
1
Fifth stage
Medicine
Lec-17
د.خالد نافع
1/5/2016
Bleeding
History
• Site of bleed
• Duration of bleed
• Precipitating causes, including previous surgery or trauma
• Family history
• Drug history
• Age at presentation
• Other medical conditions, e.g.liver disease
Examination
There are two main patterns of bleeding:
1. Mucosal bleeding
Reduced number or function of platelets (e.g. bone marrow failure or aspirin) or von
Willebrand factor (e.g.von Willebrand disease)
Skin: petechiae, bruises
Gum and mucous membrane bleeding
Fundal haemorrhage
Post-surgical bleeding
2. Coagulation factor deficiency
(e.g. haemophilia or warfarin)
Bleeding into joints (haemarthrosis) or muscles
Bleeding into soft tissues
Retroperitoneal haemorrhage
Intracranial haemorrhage
Post-surgical bleeding

2
Vessel wall abnormalities
congenital, such as hereditary haemorrhagic telangiectasia
• acquired, as in a vasculitis or scurvy
Hereditary haemorrhagic telangiectasia(HHT)
Autosomal dominant .
Telangiectasia and small aneurysms are found on the fingertips, face and tongue,
and in the nasal passages,lung and gastrointestinal tract.
Pulmonary arteriovenous malformations (PAVMs) that cause arterial hypoxaemia
due to a right-to-left shunt. These predispose to paradoxical embolism, resulting
in stroke or cerebral abscess. =
All patients with HHT should be screened for PAVMs; if these are found, ablation
by percutaneous embolisation should be considered.
Recurrent bleeds, particularly epistaxis, or with iron deficiency due to occult
gastrointestinal bleeding.
TREATMENT
1-Iron replacement for IDA
2-Local cautery or laser therapy may prevent single lesions from bleeding
Platelet function disorders
Primary Hemostasis
Platelet Plug formation
Dependent on normal platelet number & function
Initial manifestation of clot formation
1-Thrombocytopenia
2-Thrombasthenia

3
Laboratory Tests for Primary
Hemostasis Function
Platelet count
Bleeding time
Platelet Aggregation Studies
clot retraction
Flow cytometric studies for Glycoproteins
Glanzmann thrombasthenia
Background:
Thrombasthenia was first describe in 1918 by Glanzmann
when he noted purpuric bleeding in patients with normal
platelet counts
Typically, thrombasthenia is diagnosed at an early age
Pathophysiology:
Autosomal recessive trait
The production and assembly of the platelet membrane glycoprotein IIb-IIIa is altered,
preventing the aggregation of platelets and subsequent clot formation
Treatment
1-local measure.2-antifibrinolytic agent such as tranexamic acid
.3-platelet transfusion.4-Recombinant factor VII
Idiopathic thrombocytopenic purpura
Autoantibodies, most often directed against the platelet membrane glycoprotein IIb/IIIa,
which sensitise the platelet, resulting in premature removal from the circulation by cells of
the reticulo-endothelial system.
1-Isolated condition.
2-Association with connective tissue diseases,HIV infection,
B cell malignancies, pregnancy and certain drug therapies.
Clinical features
Purpura and
haematomas
Mucosal bleeding
Classification of ITP disease phases
ITP phase Definition
Newly diagnosed Within 3 months of diagnosis
Persistent 3 to 12 months from diagnosis
Chronic > 12 months from diagnosis
In adults, ITP usually has an insidious onset, with no preceding illness.

4
Nearly one-quarter of patients present asymptomatically and
receive a diagnosis of ITP through incidental routine blood
tests
Petechiae or purpura • Unusual or easy bruising (haematoma)•
Persistent bleeding symptoms from cuts or other injuries• Mucosal
bleeding• Frequent or heavy nose bleeds (epistaxis)
• Haemorrhage from any site (usually gingival or menorrhagia in
womenn
Petechie
Recommended diagnostic approaches for ITP
Patient history
Family history
Physical examination
Complete blood count and reticulocyte count
Peripheral blood smear
Quantitative immunoglobulin level measurement*
Bone marrow examination (in selected patients)
Blood group (rhesus)
Direct antiglobulin test
Helicobacter pylori
Human immunodeficiency virus (HIV)
Hepatitis C virus (HCV)
Bone marrow aspiration
is indicated in older patients (particularly those over 60 years of age to exclude
myelodysplastic syndrome),
in those with an atypical presentation (e.g. abnormalities observed on
peripheral blood smear suggestive of other haematological disorders),
in those with a poor response to first-line therapy
and in those being considered for splenectomy..
