENETICS
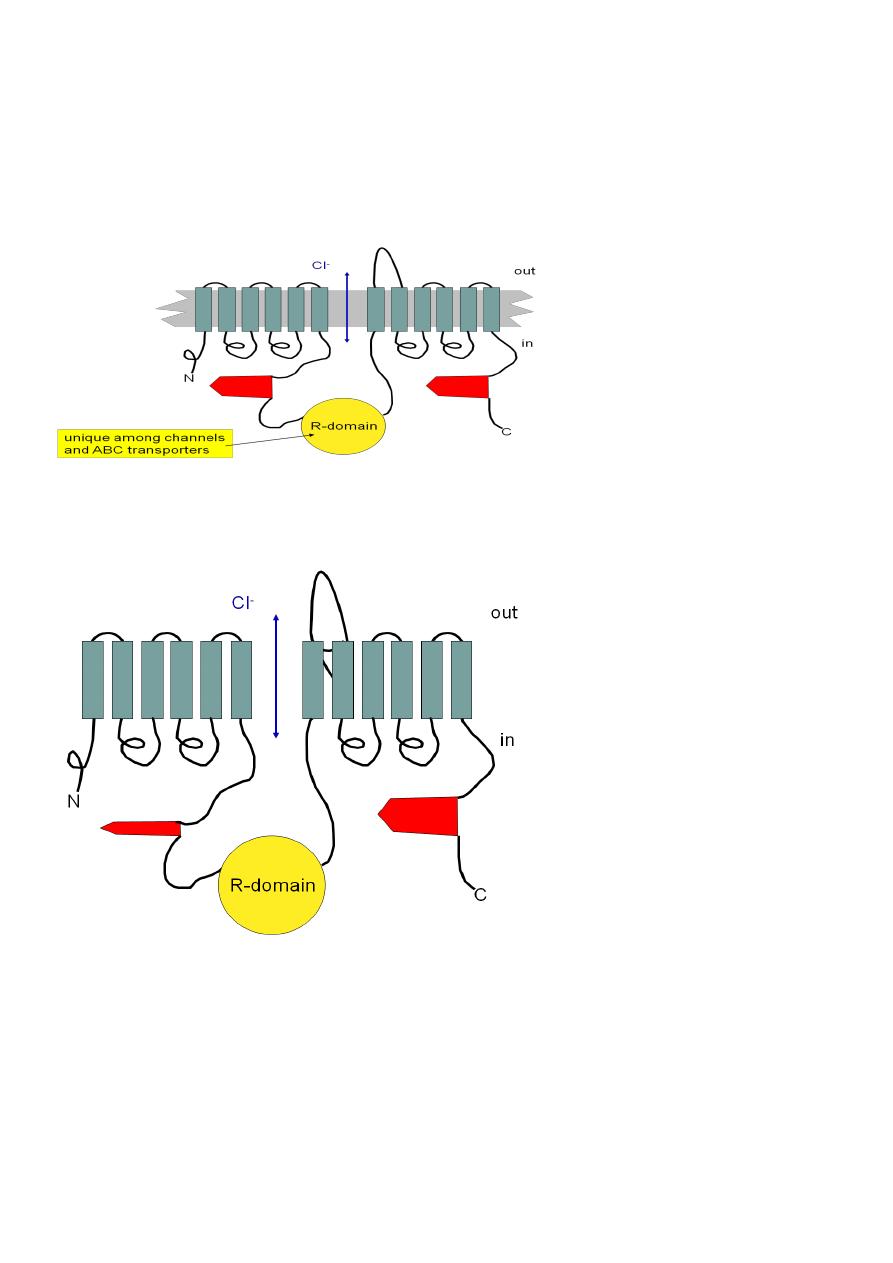
• The CF gene codes for a protein of 1,480 amino acids called the CF transmembrane
regulator
(CFTR). CFTR is expressed largely in epithelial cells of airways, the gastrointestinal tract
(including the pancreas and biliary system), the sweat glands, and the genitourinary system.
Cystic fibrosis transmembrane
conductance regulator (CFTR) gene
The most comm CFTR gene is located on the long arm of chromosome 7.
There are >1500 mutations in CFTR.
Commonest mutation is Δ F508-- -70% CF alleles in caucasians(deletion of a single
phenylalanine residue at amino acid 508 )
6
Cystic Fibrosis Transmembrane
Conductance Regulator (CFTR)
• A Cl- channel
• 2 sets of 6 transmembrane domains
• 2 “ATP-binding cassettes” (thus “ABC transport protein”)
ATP
ATP
What’s wrong with DF508?
