1
Forth stage
Surgery
Lec-5
د.نشوان
16/4/2016
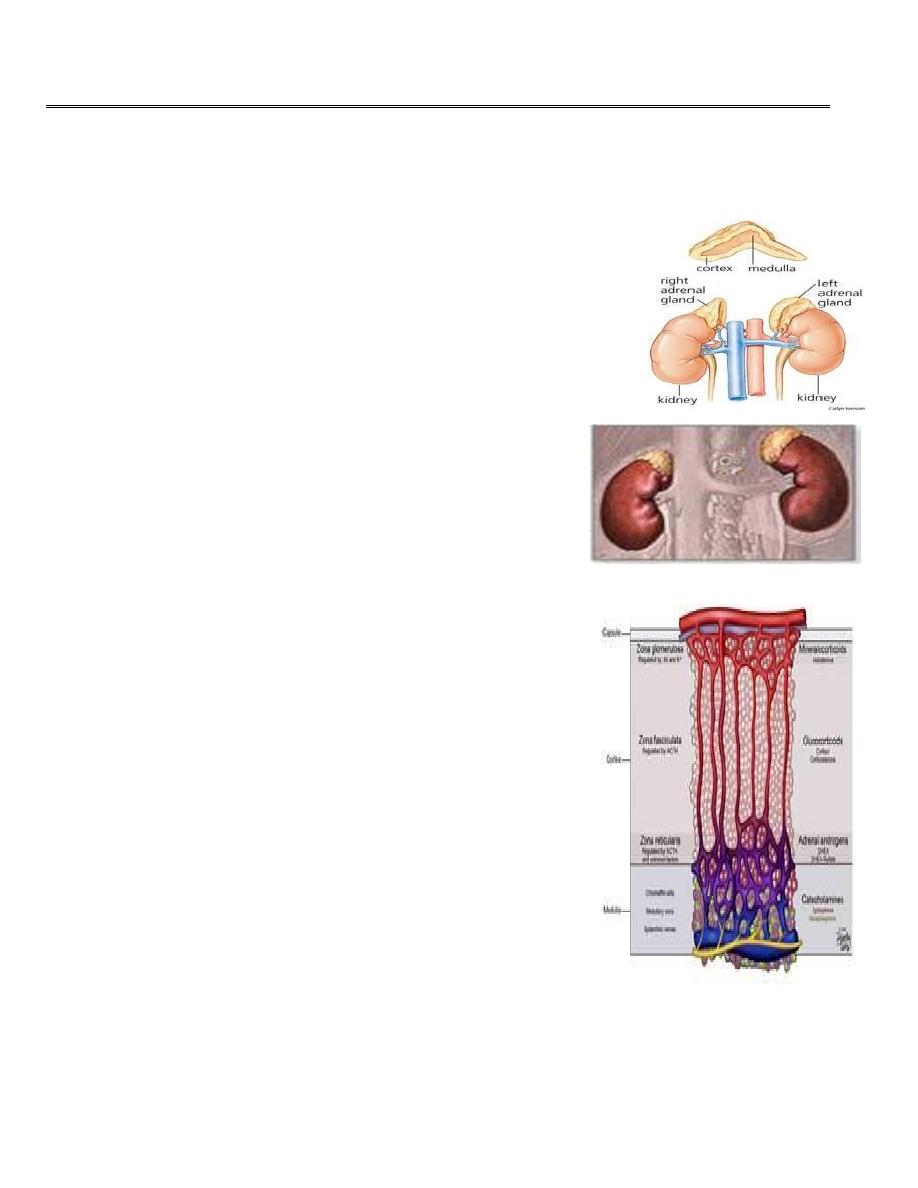
The adrenal glands
The adrenals are pair of glands of about 4 grams situated at
the upper poles of kidneys in the retro peritoneum within
Gerota’s capsule.
