
Unit 6: Genetic and Pediatric Diseases
201
GENETIC DISEASES
Nature of genetic abnormalities
contributing to human disease
Mutation
It refers to permanent changes in the DNA.
a- Those that affect germ cells are transmitted to the
progeny and may give rise to inherited diseases.
b- Mutations in somatic cells are not transmitted to the
progeny but are important in the causation of cancers
and some congenital malformations.
Point mutations result from the substitution of a single
nucleotide base by a different base,
Missense mutations it is a point mutation resulting in the
replacement of one amino acid by another in the protein
product. The mutation giving rise to sickle cell anemia is
an excellent example of a point mutation that alters the
meaning of the genetic code
Nonsense" mutation it is point mutation that results in
change an amino acid codon to a chain termination codon,
or stop codon. This mutation will interrupt translation,
and result in truncated proteins that are rapidly degraded.
Frameshift mutations occur when the insertion or
deletion of one or two base pairs alters the reading frame
of the DNA strand.
Trinucleotide repeat mutations belong to a special
category, because these mutations are characterized by
amplification of a sequence of 3 nucleotides.
For example, in fragile X syndrome, prototypical of this
category of disorders, there are 200 to 4000 tandem
repeats of the sequence CGG within a gene called FMR1.
In normal populations, the number of repeats is small,
averaging 29. The expansions of the trinucleotide
sequences prevent normal expression of the FMR1 gene,
thus giving rise to mental retardation.
Another distinguishing feature of trinucleotide repeat
mutations is that they are dynamic (i.e., the degree of
amplification increases during gametogenesis)
Genetic Disorders
Three major categories of genetic disorders:
1) Those related to mutant genes of large effect, sometimes
referred to as mendelian disorders, includes many
uncommon conditions, such as the storage diseases and
inborn errors of metabolism, all resulting from single-
gene mutations of large effect. Most of these conditions
are hereditary and familial.
2) There are heterogeneous group of genetic disorders
that, like mendelian disorders, involve single genes but do
not follow simple mendelian rules of inheritance. These
single-gene disorders with non-classic inheritance include
those resulting from
a) triplet repeat mutations,
b) those arising from mutations in mitochondrial DNA, and
c) Those in which the transmission is influenced by an
epigenetic phenomenon called genomic imprintin.
3) Diseases with multifactorial (polygenic) inheritance,
The second category includes some of the most common
disorders of humans, such as hypertension and diabetes
mellitus. Multifactorial, or polygenic, inheritance implies
that both genetic and environmental influences condition
the expression of a phenotypic characteristic or disease.
4) Those arising from chromosomal aberrations includes
disorders that are the consequence of numeric or
structural abnormalities in the chromosomes.
I- Mendelian disorders (diseases
caused by single-gene defects)
A- Diseases Caused by Mutations in
Structural Proteins
1- Marfan Syndrome
It is an autosomal dominant disorder of connective tissues.
There is Mutations in the FBN1 which is located in
chromosome 15q21 . FBN1 gene encode for Fibrillin 1, is
a glycoprotein secreted by fibroblasts, is the major
component of microfibrils found in the extracellular
matrix. Microfibrils serve as scaffolding for the
deposition of elastin and are considered integral
components of elastic fibers.
Many of the abnormalities in Marfan syndrome can be
explained on the basis of structural failure of connective
tissues, these are:
Skeletal abnormalities are the most obvious feature of
Marfan syndrome.
Patients have a slender, elongated habitus with
abnormally long legs, arms, and fingers (arachnodactyly);
a high-arched palate; and hyperextensibility of joints. A
variety of spinal deformities, such as severe
kyphoscoliosis, may appear. The chest is deformed,
exhibiting either pectus excavatum (i.e., deeply depressed
sternum) or a pigeon-breast deformity.
Unit 6: Genetic and Pediatric Diseases
201
Ocular change is bilateral dislocation, or subluxation, of
the lens owing to weakness of its suspensory ligaments.
Cardiovascular system.
Fragmentation of the elastic fibers in the tunica media of
the aorta predisposes to aneurysmal dilation & aortic
dissection. Death from aortic rupture may occur at any age
and is the most common cause of death.
2- Ehlers-Danlos Syndromes
Ehlers-Danlos syndromes (EDSs) are characterized by
defects in collagen synthesis or structure. All are single-
gene disorders, but the mode of inheritance encompasses
all three of the mendelian patterns.
