
Dr. Bilal lec-6
Bronchiectasis
• Bronchiectasis means abnormal dilatation of the bronchi. Chronic suppurative airway infection with
sputum production, progressive scarring and lung damage are present, whatever the cause.
• Aetiology and pathogenesis
• Bronchiectasis may result from a congenital defect affecting airway ion transport or ciliary function,
such as cystic fibrosis, or be acquired secondary to damage to the airways by a destructive infection,
inhaled toxin or foreign body.
• The result is chronic inflammation and infection in airways,of which tuberculosis is the most
commonworld-wide.
• Localised bronchiectasis may occur due to the accumulation of pus beyond an obstructing
bronchial lesion, such as enlarged tuberculous hilar lymph nodes, a bronchial tumour or an inhaled
foreign body (e.g. an aspirated peanut).
•
Pathology
• The bronchiectatic cavities may be lined by granulation tissue, squamous epithelium or normal ciliated
epithelium.
• There may also be inflammatory changes in the deeper layers of the bronchial wall and hypertrophy of
the bronchial arteries.
• Chronic inflammatory and fibrotic changes are usually found in the surrounding lung tissue, resulting in
progressive destruction of the normal lung architecture in advanced cases.
• Causes of bronchiectasis
• Congenital
• Cystic fibrosis • Ciliary dysfunction syndromes
• Primary ciliary dyskinesia (immotile cilia syndrome)
• Kartagener'ssyndrome (sinusitis and transposition of the viscera)
• Primary hypogammaglobulinaemia
• Acquired: children
• Pneumonia (complicating whooping cough or measles) • Primary TB
• Inhaled foreign body

• Acquired: adults
• Suppurative pneumonia • Pulmonary TB
• Allergic bronchopulmonary aspergillosis complicating asthma • Bronchial tumours
• Clinical features
• Symptoms of bronchiectasis
• Cough
• Chronic productive cough due to accumulation of pus in dilated bronchi; usually worse in mornings and
often brought on by changes of posture. Sputum often copious and persistently purulent in advanced
disease. Halitosis is a common accompanying feature
• Pneumonia and pleurisy
• Due to inflammatory changes in lung and pleura surrounding dilated bronchi when spread of infection
occurs: fever, malaise and increased cough and sputum volume, which may be associated with pleurisy.
Recurrent pleurisy in the same site often occurs in bronchiectasis
•
Haemoptysis
• Can be slight or massive and is often recurrent. Usually associated with purulent sputum or an increase
in sputum purulence. Can, however, be the only symptom in so-called 'dry bronchiectasis'
• Poor general health
• When disease is extensive and sputum persistently purulent, there may be associated weight loss,
anorexia, lassitude, low-grade fever, and failure to thrive in children. In these patients, digital clubbing
is common
• Clinical features :Physical signs
• Physical signs in the chest may be unilateral or bilateral.
• If the bronchiectatic airways do not contain secretions and there is no associated lobar collapse, there
are no abnormal physical signs.
• When there are large amounts of sputum in the bronchiectatic spaces, numerous coarse crackles may
be heard over the affected areas.
• Collapse with retained secretions blocking a proximal bronchus may lead to locally diminished breath
sounds, while advanced disease may lead to scarring and overlying bronchial breathing.
• Acute haemoptysisis an important complication of bronchiectasis.
• Investigations
• Bacteriological and mycological examination of sputum In addition to common respiratory pathogens,
sputum culture may reveal Pseudomonas aeruginosa, fungi such as Aspergillus and various mycobacteria.
• Frequent cultures are necessary to ensure appropriate treatment of resistant organisms.