The arterial blood supply from the aorta, phrenic and renal
arteries. A large adrenal vein drains on the right side
into the inferior vena cava and on the left side into the
renal vein.
There are two distinct components of the gland the
inner adrenal medulla which is a mesodermal cells and
the outer adrenal cortex which is a neuroectodermal
cells.
The cortex is under control of ACTH from the pituitary
gland which is under control of hypothalamic CRH.
The adrenal cortex compsed of 3 zones
The outer
zona glomerulosa
produce aldosterone, that
regulates
sodium–potassium homeostasis
.
The zona fasciculata
produce Cortisol
that has
numerous metabolic and immunological effects.
the inner
zona reticularis
produce
adrenal androgens
dehydroepiandrosterone
(DHEA)
. Responseble for
secondery sexsual criteria.
2
The adrenal medulla consists of a thin layer of large chromaffin cells, which store
catecholamine granules. It activate the cardiovascular system, resulting in increase blood
pressure and heart rate; vasoconstriction of i the splanchnic vessels and vasodilatation of
muscles vessels; bronchodilatation; and increased glycogenolysis in liver and muscles.
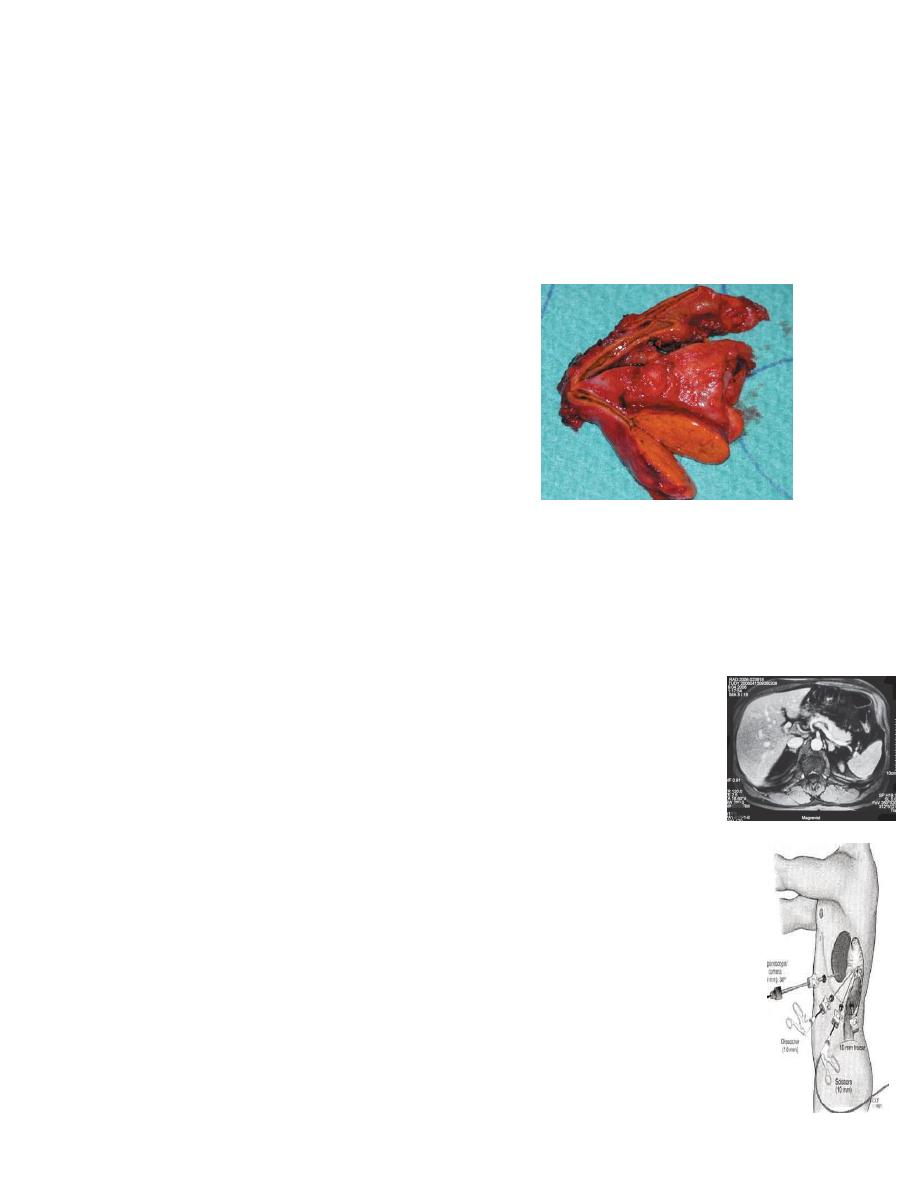
Primary hyperaldosteronism (Conn’s syndrome):
Primary hyperaldosteronism (PHA) is defined by
hypertension, hypokalaemia and hypersecretion of
aldosterone.