At least six clinical and genetic variants of EDS are
recognized.
Clinical features
Skin is hyperextensible and joints are, and rupture of
internal organs like colon, cornea, and large arteries.
Wound healing is poor.
B- Diseases Caused by Mutations in
Receptor Proteins or Channels
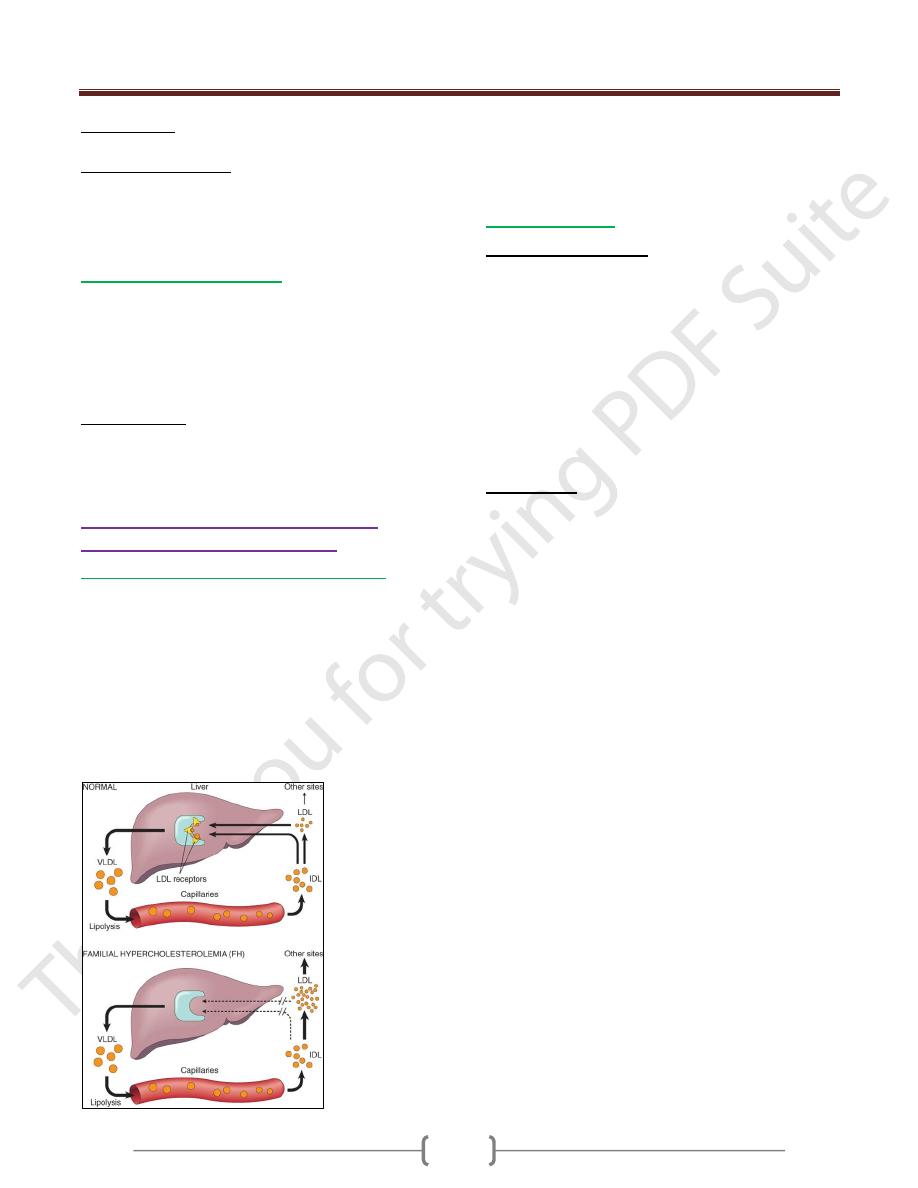
1- Familial Hypercholesterolemia (figure 1)
Familial hypercholesterolemia is an autosomal dominant
disorder caused by mutations in the LDL receptor
gene.Patients develop hypercholesterolemia due to
impaired transport of LDL into the cells.In heterozygotes,
elevated serum cholesterol greatly increases the risk of
atherosclerosis and resultant coronary artery disease;
homozygotes have an even greater increase in serum
cholesterol and occurrence of ischemic heart disease.