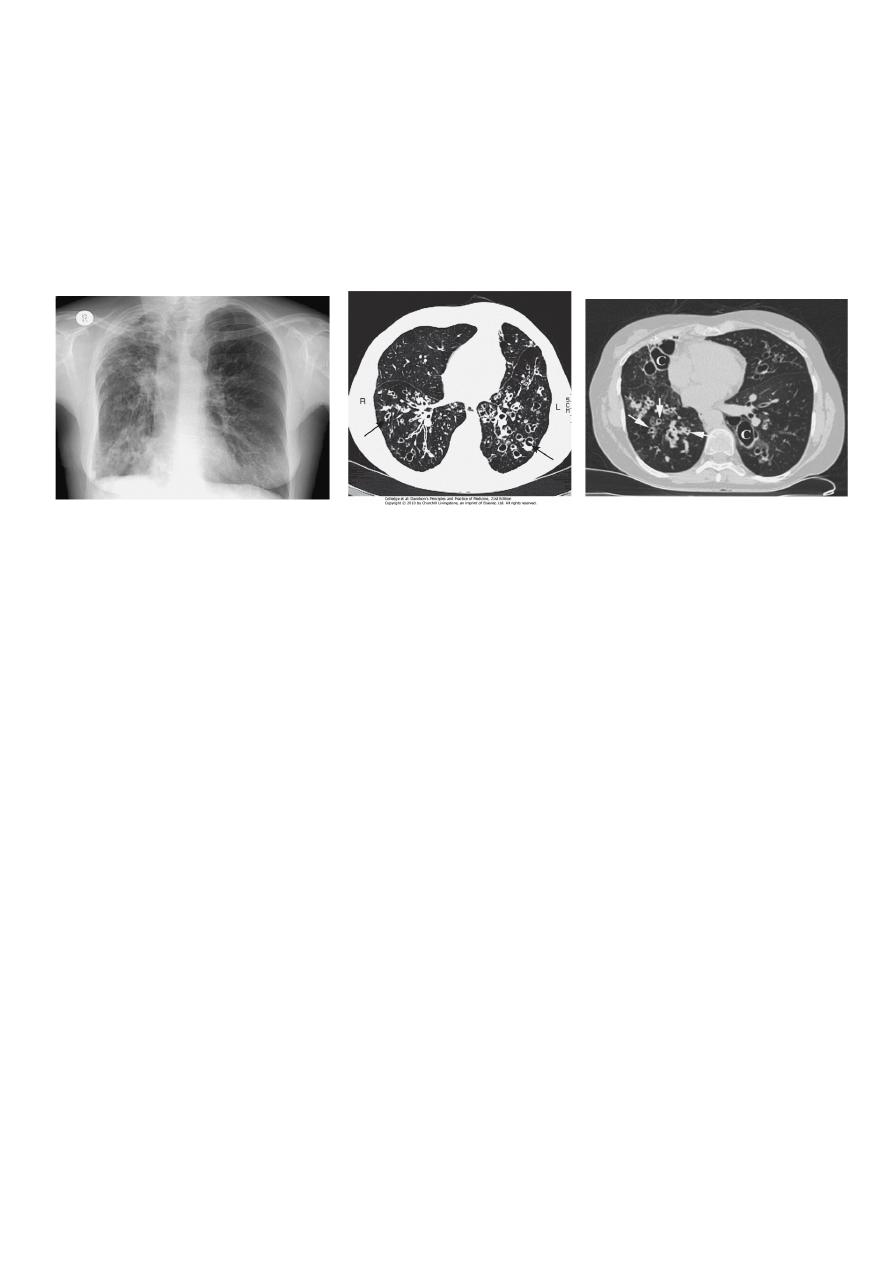
•
Radiological examination
• Bronchiectasis, unless very gross, is not usually apparent on a chest X-ray.
• In advanced disease, thickened airway walls, cystic bronchiectatic spaces, and associated areas of
pneumonic consolidation or collapse may be visible.
• CT is much more sensitive, and shows thickened dilated airways.
• Assessment of ciliary function
• A screening test can be performed in patients suspected of having a ciliary dysfunction syndrome by
measuring the time taken for a small pellet of saccharin placed in the anterior chamber of the nose to
reach the pharynx, when the patient cantaste it.