The most common mutation (70% of mutants): A phenylalanine (F) is deleted at position
508 in ATP-binding cassette #1
PATHOGENESIS
• Four long-standing observations are of fundamental pathophysiologic importance:
failure to clear mucous secretions a paucity of water in mucous secretions an elevated salt
content of sweat and other serous secretions chronic infection limited to the respiratory
tract.
CFTR helps to control bulk water flow across epithelia
1. Na+ channels are usually open, but extensive Na+ flux requires a counterion.
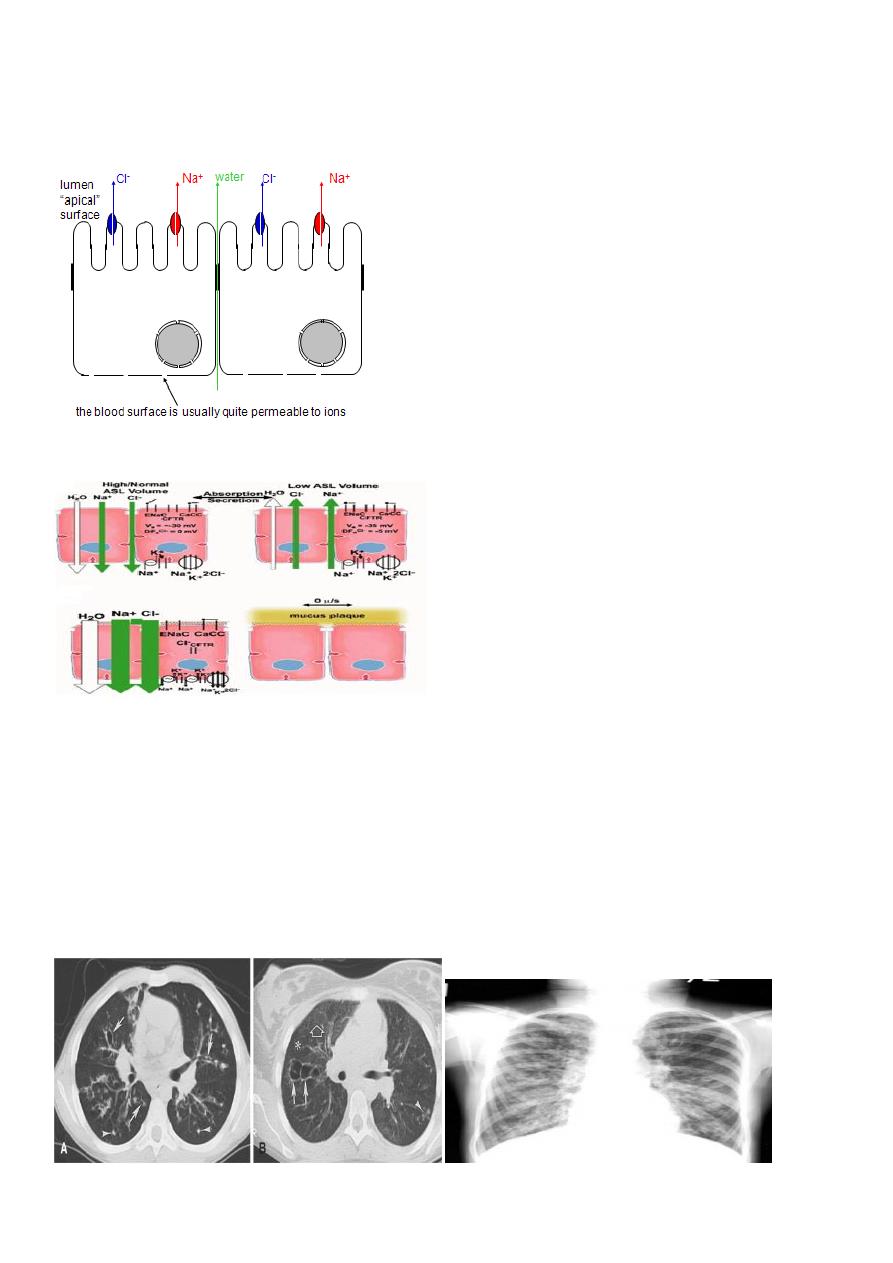
7
2. If CFTR is open, Clbecomes the counterion.
Therefore NaCl flows across the membrane.