Treatment
when to treat?

5
If a patient has two relapses,or primary refractory disease, splenectomy is considered.
Splenectomy produces complete remission in about 70% of patients and improvement in
a further 20–25%, so that,following splenectomy, only 5–10% of patients require further
medical therapy.
Second-line therapy with the thrombopoietin analogue romiplostim or the thrombopoietin
receptor agonist eltrombopag
Rituximab, ciclosporin and tacrolimus should be consideredin cases where the approaches
above are ineffective.
Coagulation disorders

6
Haemophilia A the "ROYAL DISEASE"
Epidemiology:more than 400.000persons affected
About 1 in 10.000 people is born with heamphilia A
Clinical features
A prolonged aPTT: a normal aPTT does not exclude mild hemophilia A because the aPTT
may not be sufficiently sensitive to detect slightly reduced levels of FVIII-C in the
approximate 20-30% range
Normal PT
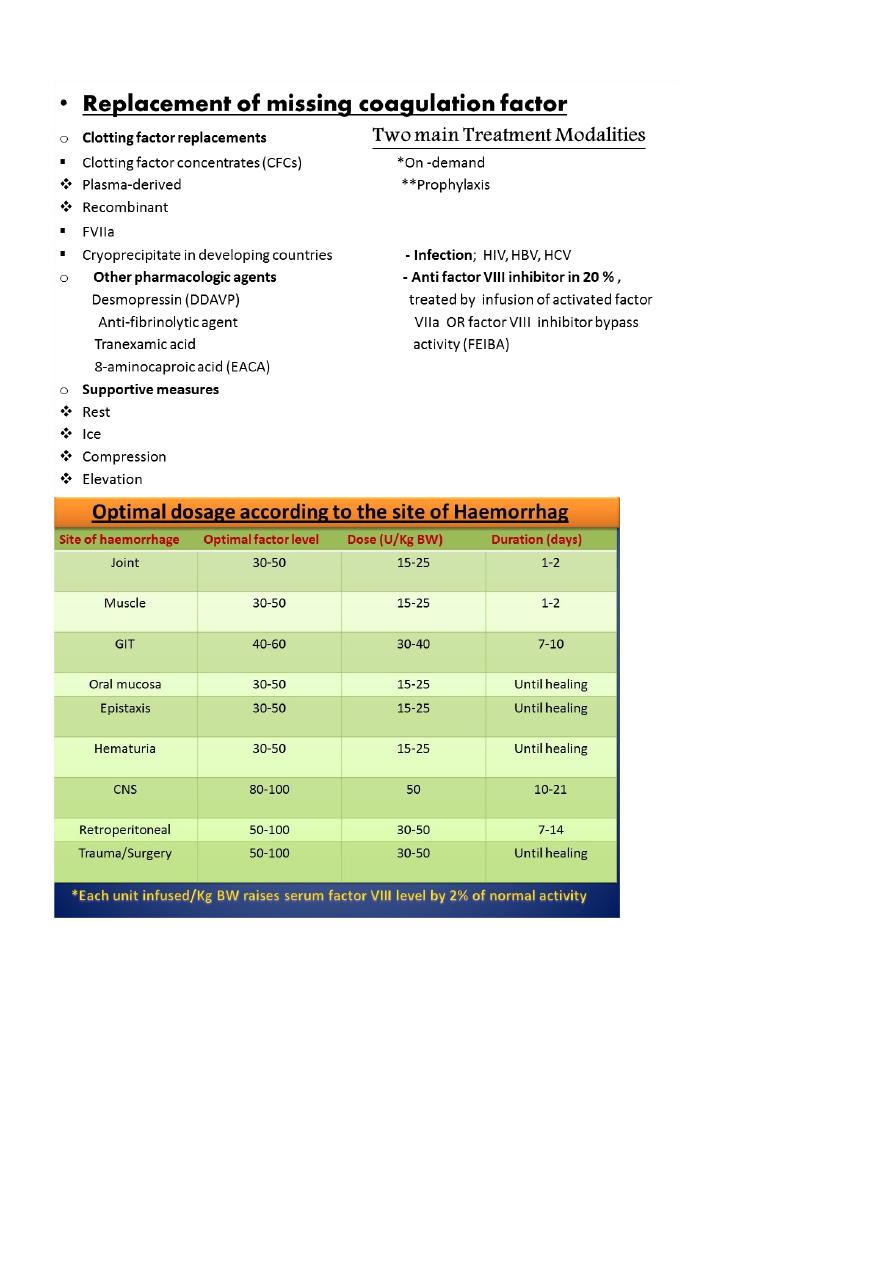
7
Treatment
Haemophilia B (Christmas disease)
• Due to deficiency of factor IX .