Cholesterol also deposits along tendon sheaths to produce
xanthomas.
Figure -1: Low-density lipoprotein (LDL) metabolism & the role
of the liver in its synthesis and catabolism, in normal persons and
those with familial hypercholesterolemia. IDL, intermediate-
density lipoprotein; VLDL, very-low-density lipoprotein.
2- Cystic fibrosis
Cystic fibrosis (CF) is the most common lethal genetic
disease that affects Caucasian populations. It is
uncommon among Asians (1 in 31,000 live births) and
African Americans (1 in 15,000 live births).
CF follows simple autosomal recessive transmission, and
does not affect heterozygote carriers.
It is a widespread disorder of epithelial transport affecting
fluid secretion in exocrine glands& the epithelial lining of
the respiratory, gastrointestinal (GI) & reproductive tracts.
a high level of sodium chloride in the sweat is a consistent
& characteristic biochemical abnormality in CF.
Pathogenesis
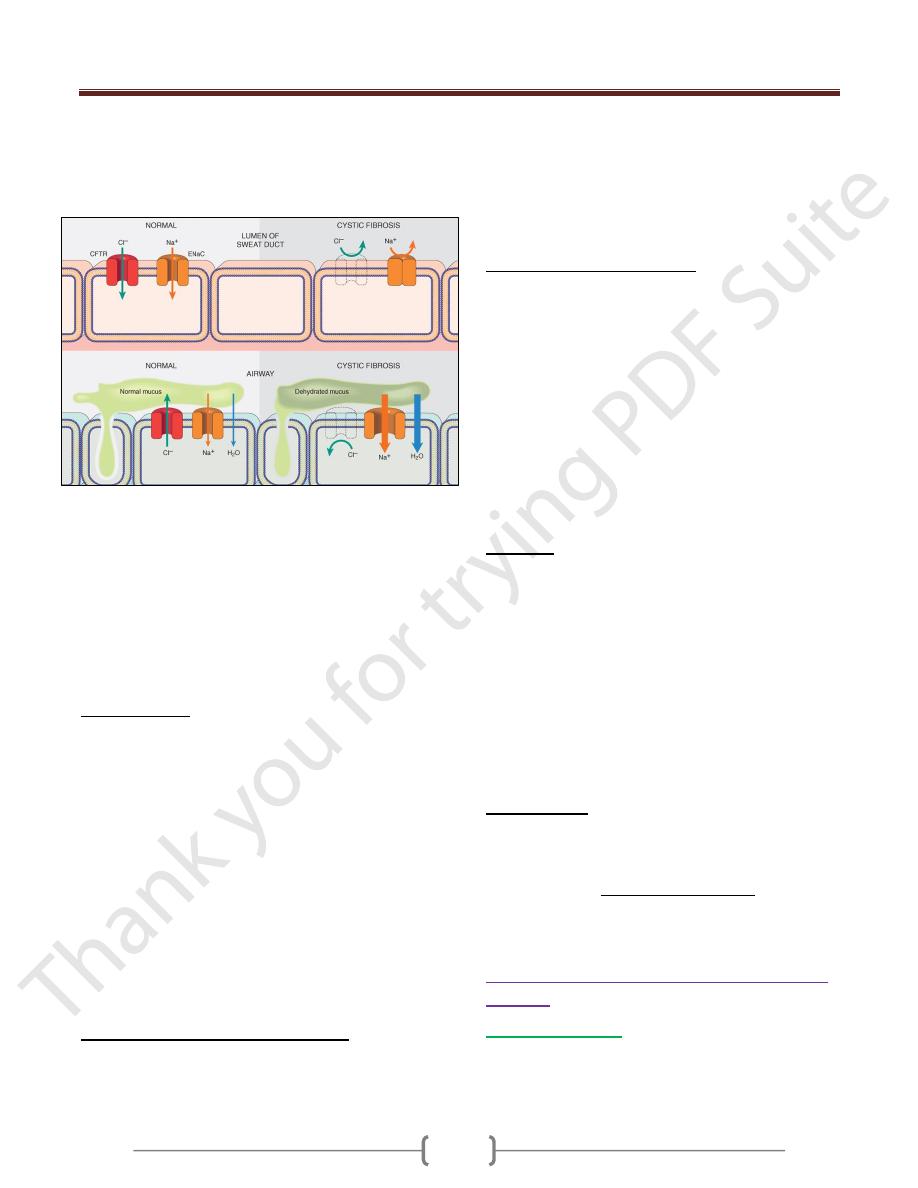
The primary defect in CF is abnormal function of an
epithelial chloride channel protein encoded by the CF
transmembrane conductance regulator (CFTR) gene on
chromosome 7q31.2. The changes in mucus are
considered secondary to the disturbance in transport of
chloride ions.