3. Water then flows around the cells to maintain osmotic pressure.
4. Result: isotonic NaCl solution flows from the blood to the lumen (or vice-versa)
CFTR and Airway Surface Liquid
Clinical features of Cystic Fibrosis
Chronic Sino-Pulmonary Disease
Nutritional deficiency/GI abnormality
Obstructive Azoospermia
Electrolyte abnormality
CF in a first degree relative
Bronchiectasis
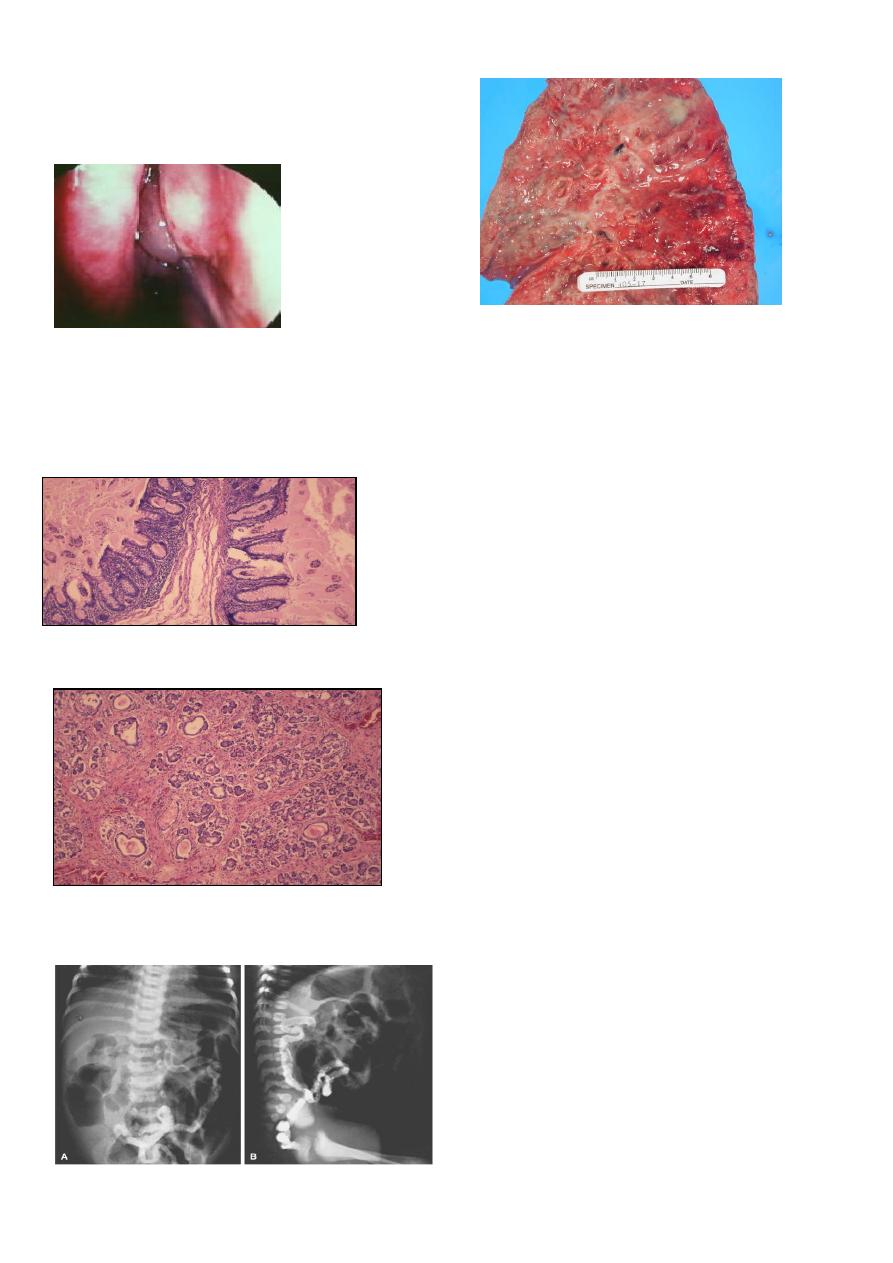
8
Mucous in the airways cannot be easily cleared from
the lungs
Nasal Polyps
Digital Clubbing
Schamroth sign +v
Colon Sticky mucus secretion
Pancreas Ducts are filled with sticky mucus. Scaring of tissue.
Meconium ileus

9
Diagnostic Methods
Sweat chloride
• Chemical that stimulates sweating placed under electrode pad; saline under other
electrode pad on arm
• Mild electric current is passed between electrodes
• Sweat collected Positive Sweat chloride: > 60 meq/L
• Genetic testingDNA Testing for most common CFTR mutations
Other Diagnostic Tests
• increased potential differences across nasal epithelium
• Prenatal diagnosis is possible by Amniotic fluid or Chorionic villous sampling
• Newborn Screening for CF:
– Immunoreactive trypsinogen usually first followed by either sweat or DNA testing
Diagnostic Criteria for Cystic
Fibrosis
TREATMENT
Multidisciplinary
• Pulmonary Therapy.
• Infection
• Nutrition
• Gastrointestinal
• Infertility
• Social Issues
Nutrition
• High calorie supplemental
• Vitamin supplementation
• Pancreatic enzymes