
1
Fifth stage
Medicine
Lec-12
د.بشار
17/4/2016
Diseases of Peripheral Nerves
Numerous inherited and acquired pathological processes may affect the nerve roots
(radiculopathy), the nerve plexuses (plexopathy) and/or the individual nerves (neuropathy).
Cranial nerves 3-12 share the same tissue characteristics as peripheral nerves elsewhere and
are subject to the same range of diseases. Nerve fibres of different types (motor, sensory or
autonomic) and of different sizes may be variably involved. Disorders may be primarily
directed at the axon, the myelin sheath (Schwann cells) or the vasa nervorum
PATTERN OF INVOLVMENT:
Mononeuropathy Simplex=Single Nerve
Mononeuropathy Multiplex=Several Nerves Randomly &Noncontiguously
Polyneuropathy( Peripheral Neuropathy)=Dysfunction of Numerous Peripheral Nerves at the
Same Time leading to predominantly distal & symmetrical deficit usually affecting lower
more than upper limbs
Types &Causes of peripheral neuropathy لالطالع

2
Diseases of Peripheral Nerves
SYMPTOMS &SIGNS
1.Sensory Disturbances
A.Numbness, Hyperpathia, Impaired Sensation &
SPONTANEOUS PAIN esp. in SMALL FIBER
involvement ;D. M. , Porphyria, AIDS, Alcoholic,
Entrapment …..
B. Dissociated Sensory Loss
Small Fib. Pain &Temp.
Large Fib. Touch, Vib. & Position
2. MOTOR DEFICITS
Weakness , Wasting , Fasciculation
Diminished or Absent Reflexes
i.e. LMNL
3.AUTONOMIC DISTURBANCES
Post. Hypotension, Coldness, Imp. Sweating, Impotence….esp. GBS, Diabetes, Renal
Failure, Porphyria….
4. ENLARGED NERVES
Leprosy, Amyloidosis, HSMN, Refsum Dis., Acromegaly..
Causes of P. N:
1. Inflammatory: GBS , CIPD
2. Metabolic &Nutritional :D.M. ,Uremia, Liver Failure, Hypothyroidism, Acromegaly, B12
Deficiency….
3.Infectious &Granulomateous:AIDS, Leprosy, Diphtheria, Sarcoidosis…
4.Vasculitis: PAN, SLE, Rh.Arthritis…

