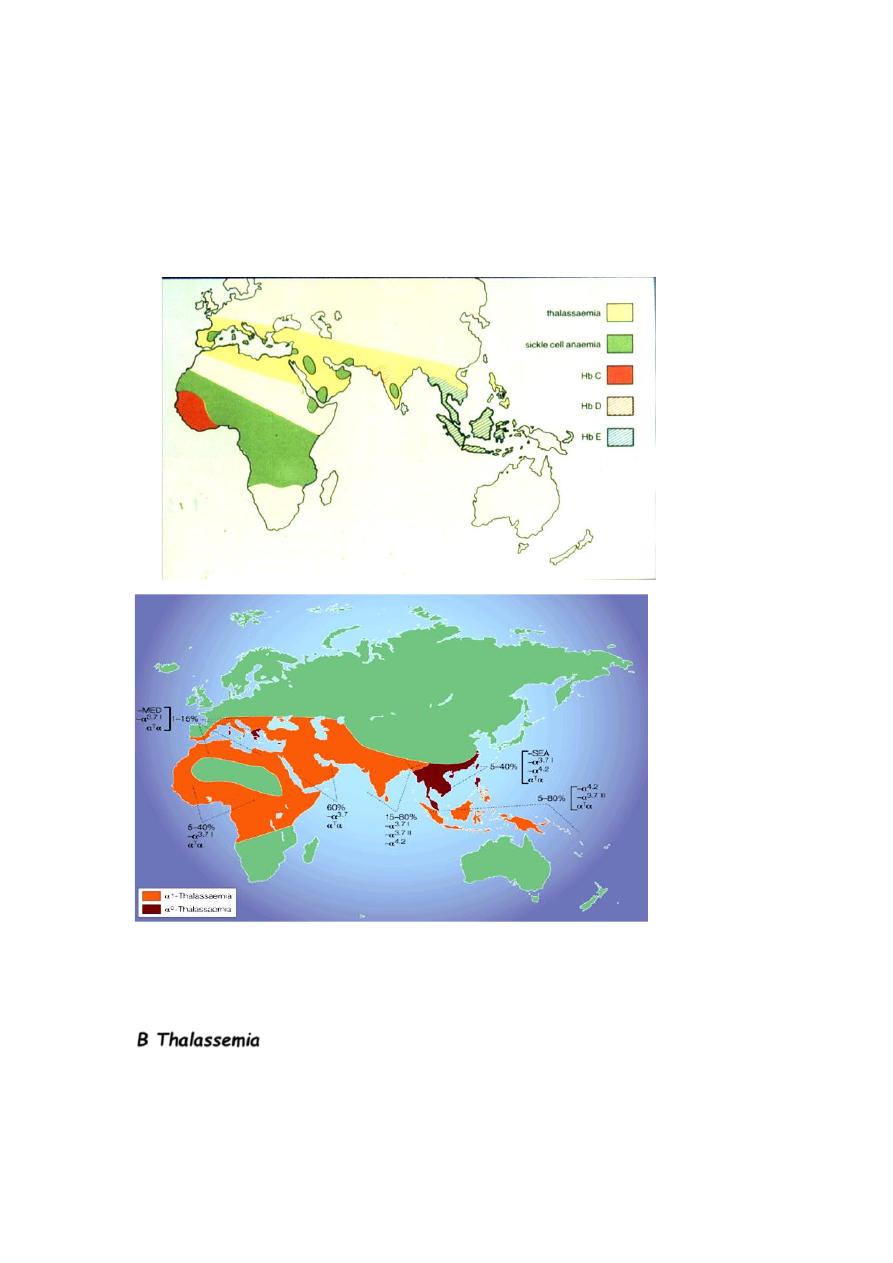
Thalassemia
n
Syndromes arising form decreased rate or absence of globin
chain synthesis.
n
The resulting imbalance-globin chain synthesis takes place,
giving rise to the excess amount of the normally synthesized
globin chain.
Common types of thalassemia
n
-thalassemia
n
-thalassemia
B
Thalassemia
• They are the most important types of thalassemias because they
are so common and usually produce severe anemia in their
homozygous and compound heterozygous states (compound=
when combined with other hemoglobinopathies or thalassemias)

• thalassemias are autosomal inherited disorders of globin
synthesis. In most, globin structure is normal but the rate of
production is reduced because of decrease in transcription of
DNA, abnormal processing of pre-mRNA, or decreased translation
of mRNA leading to decreased Hb-A production (A=Adult).
n Usually and mostly they are caused by gene mutations in the
gene in chromosome# 11, although deletions do occur.
n Hundreds of mutations possible in the globin gene, therefore
thalassemia is more diverse disease in its presentation (the
presentation differs between people depending on the type of
mutation).
n This results in excess alpha chains, because they cannot find
their counterparts (the beta chains) to bind to.
Thalassemia inheritance