• X-linked
• Clinical manifestation indistinguishable from haemophilia A
• Treatment ; factor IX concentrate , indication and dosing same as to haemophilia A.
• Complication of therapy similar to haemophilia A regarding transmission of infection BUT
the incidence of inhibitor is < 1%. Von Willebrand disease
8
von Willebrand factor
– Synthesis in endothelium and megakaryocytes
– Forms large multimer
– Carrier of factor VIII
– Anchors platelets to subendothelium
– Bridge between platelets
VWD
VWD is the most common inherited bleeding disorder
Males and females are affected equally
Deficiency of VWF results in defective platelet adhesion and causes a secondary deficiency
in factor VIII
The result is that VWF deficiency can cause bleeding that appears similar to that caused
by platelet dysfunction or haemophilia
The most common symptoms include nosebleeds, skin bruises.
Prolonged bleeding from trivial wounds, oral cavity bleeding, and excessive menstrual
bleeding are common.
Lab Studies:
Screening tests typically include
prothrombin time (PT)
activated partial thromboplastin time (aPTT),
FVIII level
ristocetin cofactor (RCoF) activity
vWF antigen (vWF:Ag).
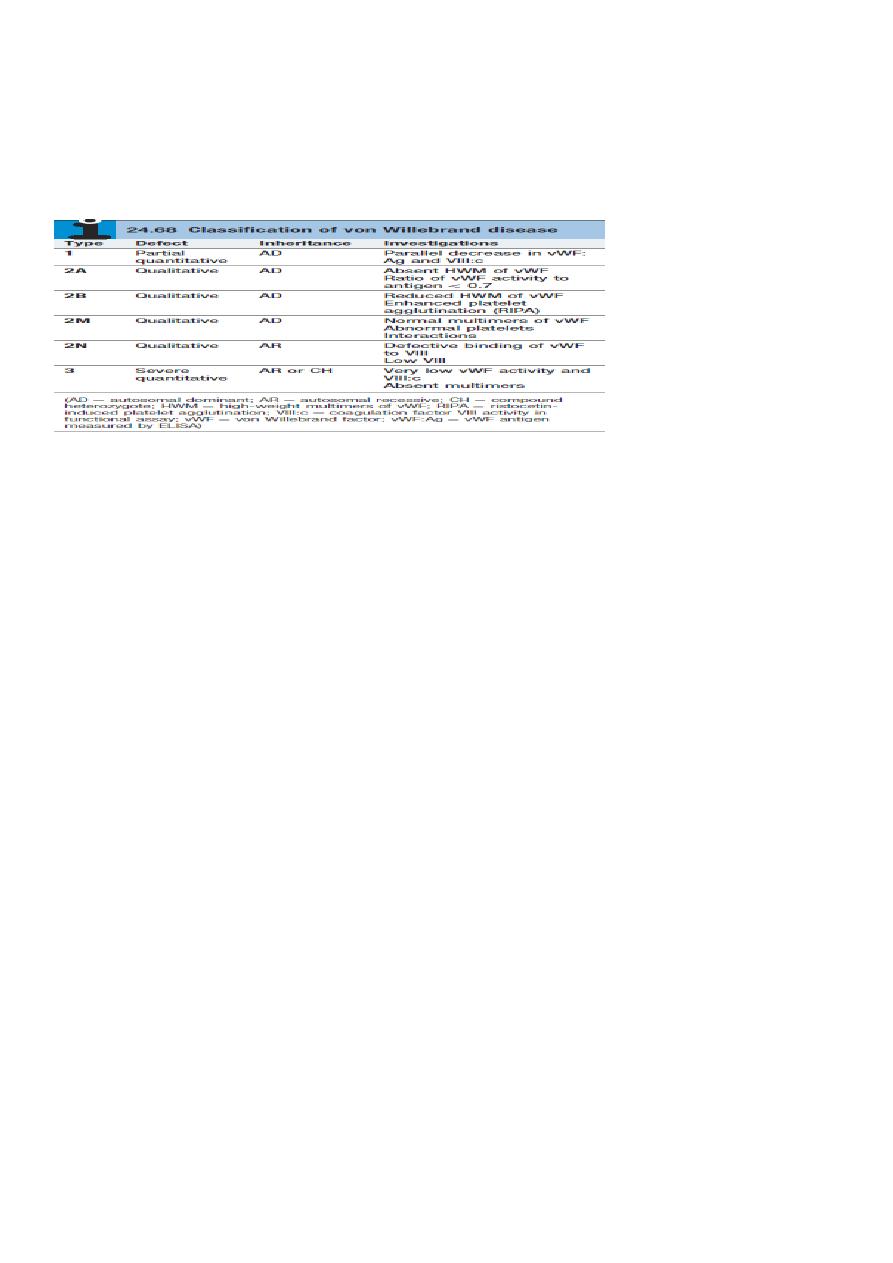
Laboratory evaluation of von Willebrand disease