In normal epithelia the transport of chloride ions across the
cell membrane occurs through transmembrane proteins
such as CFTR that form chloride channels. Mutations in the
CFTR gene render the epithelial membranes relatively
impermeable to chloride ions. However, the impact of this
defect on transport function is tissue-specific. The major
function of the CFTR protein in the sweat gland ducts is to
reabsorb luminal chloride ions and augment sodium
reabsorption. Therefore, in the sweat ducts, loss of CFTR
function leads to decreased reabsorption of sodium
chloride and production of hypertonic sweat.
In contrast to the sweat glands, CFTR in the respiratory &
intestinal epithelium forms one of the most important
avenues for active luminal secretion of chloride. At these
sites, CFTR mutations result in loss or reduction of chloride
secretion into the lumen. Active luminal sodium absorption
is also increased, and both of these ion changes increase
passive water reabsorption from the lumen, lowering the
water content of the surface fluid layer coating mucosal
cells. Thus, unlike the sweat ducts, there is no difference in
the salt concentration of the surface fluid layer coating the
respiratory and intestinal mucosal cells in normal versus
individuals with CF. Instead, the pathogenesis of
respiratory and intestinal complications in CF seems to
Unit 6: Genetic and Pediatric Diseases
201
stem from an isotonic but low-volume surface fluid layer.
In the lungs, this dehydration leads to defective mucociliary
action and the accumulation of concentrated, viscid
secretions that obstruct the air passages and predispose to
recurrent pulmonary infections.
The most common CFTR gene mutation leads to a deletion
(Δ) of 3 nucleotides coding for phenylalanine (F) at amino
acid position 508 (ΔF508). This is an example of a "severe"
mutation. Worldwide, ΔF508 mutation can be found in
approximately 70% of CF patients. Since CF is an
autosomal recessive disease, affected individuals harbor
mutations on both alleles. As discussed later, the
combination of mutations on the two alleles influences the
overall phenotype, as well as organ-specific manifestations
Clinical Course
The symptoms are extremely varied and may be:
1- mild to severe,
2- onset at birth to onset years later,
3- Involvement of one organ system to involvement of many.
Approximately 5% to 10% of the cases come to clinical
attention at birth or soon after because of an attack of
meconium ileus.
Exocrine pancreatic insufficiency occurs in the majority
(85% to 90%) of patients with CF and is associated with
"severe" CFTR mutations on both alleles (e.g.,
ΔF508/ΔF508), whereas
10% to 15% of patients with one "severe" and one "mild"
CFTR mutation, or two "mild" CFTR mutations, retain
sufficient pancreatic exocrine function so as not to require
enzyme supplementation (pancreas-sufficient phenotype).
Pancreatic insufficiency is associated with
1) Malabsorption of protein and fat and increased fecal loss.
Manifestations of malabsorption (e.g., large, foul stools;
abdominal distention; and poor weight gain) appear
during the first year of life. The faulty fat absorption may
induce deficiency states of the fat-soluble vitamins,
resulting in manifestations of avitaminosis A, D, or K.
2) Hypoproteinemia may be severe enough to cause
generalized edema.
3) Persistent diarrhea may result in rectal prolapse in as
many as 10% of children with CF.
Cardiorespiratory complications
o chronic cough,
o persistent lung infections,
o obstructive pulmonary disease,
o & cor pulmonale, are the most common cause of death
By 18 years of age, 80% of patients with classic CF
harbor P. aeruginosa, and 3.5% harbor B. cepacia.
Recurrent sinonasal polyps can occur in as many as 10%
to 25% of patients with CF, and hence, children who
present with this finding should be tested for
abnormalities of sweat chloride.
Significant liver disease occurs late in the natural history
of CF.
Diagnosis
The diagnosis of CF is based on
1) persistently elevated sweat electrolyte concentrations
(often the mother makes the diagnosis because her infant
tastes salty),
2) characteristic clinical findings (sinopulmonary disease
and GI manifestations),
3) or a family history.