• This time should not exceed 20 minutes but is greatly prolonged in patients with ciliary
dysfunction.
• Ciliary beat frequency may also be assessed using biopsies taken from the nose.
• Structural abnormalities of cilia can be detected by electron microscopy.
• Management
• In patients with airflow obstruction, inhaledbronchodilators and corticosteroids should be used to
enhance airway patency.
• Physiotherapy
• Patients should be instructed on how to perform regular daily physiotherapy to assist the drainage of
excess bronchial secretions.
• Efficiently executed, this is of great value both in reducing the amount of cough and sputum and in
preventing recurrent episodes of bronchopulmonary infection.
• Patients should adopt a position in which the lobe to be drained is uppermost.
• Deep breathing followed by forced expiratory manuvres (the 'active cycle of breathing' technique) is of
help in moving secretions in the dilated bronchi towards the trachea, from which they can be cleared by

vigorous coughing.
•
'Percussion'
of the chest wall with cupped hands may help to dislodge sputum, but does not suit all
patients.
• Devices which increase airway pressure, either by a constant amount (positive expiratory pressure
mask) or in an oscillatory manner (flutter valve), aid sputum clearance in some patients, and a variety of
techniques should be tried to find one that suits the individual.
• The optimum duration and frequency of physiotherapy depend on the amount of sputum,
but 5-10 minutes once or twice daily is a minimum for most patients
• Antibiotic therapy
• For most patients with bronchiectasis, the appropriate antibiotics are the same as those used in COPD;
however, in general, larger doses and longer courses are required, while resolution of symptoms is often
incomplete.
• When secondary infection occurs with staphylococci and Gram-negative bacilli, in particular
Pseudomonas species, antibiotic therapy becomes more challenging and should be guided by the
microbiological sensitivities.
• For Pseudomonas, oral ciprofloxacin (250-750 mg 12-hourly) or ceftazidime by intravenous injection or
infusion (1-2 g 8-hourly) may be required.
• Haemoptysis in bronchiectasis often responds to treating the underlying infection, although in severe
cases
percutaneous embolisation of the bronchial circulation by an interventional radiologist may be
necessary.
• Surgical treatment
• Excision of bronchiectatic areas is only indicated in a small proportion of cases. These are usually
young patients in whom the bronchiectasis is unilateral and confined to a single lobe or segment on
CT.
• Unfortunately, many of the patients in whom medical treatment proves unsuccessful are also
unsuitable for surgery because of either extensive bronchiectasis or coexisting chronic lung disease.
• In progressive forms of bronchiectasis, resection of destroyed areas of lung which are acting as a
reservoir of infection should only be considered as a last resort.
• Prognosis
• The disease is progressive when associated with ciliarydysfunction and cystic fibrosis, and
eventually causes respiratory failure.
• In other patients the prognosis can be relatively good if physiotherapy is performed regularly and
antibiotics are used aggressively.
•
Prevention
• As bronchiectasis commonly starts in childhood following measles, whooping cough or a primary
tuberculousinfection, it is essential that these conditions receive adequate prophylaxis and
treatment.
• The early recognition and treatment of bronchial obstruction is also important.
.............................................................................
• Cystic fibrosis
•
Genetics, pathogenesis and epidemiology
• Cystic fibrosis (CF) is the most common fatal genetic disease in Caucasians, with autosomal
recessive inheritance, a carrier rate of 1 in 25 and an incidence of about 1 in 2500 live births.