It represent 2% of hypertensive patients.
Its due to:
1- unilateral adrenocortical adenoma 60-80%
2- bilateral micronodular hyperplasia in 20–40%.
Clinical features
Most patients are between 30 and 50 years with a female
predominance.
Apart from hypertension and hypokalaemia, patients complain of
non-specific symptoms: headache, muscle weakness, cramps,
intermittent paralysis, polyuria, polydypsia and nocturia
Investigation
The key of the biochemical diagnosis is the assessment of potassium level
and the aldosterone level.
MRI or CT should be performed to distinguish unilateral from bilateral
disease. Conn’s adenomas usually measure between 1 and 2 cm and are
detected by CT with a sensitivity of 80–90%

3
Treatment
The first-line therapy for PHA with bilateral hyperplasia is medical treatment with
spironolactone.
Unilateral laparoscopic adrenalectomy is an effective therapy in patients with clear
evidence of unilateral disease which can be done by traditional way.
Cushing’s syndrome
Hyper secretion of cortisol caused by endogenous production or
excessive use of corticosteroids.
It either ACTH-dependent (85%) or ACTH-independent(15% )in origin.
The most common cause of ACTH-dependent Cushing’s syndrome is Cushing’s disease
resulting from a pituitary adenoma secreting ACTH.
Or due to Ectopic ACTH-producing tumours (small cell lung cancer, foregut carcinoid)
ACTH-independent Cushing’s syndrome (low ACTH levels) is caused by
1- Unilateral adrenocortical adenoma.
2- Adrenocortical carcinoma .
3- Bilateral macronodular or micronodular hyperplasia
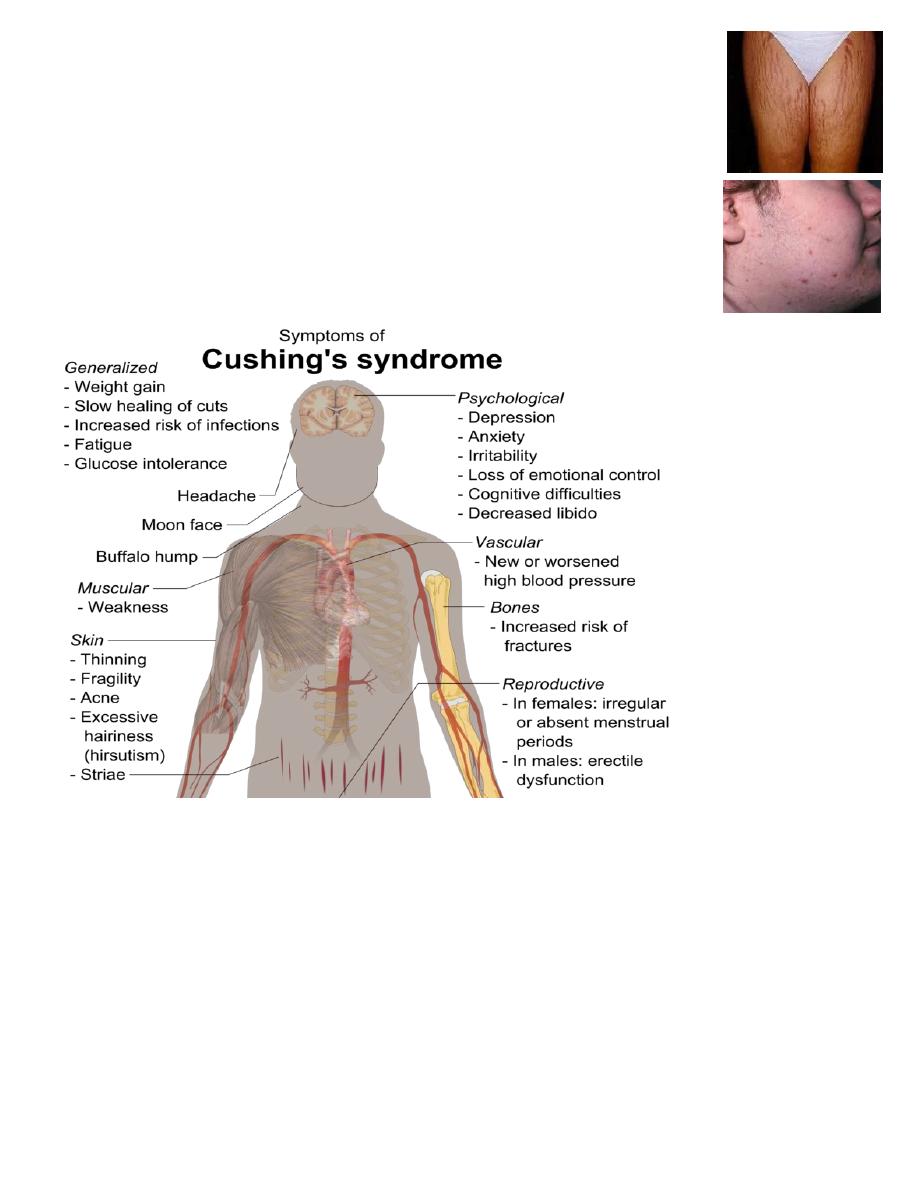