Classification
– Type 1 Partial quantitative deficiency
– Type 2 Qualitative deficiency
– Type 3 Total quantitative deficiency
*Bleeding time ↑
*aPTT ↑
vWD type
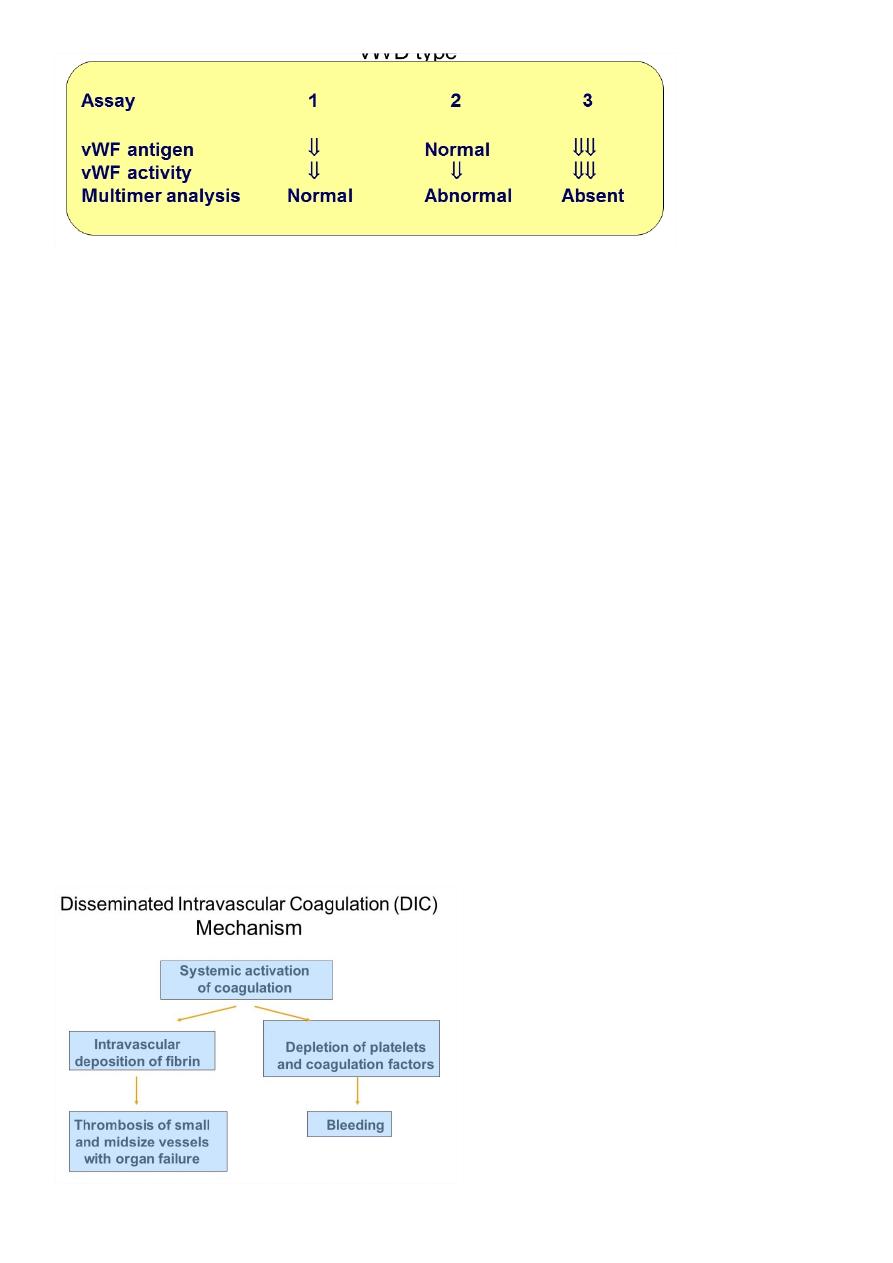
9
Treatment of von Willebrand Disease
Cryoprecipitate
– Source of fibrinogen, factor VIII and VWF
– Only plasma fraction that consistently contains VWF multimers
DDAVP (des -demino-8-arginine vasopressin)
–
plasma VWF levels by stimulating secretion from endothelium
– Duration of response is variable
– Not generally used in type 2 disease
– Dosage 0.3 µg/kg q 12 hr IV
Factor VIII concentrate (Intermediate purity)
– Virally inactivated product
– The treatment of choice for patients with vWD type III
is virus-inactivated, vWF-containing FVIII concentrates
Disseminated intravascular coagulation (DIC)
DIC is a clinicopathologic syndrome in which widespread intravascular coagulation is
induced by procoagulant that are introduce or produce in circulation and overcome the
natural anticoagulant mechanisms.
DIC may cause tissue ischemia from occlusive microthrombi as well as bleeding from
both consumption of platelet and coagulation factor and anticoagulation effect of product
of secondary fibrinolysis
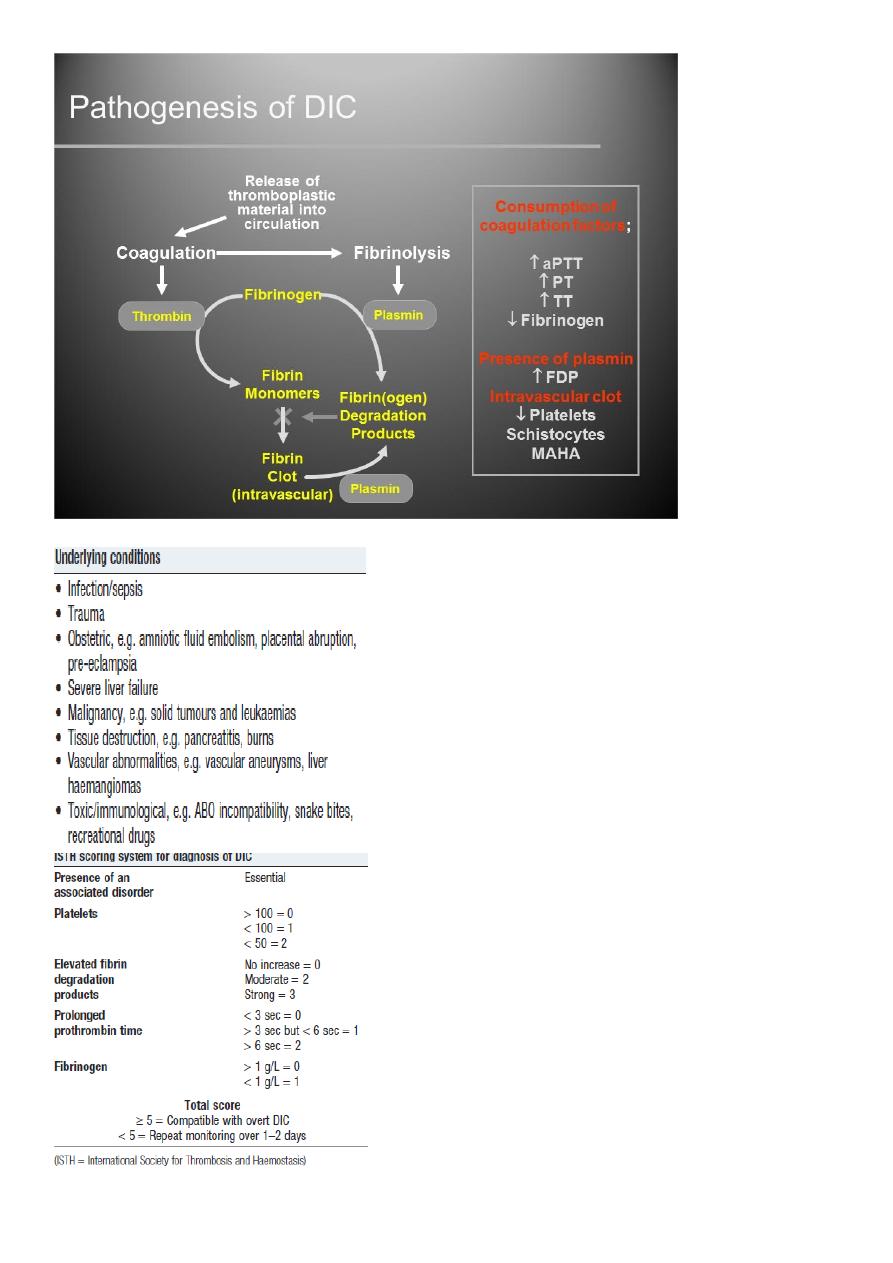
11
DIC