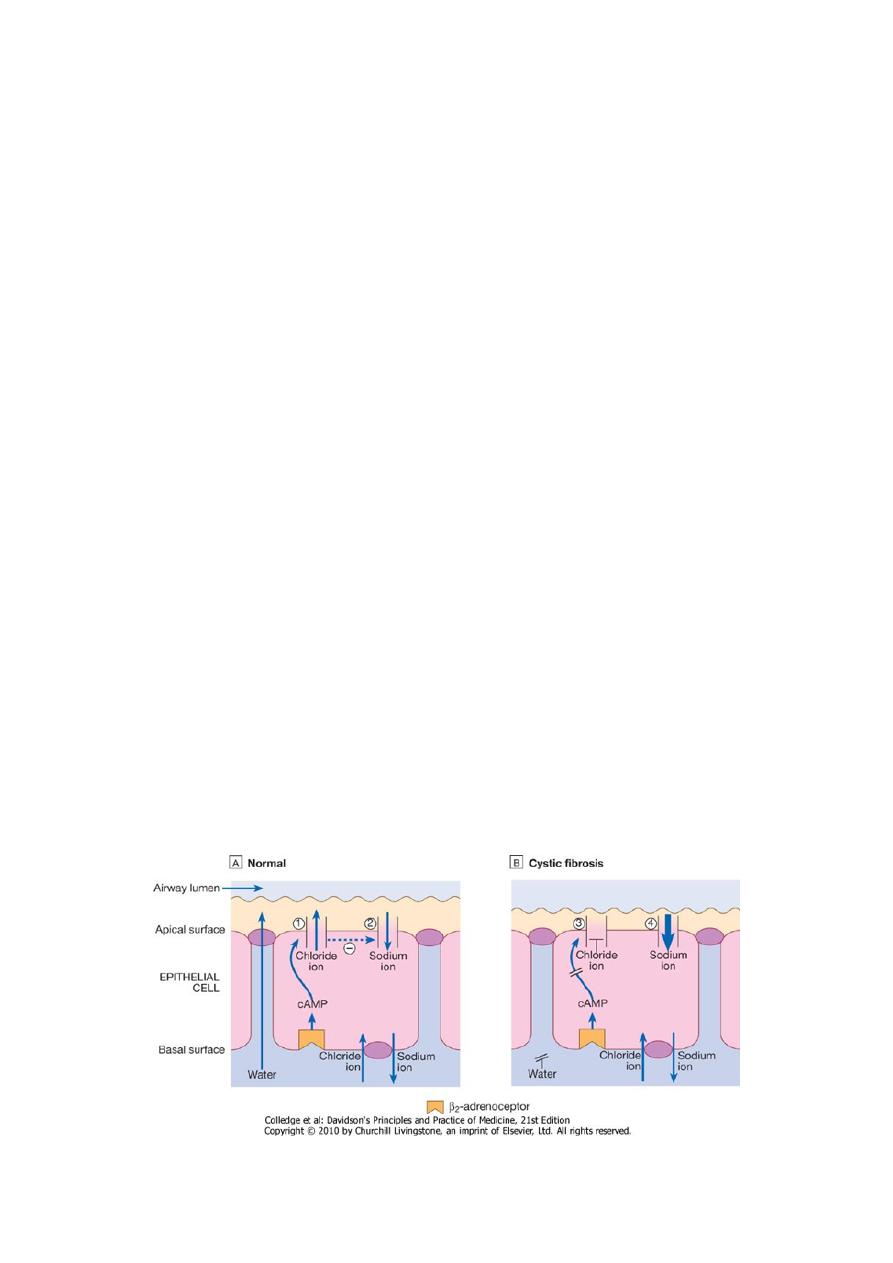
• CF is the result of mutations affecting a gene on the long arm of chromosome 7 which codes for a
chloride channel known as cystic fibrosis transmembrane conductance regulator (CFTR), that
influences salt and water movement across epithelial cell membranes.
• The most common CFTR mutation in northern European and American populations is ΔF508, but
over one thousand mutations of this gene have now been identified.
• The genetic defect causes increased sodium and chloride content in sweat and increased
resorption of sodium and water from respiratory epithelium.
• Relative dehydration of the airway epithelium is thought to predispose to chronic bacterial
infection and ciliary dysfunction, leading to bronchiectasis.