Clinical features of Cushing’s syndrome
Weight gain – particularly around the belly.
"Moon face" – a rounded shape of the face.
Easy bruising skin, with thinning of the skin.
Acne volgaris.
Ruddy complexion (plethora) – a reddening of the face or cheeks.
"Buffalo hump" a mound of fat at the back of the neck.
Abnormal hair growth on the face or abdomen.
4
Edema due to excess fluid buildup in the lower legs.
Stretch marks(purple striae) at lower abdomen and thigh.
Muscle weakness the arms and legs may become skinny like twigs
from muscle wasting.
Menstrual disturbances.
Osteoporoses. Hypertension, Diabetes, Mood changes.
Hypokalaemia.
Investigations
1- Morning and midnight plasma cortisol levels are elevated, possibly with loss of diurnal
rhythm.
2- Dexamethasone fails to suppress 24-hour urinary cortisol excretion.
3- Serum ACTH levels discriminate ACTH-dependent from ACTH-independent disease.
5
Elevated or normal ACTH levels provide evidence for an ACTH producing pituitary tumour
(85%) or ectopic ACTH production.
Therefore, in patients with elevated ACTH, MRI of the pituitary
gland must be performed.
If MRI is negative, a CT scan of the chest and abdomen is warranted to detect an ectopic
cortisol-producing tumour.
In patients with suppressed ACTH levels, a CT or MRI scan is performed
to assess the adrenal glands.
Treatment
Medical therapy with metyrapone or ketoconazole reduces steroid
synthesis and secretion and is used in patients with severe
hypercortisolism or if surgery is not possible.
ACTH-producing pituitary tumours are treated by trans-sphenoidal
resection or radiotherapy.