11
Treatment
Treatment of underlying disorder
Platelet transfusion (6-10 U plat (ideally rise to more than 50000-100000
Fresh frozen plasma;1-2 unit For coagulation factor depletion
Hypofibrinogenaemia; 8-10 U cryopercipitate
Anticoagulation with heparin; unless there is a clear contraindication
Coagulation inhibitor concentrate (ATIII)
Patients with DIC should not be treated with antifibrinolytic therapy, e.g.tranexamic
acid.
Thrombotic thrombocytopenic purpura(TTP)
thrombosis is accompanied by paradoxical thrombocytopenia,
TTP is characterised by a pentad of findings, although few patients have all five
components:
• thrombocytopenia
• microangiopathic haemolytic anaemia(MAHA)
• neurological sequelae
• fever
• renal impairment
TTP-Cont.
It is an acute autoimmune disorder mediated by antibodies against ADAMTS-13 (a
disintegrin and metalloproteinase with a thrombospondin type-1 motif).
It is a rare disorder (1 in 750 000 per annum), which may occur alone or in association with
drugs (ticlopidine, ciclosporin), HIV, shiga toxins and malignancy.
It should be treated by emergency plasma exchange.
Corticosteroids, aspirin and rituximab also have a role in management
Untreated mortality rates are 90% in the first 10 days, and even with appropriate