•
The gene defect also causes disorders in the gut epithelium, pancreas, liver and reproductive tract
• In the 1960s, few patients with CF survived childhood, yet with aggressive treatment of airway
infection and nutritional support, life expectancy has improved dramatically, such that there are
now more adults than children with CF in many developed countries.
• Until recently, the diagnosis was most commonly made from the clinical picture (bowel
obstruction, failure to thrive, steatorrhoea and/or chest symptoms in a young child) supported by
sweat electrolyte testing (greater than 60 mEq/L on at least two occasions) and genotyping.
•
Patients with unusual phenotypes were commonly missed, however, and late diagnosis led to
poorer outcomes.
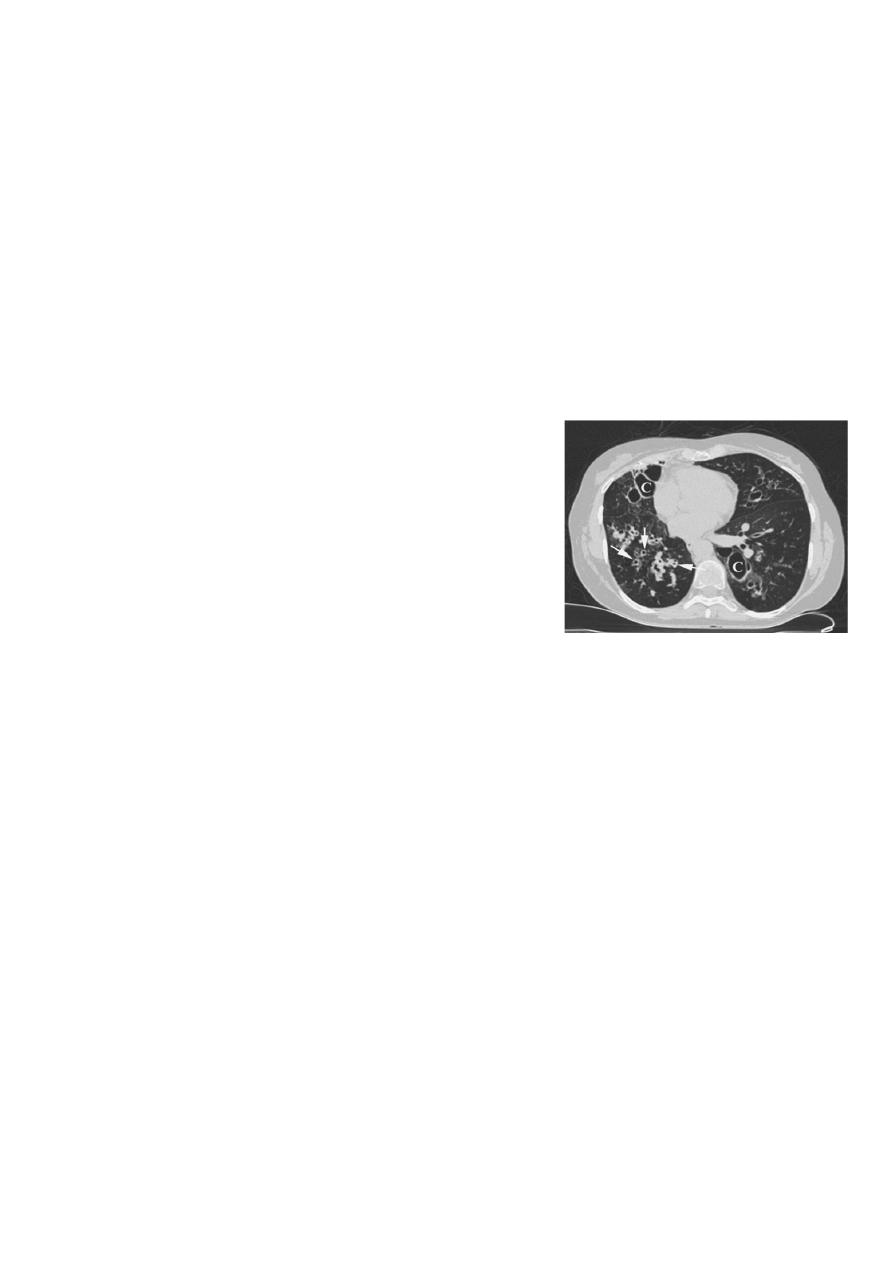
• Neonatal screening for CF using immunoreactivetrypsin and genetic testing of newborn blood
samples is now routine in the UK, and should reduce delayed diagnosis and improve outcomes.