4) Sequencing the CFTR gene is of course the "gold standard"
for the diagnosis of CF. Therefore, in patients with clinical
findings or family history (or both) suggesting this
diagnosis, genetic analysis may be warranted.
Manangement
The Advances in management of CF, more patients are
now surviving to adulthood; the median life expectancy
approaches 30 years and continues to increase.
Clinical trials with gene therapy in humans are still in
their early stages but provide a source of encouragement
for millions of CF patients worldwide.
C- Diseases Caused by Mutations in Enzyme
Proteins
1- Phenylketonuria
It is an autosomal recessive disorder of inborn error of
metabolism, which affects 1 in 12,000 live-born
Caucasian infants.
Unit 6: Genetic and Pediatric Diseases
201
The most common form, referred to as classic
phenylketonuria (PKU) have a severe lack of phenylalanine
hydroxylase, leading to hyperphenylalaninemia & PKU. ,
is quite common in persons of Scandinavian descent and is
distinctly uncommon in blacks and Jews.
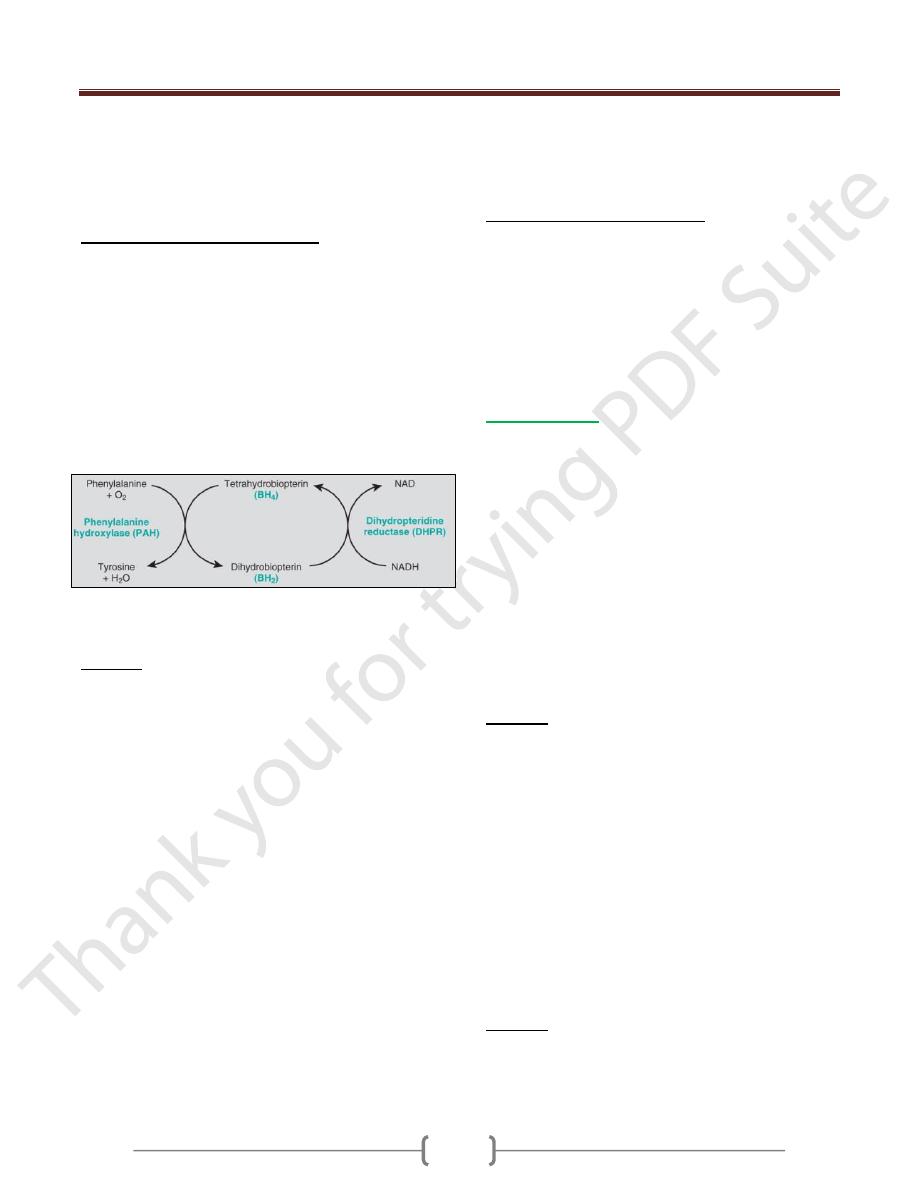
Biochemical abnormalities (Figure 2)
The biochemical abnormality in PKU is an inability to
convert phenylalanine into tyrosine because of severe lack
of phenylalanine hydroxylase.