If an ectopic ACTH source is localized, resection will cure
hypercortisolism.
Patients with an ectopic ACTH-dependent Cushing’s syndrome and an
irresectable or un localized primary tumor treated by bilateral
adrenalectomy.
A unilateral adenoma is treated by adrenalectomy.
In cases of bilateral ACTH-independent disease, bilateral adrenalectomy is the primary
treatment
Preoperative management
Patients with Cushing’s syndrome are at an increase risk of
1-Hospital-acquired infection.
2- Thromboembolic condition.
3- Myocardial complications.
6
4- Post operative Addison crises
Therefore, prophylactic anti-coagulation and the use of prophylactic antibiotics are essential
with post operative steroid supplement.
Postoperative management
Supplemental cortisol should be given after surgery. In total, 15 mg/ h IV for the first 12
hours followed by a daily dose of 100 mg for 3 days, which is gradually reduced
thereafter.
After unilateral adrenalectomy, the contralateral suppressed gland needs up to 1 year to
recover adequate function.
In 10% of patients with Cushing’s disease who undergo a bilateral adrenalectomy, the
pituitary adenoma converts into an aggressive tumor (Nelson’s syndrome).
Congenital adrenal hyperplasia (adrenogenital syndrome) (CAH)
This is an autosomal recessive disorder with incidence of 1 in
5000 live births, caused by a variety of enzymatic defects in the
synthetic pathway of cortisol and other steroids from Cholesterol.
Virilisation and adrenal insufficiency in children are
pathognomonic of congenital adrenal hyperplasia.
CAH may present in girls at birth with ambiguous genitalia.
Hypertension and short stature, caused by the premature
epiphyseal plate closure, are common symptoms.
Affected patients are treated by replacement of cortisol and
with fludrocortisone.
Adrenal insufficiency
Primary adrenal insufficiency is caused by the loss of function of the adrenal cortex.
Symptoms are only evident when about 90% of the adrenal cortex is destroyed.

7
Diseases associated with adrenal insufficiency:
■ Tuberculosis
■ After bilateral adrenalectomy
■ Hemorrhage into the glands
■ secondary metastases
■ Systemic diseases (Boeck’s disease, amyloidosis, Wilson’s disease)
■ Adrenogenital syndrome
■ HIV infection
■ Polyglandular autoimmune syndrome
Secondary adrenal insufficiency is defined as a deficiency of pituitary ACTH secretion.
Tertiary adrenal deficiency is due to loss of hypothalamic CRH secretion , therapeutic
glucocorticoid administration, brain tumor or irradiation.
Acute adrenal insufficiency
Acute adrenal insufficiency usually presents as shock with fever, nausea,
vomiting, abdominal pain, hypoglycaemia and electrolyte imbalance
(Addisonian crisis).
The Waterhouse–Friderichsen syndrome is a bilateral adrenal infarction
associated with meningococcal sepsis and is rapidly fatal unless immediately
treated.
Treatment
1- Intravenous administration of hydrocortisone, 100 mg every 6 hours, 2- 3 liters of saline is
given in 6 hours under careful cardiovascular monitoring.
3- Concomitant infections, which are frequently present, require aggressive treatment.

8
Chronic adrenal insufficiency
Primary type:
Anorexia, weakness and nausea. Hypotension, hyponatraemia, hyperkalaemia and
hypoglycaemia.
As a result of negative feedback, ACTH level increase and cause hyperpigmentation of
the skin and mucosa.
INVESTEGATION
Basal ACTH levels are found to be high with cortisol levels decreased.
Treatment
Replacement therapy with daily oral hydrocortisone (10 mg m–2 body surface area) and
fludrocortisone (0.1 mg).
To prevent an Addisonian crisis, patients must be aware of the need to adjust the dose
in case of illness or stress.
If patients with adrenal insufficiency are scheduled for surgery, appropriate steroid
cover must be administrated.