• Prenatal screening by amniocentesis may be offeredto those known to be at high risk
• Clinical features
• The lungs are macroscopically normal at birth, but bronchiolar inflammation and infections usually
lead to bronchiectasis in childhood.
• At this stage, the lungs are most commonly infected with Staphylococcus aureus; however, many
patients become colonised with Pseudomonas aeruginosa by the time they reach adulthood.
•
Recurrent exacerbations of bronchiectasis, initially in the upper lobes but subsequently
throughout both lungs, cause progressive lung damage resulting ultimately in death from
respiratory failure
• Most men with CF are infertile due to failure of development
of the vas deferens, but microsurgical sperm aspiration and in
vitro fertilisation are now possible.
• Genotype is a poor predictor of disease severity in
individuals; even siblings with matching genotypes may have
quite different phenotypes.
• This suggests that other 'modifier genes', as yet unidentified,
influence clinical outcome.
• Complications of cystic fibrosis
• Respiratory
Others
• Infective exacerbations of • Diabetes (25% of adult)
bronchiectasis
• Spontaneous pneumothorax • Delayed puberty
• Haemoptysis • Male infertility
• Nasal polyps • Stress incontinence du
• Respiratory failure to repeated forced cough
• Cor pulmonale • Psychosocial problems
• Lobar collapse due to secretions • Osteoporosis
• Gastrointestinal
• Arthropathy
• Malabsorption and steatorrhoea • Cutaneous vasculitis
• Distal intestinal obstruction syndrome
• Biliary cirrhosis and portal hypertension

• Gallstones
• Management
• Treatment of CF lung disease
• The management of CF lung disease is that of severe bronchiectasis. All patients with CF who
producesputum should perform regular chest physiotherapy, and should do so more frequently during
exacerbations.
• While infections with Staph. aureus can often be managed with oral antibiotics, intravenous treatment
is usually needed for Pseudomonasspecies.
• Regular nebulised antibiotic therapy (colomycin or tobramycin) is used between exacerbations in an
attempt to suppress chronic Pseudomonasinfection.
• Unfortunately, the bronchi of many CF patients eventually become colonised with pathogens which are
resistant to most antibiotics.
• Resistant strains of P. aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia are the main
culprits, and may require prolonged treatment with unusual combinations of antibiotics. Aspergillus and
'atypical mycobacteria' are also frequently found in the sputum of CF patients.
• Some patients have coexistent asthma, which is treated with inhaled bronchodilators and
corticosteroids; allergic bronchopulmonary aspergillosis also occurs occasionally in CF.
• Three maintenance treatments have been shown to cause modest rises in lung function and/or to
reduce the frequency of chest exacerbations in CF patients .
Treatments that may reduce chest exacerbations and/or improve lung
function in CF
Therapy Patients treated
Nebulised recombinant Age ≥ 5, FVC > 40%predicted
human DNase 2.5 mg daily
Nebulised tobramycin Patients colonised with Pseudomonas aeruginosa
300 mg 12-hourly, given
in alternate months
Regular oral azithromycin Patients colonised with Pseudomonas aeruginosa
500 mg three times/week

• For advanced CF lung disease, home oxygen and NIV may be necessary to treat respiratory failure.
• Ultimately, lung transplantation can produce dramatic improvements but is limited by donor organ
availability.
• Treatment of non-respiratory manifestations of CF
• There is a clear link between good nutrition and prognosis in CF.