In normal children, less than 50% of the dietary intake of
phenylalanine is necessary for protein synthesis. The
remainder is converted to tyrosine by the phenylalanine
hydroxylase system.
It is believed that excess phenylalanine or its metabolites
contribute to the brain damage in PKU. Concomitant lack
of tyrosine, a precursor of melanin, is responsible for the
light color of hair and skin.
Figure 2: The phenylalanine hydroxylase system. NAD(H),
Nicotinamide adenine dinucleotide (reduced form).
Clinically
1) Affected infants are normal at birth but within a few
weeks develop a rising plasma phenylalanine level, which
in some way impairs brain development.
2) Usually by 6 months of life severe mental retardation
becomes all too evident; fewer than 4% of untreated
phenylketonuric children have IQs greater than 50 or 60.
3) About one-third of these children are never able to walk,
and two-thirds cannot talk.
4) Seizures, other neurologic abnormalities,
5) decreased pigmentation of hair and skin, and
6) eczema
7) strong musty or mousy odor to affected infants because
when phenylalanine metabolism is blocked as a result of a
lack of phenylalanine hydroxylase, minor shunt pathways
come into play, yielding several intermediates that are
excreted in large amounts in the urine and in the sweat.
Hyperphenylalaninemia and the resultant mental
retardation can be avoided by restriction of phenylalanine
intake early in life. Hence, several screening procedures
are routinely performed to detect PKU in the immediate
postnatal period.
Female PKU patients who discontinue dietary treatment
can give birth to mentally retarded children with
malformations due to transplacental passage of
phenylalanine metabolites.
Non-PKU hyperphenylalaninemia occurs in those with
a partial deficiency of phenylalanine hydroxylase. There
is only modest elevations of phenylalanine levels occur,
and there is no neurologic damage.
It is important to recognize because affected individuals
may test positive in screening tests but do not develop the
stigmata of classic PKU. Measurement of serum
phenylalanine levels is necessary to differentiate non-
PKU hyperphenylalaninemia from PKU.
2- Galactosemia
Galactosemia is an autosomal recessive disorder of
galactose metabolism that affects one in 30,000 live-born
infants.
Normally, lactase splits lactose, the major carbohydrate of
mammalian milk, into glucose
and galactose in the
intestinal microvilli. Galactose is then converted to
glucose in several steps, in one of which the enzyme
galactose-1-phosphate uridyltransferase is required.
Lack of galactose-1-phosphate uridyltransferase
enzyme is responsible for galactosemia. As a result of this
lack of transferase, galactose 1-phosphate and other
metabolites, including galactitol, accumulate in many
tissues, including the liver, spleen, lens of the eye, kidney,
and cerebral cortex.
Clinically
1) Almost from birth, these infants fail to thrive.
2) Vomiting and diarrhea appear within a few days of milk
ingestion.
3) Jaundice and hepatomegaly usually become evident
during the first week of life.
4) Accumulation of galactose and galactose 1-phosphate in
the kidney impairs amino acid transport, resulting in
aminoaciduria.
5) There is an increased frequency of fulminant Escherichia
coli septicemia.
6) Without appropriate dietary therapy, long-term
complications such as cataracts, speech defects,
neurologic deficits, and ovarian failure may occur in
older children and adults.
Diagnosis
The diagnosis is established by assay of the transferase in
leukocytes and erythrocytes. Antenatal diagnosis is
possible by enzyme assays or DNA-based testing of
cultured amniocytes or chorionic villi.
Unit 6: Genetic and Pediatric Diseases
201
Prevention
Most of the clinical and morphologic changes can be
prevented by early removal of galactose from the diet for
at least the first 2 years of life.