β
Thalassemia :The Story in Brief
• ineffective erythropoiesis.
n The end result is an extremely rigid red cell with a shortened survival
(i.e. hemolysis).
n
The anemia is due to two main components:
n
Ineffective erythropoiesis (intramedullary).
n
Extravascular Hemolysis in RES esp. spleen
n
A third component that could contribute for the severity of
anemia is Splenomegaly that may also worsen the anemia,
because of two components: the (1) increased sequestration,
and (2) increased plasma volume caused by the splenomegaly
(dilutional).
• This occurs in utero when embryonic hemoglobins switch to HbF. Also
it occurs postnatal when HbF is switched to HbA.
• Hb switching requires coordination of numerous genetic, cellular and
signaling factors during periods of human development.
At what age could
β
Thalassemia cause its effect???
n In contrast to α globin, β globin is not necessary during fetal life (Hb-F=
α2γ2), thus the onset of
β Thalassemia isn’t apparent until a few
months after birth, when HbF is switched to HbA.
Types of βThalassemia
Three common types of
Thalassemia:
o
Thalassemia: The production of
chain is mildly reduced.
o
Thalassemia: The production of
chain is more reduced than
But NOT ABSENT.
and
are caused by mutation in
Promoter region, 5`UTR, Cap site, Consensus sites, within
Introns, 3`UTR, or Poly A site, and change in coding region.
o
Thalassemia: ABSENCE of
chain production. It is caused by
mutation in Initiation codon, Splicing at junctions, Frameshift,
Nonsense mutation.
Who is at risk?
Ethnic origin is very critical
Classical Clinical Syndromes of
Thalassemia;
thalassemia
can be presented as:
o Silent carrier state – mildest form of thal.
o thalassemia minor - heterozygous disorder resulting in mild
hypochromic, microcytic hemolytic anemia.
o thalassemia intermedia - Severity lies between the minor and
major.
o thalassemia major - homozygous disorder resulting in severe
life long transfusion-dependent hemolytic anemia.
Silent Carrier State for β Thalassemia
• Are various heterozygous (from one parent) β gene mutations that
produce only small decrease in production of β globin chains.
• Patients have nearly normal alpha/beta chain ratio and no hematologic
abnormalities.
• Have normal levels of HbA
2
.
β Thalassemia Minor (Trait)
• Caused by heterozygous (from one parent) mutations that affect β
globin synthesis.
• β Chains production and thus Hb-A production is more reduced
than the silent carrier Hb-A.
• Usually presents as mild, asymptomatic hemolytic anemia unless
patient in under stress such as pregnancy, infection, or folic acid
deficiency.
• Have one normal β gene and one mutated β gene.

•
Hemoglobin level in 10-13 g/dL range with normal or slightly
elevated RBC count
(RCC).
β Thalassemia Minor (Trait)
n Anemia usually hypochromic and microcytic with slight aniso and
poik, including target cells and elliptocytes; also may see
basophilic stippling.
n Rarely see hepatomegaly or splenomegaly.
n Have high HbA
2
levels (3.6-8.0%) and normal to slightly elevated
HbF levels.
n Normally require no treatment.
n You have to make sure are not diagnosed as IDA.
n Mentzer index: <13 (Why?).
β
Thalassemia Minor (Trait)
n 2- 6% HbF (N = < 1% after age 1 year)
n 3.6 - 8% HbA
2
(N = 2.2-3.6%)
n 87 - 95% HbA (N=95-100%)
β Thalassemia Intermedia
n Patients able to maintain minimum Hb (7 g/dL or greater) without
transfusion dependence.
n Expression of disorder falls between thalassemia minor and
thalassemia major.
n We will see increase in both HbA
2
production and HbF
production.
n Peripheral blood smear picture is similar to
thalassemia min
n oHave varying symptoms of anemia, jaundice, splenomegaly and
hepatomegaly.
n Have significant increase in bilirubin levels.