• Malabsorption is treated with oral pancreatic enzyme supplements and vitamins.
• Diabetes eventually appears in over 25% of patients and often requires insulin therapy.
• Osteoporosis secondary to malabsorption and chronic ill health should be sought and treated.
• Somatic gene therapy
• The discovery of the CF gene and the fact that the lethal defect is located in the respiratory
epithelium (which is accessible by inhaled therapy) presents an
exciting opportunity for gene therapy.
• Manufactured normal CF gene can be 'packaged' within a viral or liposome vector and delivered to the
respiratory epithelium to correct the genetic defect.
• Initial trials in the nasal and bronchial epithelium have shown some effect, and further trials of
nebulised bronchial delivery are planned.
• The prognosis:
• CF has greatly improved in the last decade, mainly because of better control of bronchial sepsis and
nutritional support.
• The median survival of patients with CF born in the 1990s is now predicted to be at least 40 years.
........................................
Lung Abscess
•Lung abscess is defined as pulmonary parenchymal necrosis and cavitation resulting from infection.
•The development of a lung abscess implies a high microorganism burden as well as inadequate microbial
clearance from the airways.
•Aspiration is the most common cause; factors that portend an increased risk of aspiration include
1. esophageal dysmotility,
2. seizure disorders, and
3. neurologic conditions causing bulbar dysfunction.
4. Other predisposing conditions for lung abscess include periodontal disease and
5. alcoholism.
Microbiology
Anaerobic bacteria are the most common causative organisms for lung abscess.
Aerobic or facultative bacteria such as S. aureus, Klebsiella pneumoniae, Nocardia sp., and gramnegative
organisms, as well as nonbacterial pathogens like fungi and parasites, may also cause abscess formation.
In the immunocompromised host, aerobic bacteria and opportunistic pathogens may predominate.
Clinical Manifestations
The symptoms of lung abscess are typical of pulmonary infection in general and may include
cough, purulent sputum production, pleuritic chest pain, fever, and hemoptysis.
and some patients may be asymptomatic.
Physical examination
is often unrevealing. Rales or evidence of consolidation may be present. Fetid