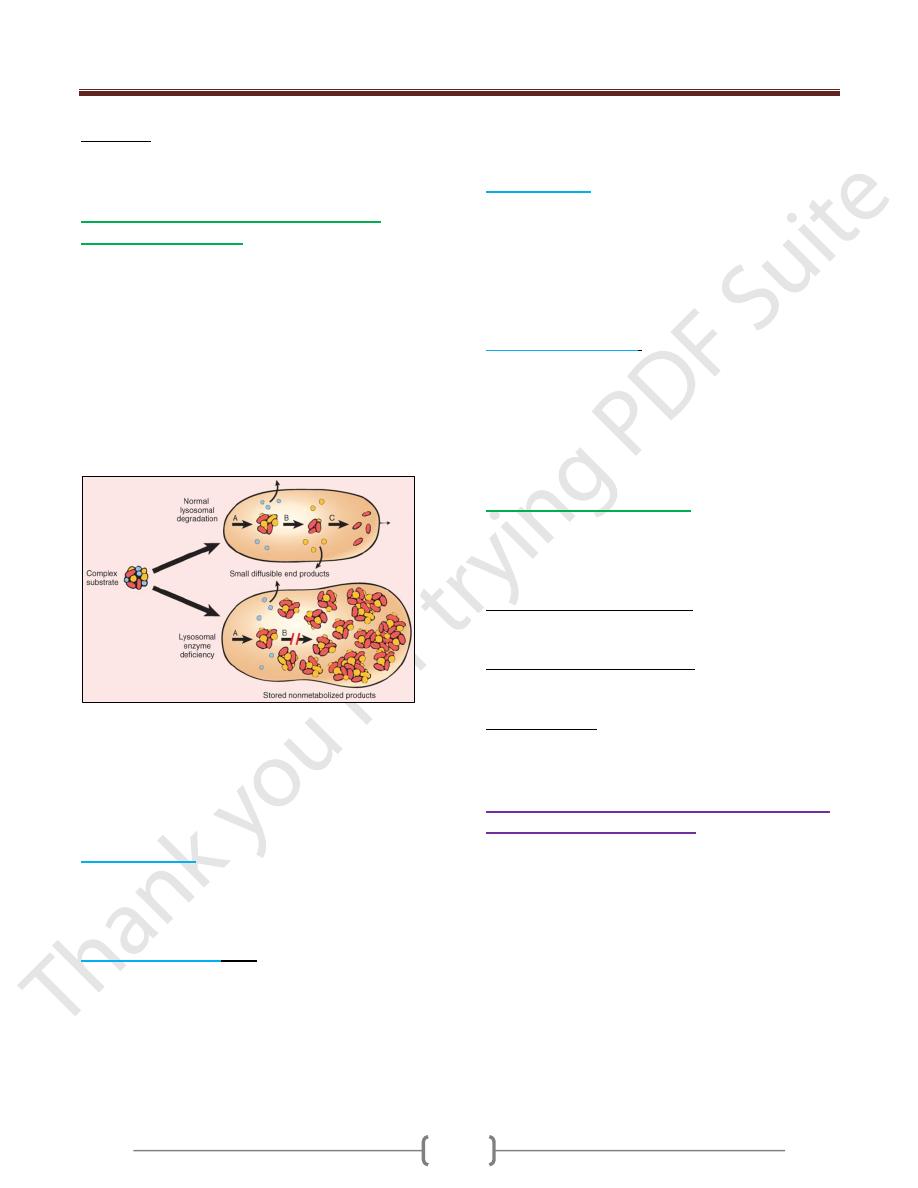
3- Lysosomal Storage Diseases (Autosomal
recessive transmission)
Lysosomes contain a variety of hydrolytic enzymes that
are involved in the breakdown of complex substrates,
such as sphingolipids and mucopolysaccharides, into
soluble end products. These large molecules may be
derived from the turnover of intracellular organelles that
enter the lysosomes by autophagocytosis, or they may be
acquired from outside the cells by phagocytosis.
With an inherited lack of a lysosomal enzyme, catabolism
of its substrate remains incomplete, leading to
accumulation of the partially degraded insoluble
metabolites within the lysosomes (Fig. 3).