Phaeochromocytoma (adrenal paraganglioma)
A tumour of adrenal medulla, derived from chromaffin cells and produces
catecholamines.
It represents 0.1–0.6% of hypertensive patients.
It is known as the ‘10% tumor’ as 10% are inherited, 10% are extra-adrenal, 10% are
malignant, 10% are bilateral and 10% occur in children.
its either hereditary which associated with several tumor syndromes and diagnosed
earlier or sporadic which occur after the fourth decade.

9
Clinical features
The cardinal pictures are headache, palpitations and sweating. Paroxysms may be
precipitated by physical training, general anesthesia and some drugs and agents
(contrast media, tricyclic antidepressive, metoclopramide and opiates).
Hypertension may occur continuously, be intermittent or absent.
Investigation
1- elevation of adrenaline, noradrenaline, metanephrine and nor
metanephrine levels in a 24-hour urine collection.
2- imaging study for the localization of the phaeochromocytoma and/or metastases. MRI is
preferred because contrast media used for CT scans can provoke paroxysms.
FNA biopsy is contraindicated.
Treatment
Laparoscopic resection is the treatment of phaeochromocytoma.
If the tumor is larger than 8–10 cm or radiological signs of malignancy are detected an
open approach should be considered.
Preoperative evaluation:
α- adrenoreceptor blocker (phenoxybenzamine) is used to block catecholamine excess
during surgery.
Additional β- blockade is required if tachycardia or arrhythmias develop; this should not
be introduced until the patient is α-blocked.
10
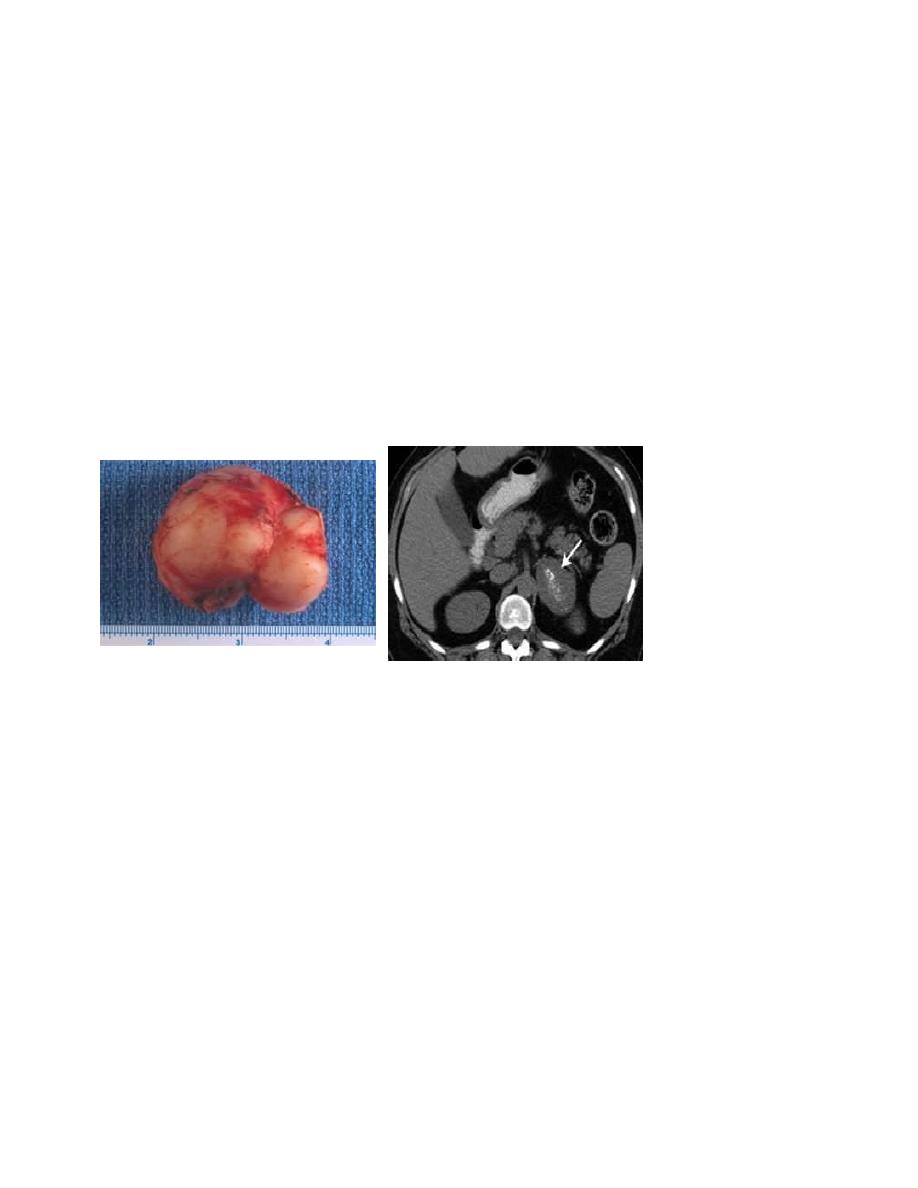
Neuroblastoma