therapy, the mortality rate is 20–30% at 6 months
THROMBOTIC DISORDERS
Virchow’s Triad
Pathogenesis of a Thrombus
Endothelial injury
Abnormal blood flow
Hypercoagulability
Genetic
acquired
12
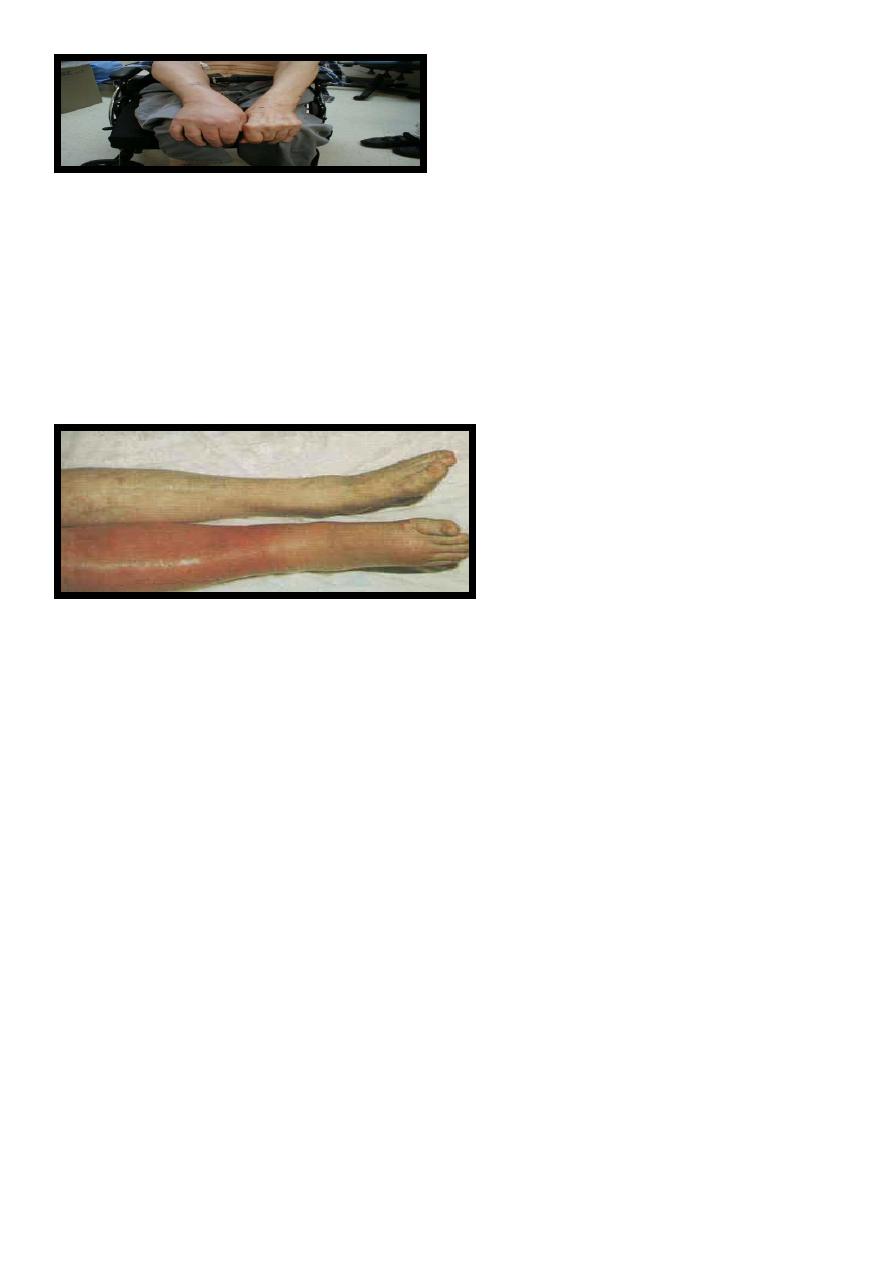
Signs & Symptoms
DVT:
50% with no clinical signs
?Edematous extremity
Plethoric,Warm,Painful extremity
PE:
Cough, SOB, Hemoptysis
Tachycardia
Thrombophilia
Physiologic Inhibitors of
coagulation
Antithrombin
Activated Protein C + protein S
InactivatesVa andVIIIa (via proteolysis)
Thrombomodulin
Binds to thrombin
activate Protein C
Hereditary Thrombophilias
Protein C pathway
Factor V Leiden
Protein C deficiency
Protein S deficiency
Prothrombin G20210A mutation
Antithrombin deficiency
Hyperhomocystinemia
C677T MTHFR mutation

13
Hereditary Thrombophilias
None of them is strongly associated with arterial thrombosis.
• All are associated with a slightly increased incidence of adverse outcome of
pregnancy,including recurrent early fetal loss, but there are no data to indicate that any
specific intervention changes that outcome.
• Apart from in antithrombin deficiency and homozygous factor V Leiden, most carriers of
these genes will never have an episode of VTE; if they do, it will be associated with the