breath and poor dentition may be diagnostic clues. Clubbing or
hypertrophic pulmonary osteoarthropathy may occur in chronic cases.
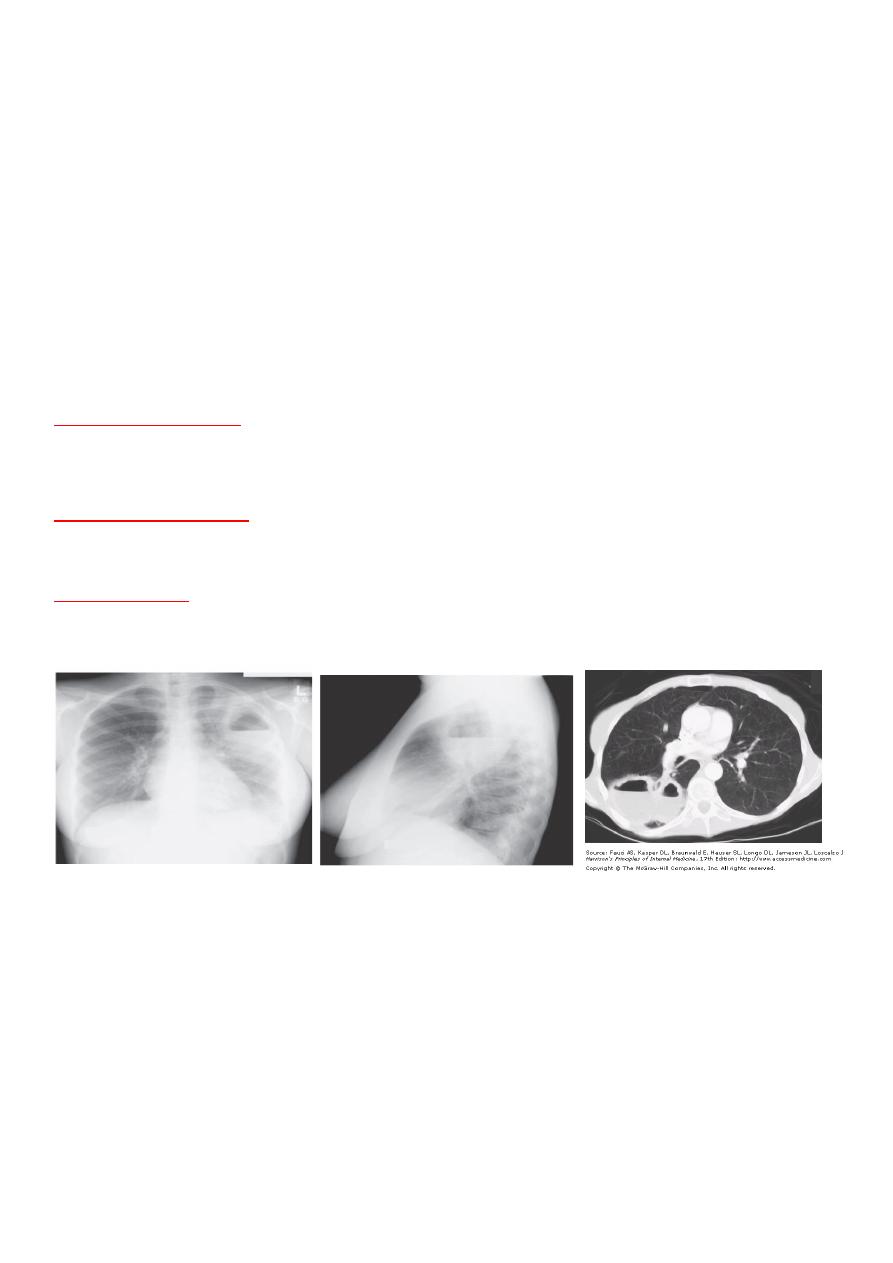
The chest radiograph
classically reveals one or two thickwalled cavities in dependent areas of the
lung, particularly the upper lobes and posterior segments of the lower lobes .An air-fluid level is often
present.
CT of the chest
is helpful in defining the size and location of the abscess, as well as to evaluate for
additional cavities and the presence of pleural disease. Laboratory studies may reveal leukocytosis,
anemia, and an elevated erythrocyte sedimentation rate.
Diagnosis
The diagnosis of lung abscess is based on clinical symptoms, identification of predisposing conditions,
and chest radiographic findings. The differential diagnosis includes mycobacterial infection, pulmonary
sequestration, malignancy, pulmonary infarction, and an infected bulla.
Lung Abscess:
Treatment
For many years, penicillin was the mainstay of empiricantibiotic therapy for lung abscess. Due to the
emergence of -lactamase–producing organisms, clindamycin (150 mg–300 mg every 6 h) is now standard
therapy. Other agents, such as carbapenems and Β-lactam/-

lactamase inhibitor combinations, may be useful in selected cases. Metronidazole alone is associated
with a high treatment failure rate. When possible, the choice of antibiotics should be guided by
microbiologic results. The duration of treatment for lung abscess is controversial. Four to six weeks of
antibiotic therapy is typically employed, though a more
extended course is favored by some experts. Treatment failure suggests the possibility of a
noninfectious etiology. While surgery has had a limited role in treatment in the antibiotic era,
indications include:
1-refractory hemoptysis,
2-inadequate response to medical therapy,
3-or the need for a tissue diagnosis when there is concern for a noninfectious etiology.
In general, outcomes for patients with classic anaerobic lung abscess are favorable, with a 90–95% cure
rate. Higher mortality rates have been reported in
immunocompromised patients, those with significant comorbidities, and infection with P. aeruginosa, S.
aureus, and K. pneumoniae.
by :hazha fatah
Edited by Mohammed Musa