It is a malignant tumor that is derived from the sympathetic nervous system. It arises from
adrenal medulla in (38%).
Clinical features
Predominantly newborn infants and young children (< 5 years of age) are affected.
Symptoms are caused by a mass in the abdomen or by metastases )70%(with proptosis,
bone pain, painless bluish skin secondaries, weakness or paralysis.
Investigation
Urinary excretion (24-hurine) of vanillylmandelic acid (VMA),levels are present in about
80%.
(The catecholamine excess is asymptomatic) .
CT/MRI of the chest and abdomen, a bone scan.
Bone marrow aspiration and core biopsies in suspicions of secondary.
Prognosis depends on the tumor stage and the age at diagnosis. Patients are classified as low,
intermediate or high risk.
Treatment
Low-risk patients are treated by surgery alone (the addition of 6–12 weeks of
chemotherapy is optional) whereas intermediate risk patients are treated by surgery
with adjuvant chemotherapy (carboplatin, cyclophosphamide, etoposide, doxorubicin).
High-risk patients receive high-dose chemotherapy followed by surgical resection in
responding tumors and myeloablative stem cell rescue.
11
Ganglioneuroma
It is a benign adrenal neoplasm arises from neural crest tissue characterized by mature sympathetic ganglion
cells and Schwann cells in a fibrous stroma.
Clinical features
It can be found in all age groups , more common before the age of 60.
it is occur anywhere along the paravertebral sympathetic plexus and in the adrenal
medulla (30%).
Most often they are identified incidentally by CT or MRI performed for other indications.
Treatment
Treatment is by surgical excision.