presence of an additional temporary risk factor.
• There is little evidence that detection of these abnormalities predicts recurrence of VTE.
• None of these conditions per se requires treatment with anticoagulants
Antiphospholipid Antibody Syndrome
Autoimmune Acquired Prothrombotic Disorder
Very High Risk for recurrent thromboembolic disease
both venous and arterial
Indefinite duration anticoagulation recommended +/- immunosuppression
Strict Diagnostic Criteria
Clinical criteria (≥1 must be present):
1. Vascular thrombosis:
- ≥ 1clinical episode of, objectively confirmed, arterial, venous, or small vessel thrombosis
2. Pregnancy morbidity:
- ≥ 1 unexplained fetal death @ ≥ 11 weeks EGA
- ≥ 1 premature birth (≤ 34th week of gestation) due to eclampsia, severe pre-eclampsia, or
placental insufficiency
- ≥ 3 unexplained consecutive spontaneous abortions @ <11 weeks EGA
Laboratory criteria (≥1 must be present):
Lupus anticoagulant {LA} (+) ≥ 2 occasions, at least 12 weeks apart, according to ISTH
guidelines:
prolonged aPTT, lack of correction with 1:1 mix, and correction with
Anticardiolipine antibody(ACLA) and/or anti-β2 glycoprotein-I antibody:
medium or high IgG and/or IgM isotype titer ≥ 2 occasions, at least 12 weeks apart
Standardized ELISA assays