3
5.Neoplastic &Paraneoplastic
6. Drugs &Toxins:Alcohol, INH, Vinicristine, Phenytoin, Heavy Metals….
7.Hereditory:HSMN, HSN, Refsum Disease,Porphyria..
8. IDIOPATHIC
Drugs causing peripheral neuropathy لالطالع
EVALUATION OF PATIENTS:
TIME COURSE=Acute; Inflamm., Infectious, Toxins…
Chronic;Hereditory, Metabolic…
AGE= Early; Hereditory
Late; Metabolic, Neoplastic
OCCUPATION= Exposure to toxins
MEDICAL HISTORY
DRUGS
FAMILY HISTORY
DIFFERENTIALDIAGNOSIS:
Diseases of muscles &n.m. junction normal sensation& tendon reflexes
Diseases of spinal cord pyramidal signs &sensory level below the lesion
4
Radiculopathies dermatomal &myotomal distribution
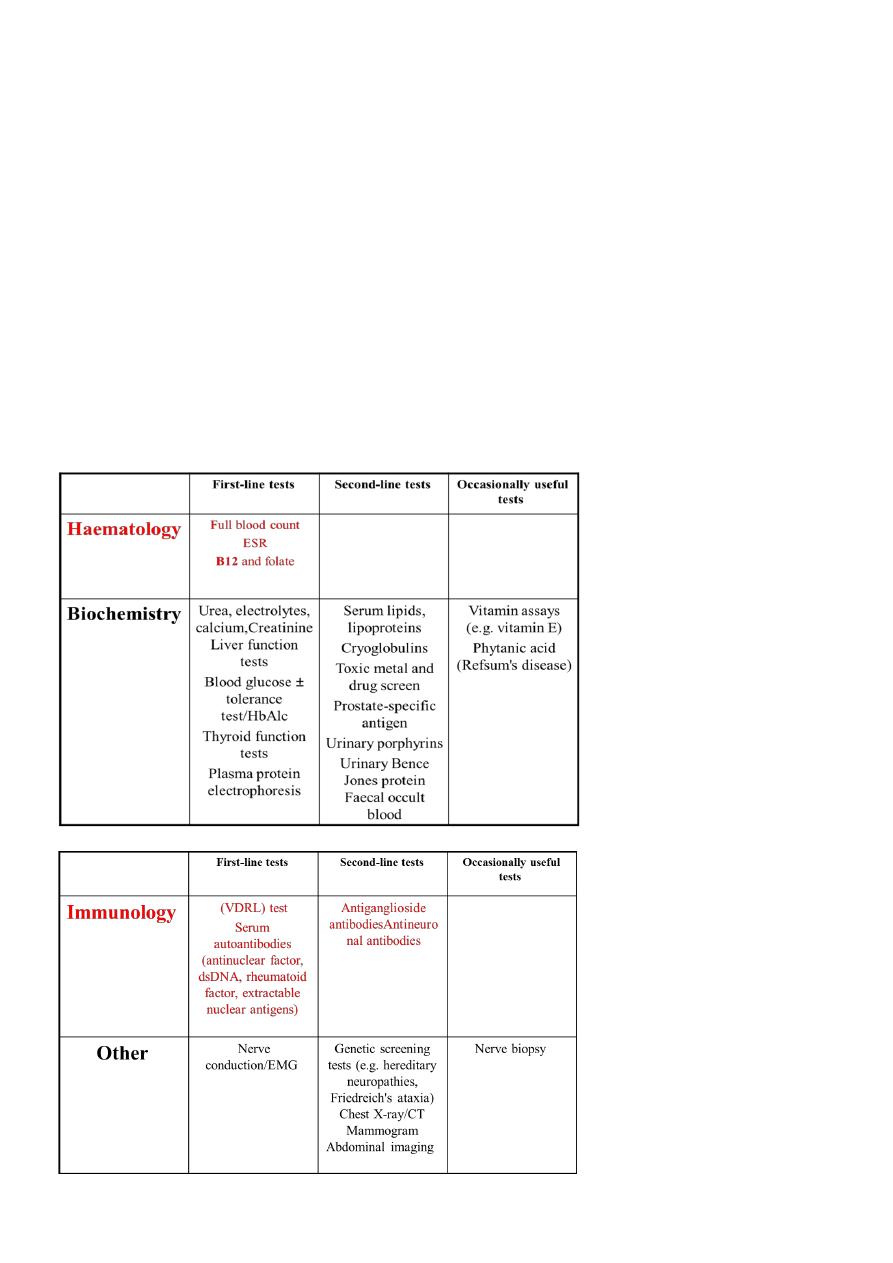
INVESTIGATIONS:
CONFIRM DIAGNOSIS
EMG = DENERVATION
ENG &NCV = Demyelination or Axonal Degeneration
REVEAL UNDERLYING CAUSE
INVESTIGATION OF PERIPHERAL NEUROPATHY لالطالع

5
TREATMENT:
UNDERLYING CAUSE
NURSING CARE ? ULCERS &CONTRACTURES
RESPIRATORY MONITORING &MANAGEMENT
CARE OF SKIN &NAILS
RELIEF OF PAIN ----Lancinating Pain-- PHENYTOIN
CARBAMAZEPINE
MEXILETINE
Constant Pain----AMITRIPTYLINE
GABAPENTINE
GUILLIAN BARIE SYNDROME
ACUTE ASCENDING POLYRADICLONEUROPATHY
This syndrome of acute paralysis develop in 70 % of patients 1-4 weeks after respiratory
infection or diarrhoea (particularly Campylobacter).
In Europe and North America, acute inflammatory neuropathy is most commonly
demyelinating ( AIDP ).
Axonal variants ,either ( AMAN ) or (ASMAN) are more common in China and Japan .
Clinical features:
Distal paraesthesia and limb pains precede a rapidly ascending muscle weakness from lower
to upper limbs , more marked proximally than distally.
Facial and bulbar weakness commonly develops , and respiratory weakness requiring
ventilatory support occurs in 20 % of cases .
In most patients weakness progresses for 1-3 weeks but rapid deterioration to respiratory
failure can develop within hours
On examination there is diffuse weakness with widespread loss of reflexes.
An unusual axonal variant described by Miller Fisher comprises the triad of ophthalmoplegia
, ataxia and areflexia .