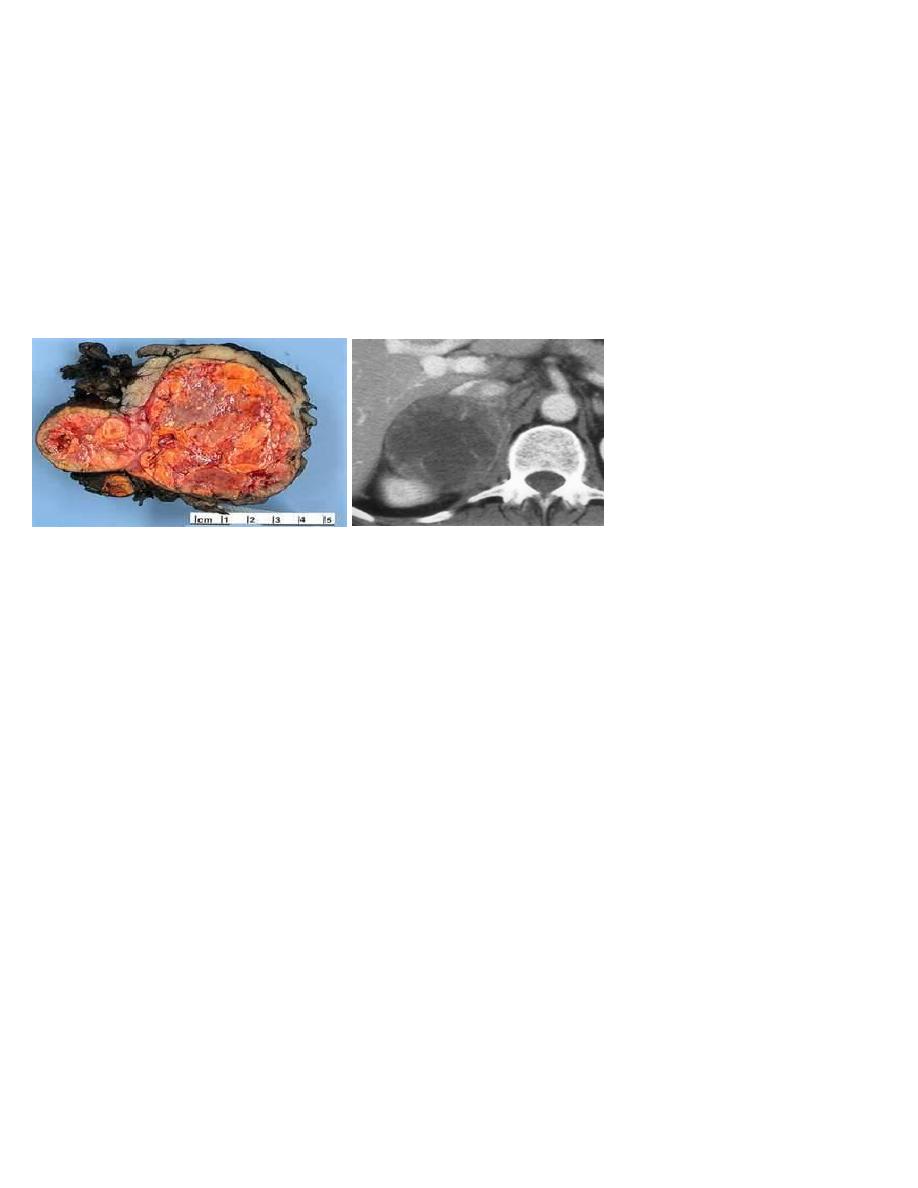
Adrenocortical carcinoma
A rare malignancy with an incidence of 1–2 cases per 1 000 000 with generally poor
prognosis.
A slight female predominance is observed (1.5:1).
The age distribution is bimodal with a first peak in childhood and a second between the
fourth and fifth decades.
Clinical presentation
Approximately 60% of patients present with evidence of steroid hormone excess
(Cushing’s syndrome).
Patients with nonfunctioning tumours complain of abdominal or back pain caused by
large tumours.
12
Diagnosis
1-measurements of DHEAS, cortisol and catecholamines to exclude a
phaeochromocytoma
2- dexamethasone suppression test.
3-MRI and CT are equally effective in distinguishing adrenocortical adenoma from
carcinoma .
Treatment
Complete tumor resection should be attempted whenever possible.
Laparoscopic adrenalectomy is associated with a high incidence of local recurrence and
not recommended.
Tumor debulking plays a role in functioning tumours to control hormone excess.
Patients should be treated postoperatively with mitotane alone or in combination with
other cytotoxic.
Adjuvant radiotherapy may reduce the rate of local recurrence.
Adrenal metastases
Adrenal metastases are discovered at autopsy in one-third of patients with
malignant disease.
The most common primary tumours are breast, lung, renal, gastric, pancreatic, ovarian
and colorectal cancer.
In selected cases an adrenalectomy can be performed.