n Anemia usually becomes worse with infections, pregnancy, or
folic acid deficiency.
n May become transfusion dependent.
n Tend to develop iron overloads as result of increased
gastrointestinal absorption.
n
Usually survive into
adulthood.
r.
β Thalassemia Major
n Characterized by very severe microcytic, hypochromic
anemia.
n Detected early in childhood:
n Hb level lies between 2 and 8 g/dL.
n Severe anemia causes marked bone changes due to
expansion of marrow space for increased erythropoiesis
(Epo is increased).
See characteristic changes in skull, long bones, and hand bones.
n Have protrusion upper teeth and Mongoloid facial features.
n Physical growth and development delayed.
n Peripheral blood shows markedly hypochromic, microcytic erythrocytes
with extreme poikilocytosis, such as target cells, teardrop cells
(WHY??) and elliptocytes. See marked basophilic stippling and
numerous NRBCs.
n MCV in range of 50 to 60 fl.
n Retic count seen (2-8%). But low for the degree of anemia. RPI<2.
n Most of Hemoglobin present is Hb F with slight increase in HbA
2
.
n
Regular transfusions usually begin around one year of age and
continue throughout life.
n
Excessive number of transfusions results in tranfusional
hemosiderosis; Without iron chelation, patient develops cardiac
disease, liver cirrhosis, and endocrine deficiencies.
n
Dangers in continuous tranfusion therapy:
n
Development of iron overload.
n
Development of alloimmunization (developing antibodies to
transfused RBCs).
n
Risk of transfusion-transmitted diseases (e.g. hepatitis,
AIDS).
n
Bone marrow transplants may be future treatment, along with
genetic engineering and new drug therapies.
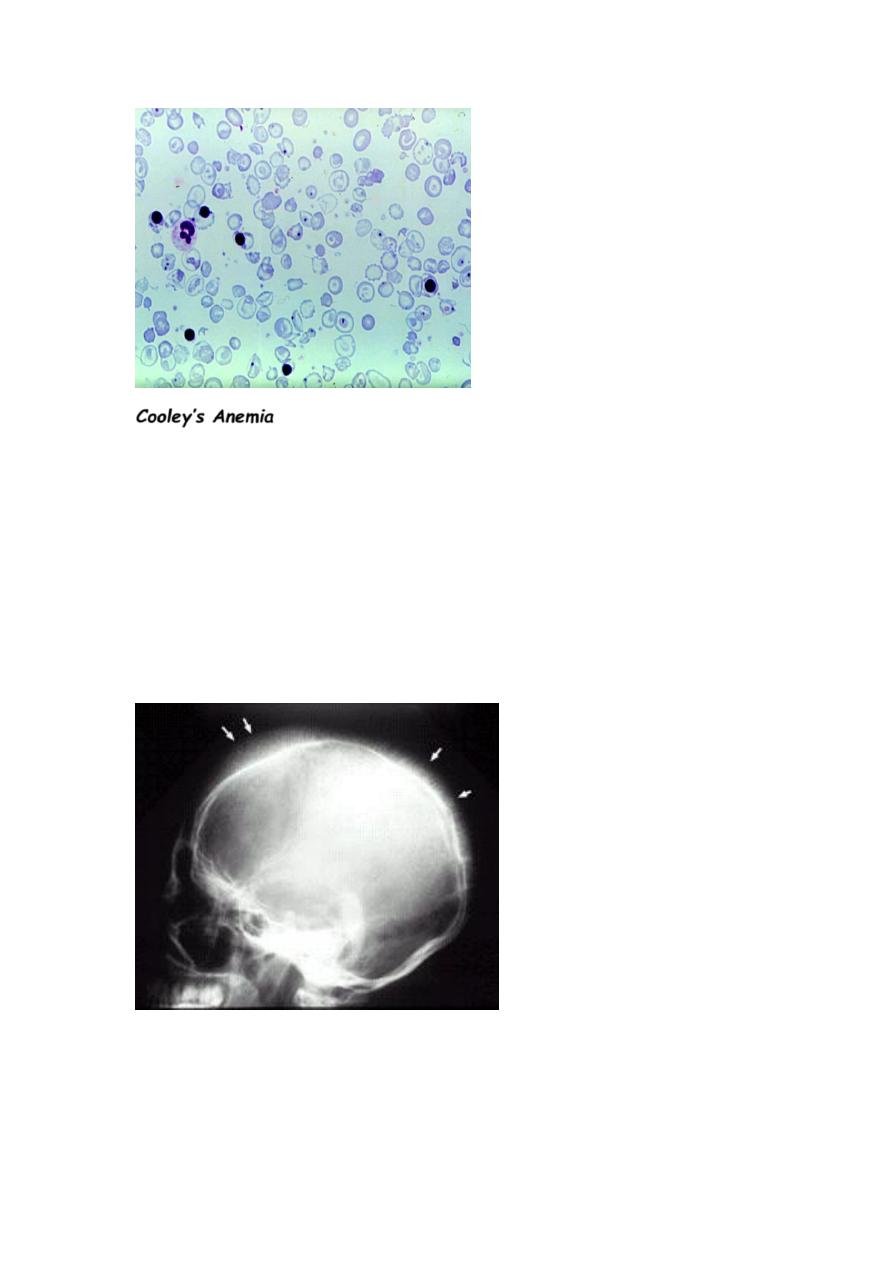
Cooley’s Anemia
n This is another name for β Thalassemia Major, because Cooley
was the first one to describe these cases.
Good point for you to know!
• In iron def. anemia the severity of anemia correlates will with the
degree of microcytosis. This means when the anemia gets more
worse the MCV gets lower and lower.
• While in thalassemia minor either beta or alpha the MCV is out of
proportion with the degree of anemia. This means that the MCV
will be much lower than expected for the minimal reduction in Hb.
Thalassaemia major-life expectancy
•
W
ithout regular transfusion
– Less than 10 years
• With regular transfusion and no or poor iron chelation

– Less than 25 years
• With regular transfusion and good iron chelation
– 40 years, or longer??