6
Overall, 80% of patients recover completely within 3-6 months, 4% die, and the remainder
suffer residual neurological disability which can be severe. Adverse prognostic features
include older age, rapid deterioration to ventilation and evidence of axonal loss on EMG.
Investigations:
The CSF protein is abnormal at some stage of the illness, but may be normal in the first 10
days. There is usually no rise in CSF cells ( lymphocytosis of > 50/ml suggests an alternative
diagnosis ).
Electrophysiological studies are often normal in the early stages but show typical changes
after a week or so , with conduction block and multifocal motor slowing , sometimes most
evident proximally as delayed F-waves.
Management:
During the phase of deterioration , regular monitoring of respiratory function ( vital capacity
and arterial blood gases) is required, as respiratory failure may develop with little warning
and require ventilatory support.
Ventilation may be needed if the vital capacity falls below 1L , but ventilation is more often
required because of bulbar weakness leading to aspiration.
General management to protect the airway and prevent pressure sores and venous thrombosis
is essential .
Corticosteroids have been shown to be ineffective .
Plasma exchange and intravenous immunoglobulin therapy shorten the duration of the illness
,reduce severity and improve prognosis provided treatment is started within 14 days of the
onset of symptoms .
Chronic demyelinating polyneuropathy لالطالع
It is either hereditary or immune-mediated.
Charcot-Marie-Tooth (CMT) disease which is of many types ;the most commom 70-80 % is
the autosomal dominant one causing distal wasting (inverted champagne bottle or stork leg )
often with pes cavus and a predominantely motor involvement
Presents with a relapsing or progressive generalized neuropathy.

7
Sensory,motor or autonomic nerves can be involved but the signs are predominantely motor .
Multifocal Motor Neuropathy MMN is a variant with motor involvement only .
CIDP لالطالع
CIDP usually responds to immunosuppressive treatment ;corticosteroids , methotrexate or
cyclophosphamide OR to immunomodulatory treatments (plasma exchange or IVIg ),which
is the best of patients with MMN .
About 10 % of patients with acquired demyelinating polyneuropathy have an abnormal
serum paraprotein , sometimes associated with a lymphoproliferative malignancy .
DIABETIC NEUROPATHY:
AMYOTROPHY = PAIN &WEAKNESS WITH ATROPHY PELVIC GIRDLE
&THIGH MUSCLES WITH ABSENT KNEE REFLEX &LITTLE SENSORY LOSS
MONONEUROPATHY = ACUTE PAINFUL CRANIAL NERVES
BOTH HAVE GOOD PROGNOSIS
Entrapment neuropathy:
Focal compression or entrapment is the usual cause of mononeuropathy . However , some
patients present with what initially appears to be a single nerve lesion and then go on to
develop multiple nerve lesions.This is termed mononeuritis multiplex .
Symptoms and signs:

8
In an entrapment neuropathy, pressure initially damages the myelin sheath, and
neurophysiology will show slowing of conduction over the relevant site. Sustained or severe
pressure damages the integrity of the axons, demonstrable as loss of the sensory action
potential distal to the site of compression.
Certain conditions increase the propensity to develop entrapment neuropathies. These
include acromegaly, hypothyroidism, pregnancy, any pre-existing mild generalised axonal
neuropathy (e.g. diabetes), and oseophytes. Patients with multiple recurrent entrapment
neuropathies, especially at unusual sites, should be screened for autosomal dominant
hereditary neuropathy with liability to pressure palsies (HNPP) .
Unless axonal loss has occurred, entrapment neuropathies will recover, provided the pressure
on the nerve is relieved, either by avoiding precipitating activities or limb positions, or by
surgical decompression.
Entrapment neuropathy CTS:
MEDIAN N. COMPRESSION AT THE WRIST
IDIOPATHIC, PREGNANCY, TRAUMA, ARTHRITIS, MYXOEDEMA,
ACROMEGALY, TENOSYNOVITIS……
PAIN, NUMBNESS IN MEDIAN N. DISTIBUTION ?SHOULDER
AT NIGHT
WEAKNESS & ATROPHY OF THENAR MUSCLES
TINEL SIGN , PHALEN MANEUVER
Rx LOCAL STEROIDS, WRIST SPLINT, SURGERY

9
Facial nerve palsy:
Idiopathic facial nerve palsy or Bells palsy is a common condition affecting all ages and both
sexes .The lesion is within the facial canal and may be due to reactivation of latent herpes
simplex virus 1 infection .Symptoms usually develop subacutely over a few hours , with pain
around the ear preceding the unilateral facial weakness. Patients often describe the face as
numb but there is no objective sensory loss (except possibly to taste).
Hyperacusis can occur if the nerve to stapedius is involved , and there may be diminished
salivation and tear secretion . Examination reveals an ipsilateral lower motor neuron facial
nerve palsy . Vesicles in the ear or on the palate indicate that the facial palsyis due to herpes
zoster rather than Bells palsy .
Prednisolone 40-60 mg daily for a week speeds recovery if started within 72 hours.Artificial
tears and ointment prevent exposure keratitis and the eye should be taped shut overnight.
About 80% of patients recover spontaneously within 12 weeks. A slow or poor recovery is
predicted by complete paralysis, older age and reduced facial motor action potential
amplitude after the first week. Recurrences can occur but should prompt further
investigation. Aberrant re-innervation may occur during recovery, producing unwanted facial
movements (e.g. eye closure when the mouth is moved) or 'crocodile tears' (tearing during
salivation).
MOTOR NEURONE DISEASE:
DEGENERATION OF MOTOR NEURONS IN SPINAL CORD MOTOR
NUCLIE OF LOWER CRANIAL NERVES ,
ONSET 30 -60 YEARS, > IN MALES, 2/ 100000,
SPORADIC, 5- 10 %FAMILIAL ( AUT. DOM. ) CHR 15
CAUSE : UNKNOWN
o ?AUTOIMMUNE
o INCREASED OFR FORMATION
o REDUCED NEUROTROPHIC FACTORS
o EXCITOTOXINS
CLINICAL TYPES &FEATURES
A. PROGRESSIVE BULBAR PALSY = LMN CRANIAL NERVES
B.PSEUDOBULBAR PALSY = UMN CRANIAL NERVES
C.SPINAL MUSCULAR ATROPHY = LMN SPINAL CORD AHC
D. PURE LATERAL SCLEROSIS = UMN IN LIMBS
E. AMYOTROPHIC LAT. SCLEROSIS = MIXED C. & D.
MAY BE WITH A. & B.

10
NO EXTRA OCULAR MUSCLES INVOLVEMENT
NO SPHINCTER INVOLVEMENT
NO SENSORY DEFICIT
NORMAL CSF
EMG
Rx = RILUZOLE 100mg /day may slow progression & reduce mortality. It is GLUTAMATE
ANTAGONIST . Side effects = fatigue, dizziness, GIT diturbance, raised liver enzymes.
Anticholinergics for drolling of saliva
Physiotherapy
FEEDING = Semisolid diet, NG tube, Gastrostomy.
Tracheostomy
Prognosis = Bad especially in Bulbar type
Progressive &fatal in 3 -5 years
MND IN CHILDREN
لالطالع
INFANTILE = WERDING HOFFMANN DIS. ONSET AT 3 MONTHS , DEATH
AT 3 YEARS, AR , DIFFICULT SUCKING , SWALLOWING&
VENTILATION, ATROPHY & FASCICULATION OF TONGUE &
LHMB MUSCLES, NO SENSORY DEFICIT , NO Rx
INTERMEDIATE = CHRONIC W-H-DISEASE
AR, 2
nd
of 1
st
year, LESS BULBAR INVOLVEMENT ,
BENIGN, SLOWLY PROGRESSIVE , KYPHOSCOLIOSIS
&CONTRACTURES , SURVIVE TO ADULTHOOD
JUVENILE = KUGELBERG – WELANDER DISEASE
AR, COULD BE AD OR X – LINKED
ONSET AT CHILDHOOD OR EARLY ADOLESCENCE
MORE IN PROXIMAL MUSCLES , LITTLE BULBAR INVOLV.
DISABILITY IN EARLY ADULT LIFE
NO Rx
SYRINGOMYELIA
لالطالع
CAVITATION OF SPINAL CORD
COMMUNICATING = CENTRAL CANAL &THE CAVITY
NON – COMMUN. = CYSTIC DILATATION OF SPINAL CORD
CLINICALLY = DISSOCIATED SENSORY LOSS

11
WEAK. & WASTING OF MUSCLES ; CERVICAL &T 1
PYRAMIDAL SIGNS &SPHINCTER DIST. BELOW
NECK & RADICULAR PAIN
HORNER SYNDROME
SYRINGOBULBIA
MAY BE ASSO. WITH TUMOR, TRAUMA , ARACHNOIDITIS OR ANMALIES LIKE
ARNOLD – CHIARI MALFORMATION
Rx = DECOMPRESSION OF DISTENDED SYRINX
SUBACUTE COMBINED DEGEN. OF THE CORD
لالطالع
B12 DEFICIENCY
PARASTHESIA &WEAKNESS OF THE EXTREMITIES = PERIPHERAL NERVES
SPASTIC PARAPARESIS = PYRAMIDAL FIBERS
SENSORY ATAXIA = POSTERIOR COLUMN TRACT
LHERMITTS SIGN
SCOTOMAS = OPTIC ATROPHY
BEHAVIOURAL OR PSYCHIATRIC SYMPTOMS
ANEMIA MEGALOPLASTIC NOT NECESSARILY PRESENT
Rx B12I. M. …….