Fig.3: Pathogenesis of lysosomal storage diseases. In this
example, a complex substrate is normally degraded by a
series of lysosomal enzymes (A, B, and C) into soluble
end products. If there is a deficiency or malfunction of
one of the enzymes (e.g., B), catabolism is incomplete,
and insoluble intermediates accumulate in the lysosomes.
a) Tay-Sachs disease
is caused by an inability to metabolize
G
M2
gangliosides due to lack of lysosomal
hexosaminidase A. G
M2
gangliosides accumulate in the
CNS and cause severe mental retardation, blindness,
motor weakness, and death by 2-3 years of age.
b) Niemann-Pick disease
types A and B are caused by a
deficiency of sphingomeylinase. In the more severe type
A variant, accumulation of sphingomyelin in the nervous
system results in neuronal damage. Lipid is also stored in
phagocytes within the liver, spleen, bone marrow, and
lymph nodes, causing their enlargement. In type B,
neuronal damage is not present.Niemann-Pick type C
disease is caused by a defect in cholesterol transport and
resultant accumulation of cholesterol and gangliosides in
the nervous system. Affected children have ataxia,
dysarthria, and psychomotor regression.
c) Gaucher disease
results from lack of the lysosomal
enzyme glucosylceramidase and accumulation of
glucosylceramide in mononuclear phagocytic cells. In the
most common, type I variant, affected phagocytes become
enlarged (Gaucher cells) and accumulate in liver, spleen,
and bone marrow, causing hepatosplenomegaly and bone
erosion. Type II and III have variable neuronal
involvement.
d) Mucopolysaccharidoses
result from accumulation of
mucopolysaccharides in many tissues including liver,
spleen, heart, blood vessels, brain, cornea, and joints.
Affected patients in all forms have coarse facial features.
In Hurler syndrome there is corneal clouding, coronary
arterial and valvular depositions, and death in childhood.
Hunter syndrome has a milder course.
4- Glycogen Storage Diseases
Inherited deficiency of enzymes involved in glycogen
metabolism can result in storage of normal or abnormal
forms of glycogen, predominantly in liver or muscles or
in all tissues.
a) Hepatic form (von Gierke disease), liver cells store
glycogen because of a lack of hepatic glucose-6-
phosphatase.
b) There are several myopathic forms, including McArdle
disease, in which muscle phosphorylase lack gives rise to
storage in skeletal muscles and cramps after exercise.
c) In Pompe disease there is lack of lysosomal acid maltase,
and all organs are affected but heart involvement is
predominant.
D- Diseases Caused by Mutations in Proteins
That Regulate Cell Growth
There are two classes of genes, proto-oncogenes and
tumor suppressor genes regulate normal cell growth and
differentiation.
Mutations affecting these genes, most often in somatic
cells, are involved in the pathogenesis of tumors. In
approximately 5% of all cancers, however, mutations
affecting certain tumor suppressor genes are present in all
cells of the body, including germ cells, and hence can be
transmitted to the offspring. These mutant genes
predispose the offspring to hereditary tumors.

Unit 6: Genetic and Pediatric Diseases
201
II- Genetic disorders arising from
chromosomal aberrations (numeric
or structural abnormalities in the
chromosomes) - Cytogenetic disorders
It is estimated that approximately one of 200 newborn
infants has some form of chromosomal abnormality. The
figure is much higher in fetuses that do not survive to
term. It is estimated that in 50% of first-trimester
abortions, the fetus has a chromosomal abnormality.
Cytogenetic disorders may result from alterations in the
number or structure of chromosomes and may affect
autosomes or sex chromosomes.
Numeric Abnormalities
In humans, the normal chromosome count is 46 (i.e., 2n = 46)
euploid. Any exact multiple of the haploid number (n.).
Chromosome numbers such as 3n and 4n are called
polyploid. Polyploidy generally results in a spontaneous
abortion.
aneuploid Any number that is not an exact multiple of n.
The chief cause of aneuploidy is
1- Nondisjunction of a homologous pair of chromosomes at
the first meiotic division or a failure of sister chromatids
to separate during the second meiotic division.
2- Failure of pairing of homologous chromosomes followed
by random assortment (anaphase lag) can also lead to
aneuploidy.
When nondisjunction occurs at the time of meiosis, the
gametes formed have either an extra chromosome (n + 1)
or one less chromosome (n - 1). Fertilization of such
gametes by normal gametes would result in two types of
zygotes: trisomic, with an extra chromosome (2n + 1), or
monosomic (2n - 1).
Monosomy involving an autosome is incompatible with
life, whereas trisomies of certain autosomes & monosomy
involving sex chromosomes are compatible with life
Mosaicism is a term used to describe the presence of two
or more populations of cells in the same individual.
Structural abnormalities
I) Cytogenetic Disorders Involving Autosomes
Trisomy 21 (Down Syndrome)
Is the most common of the chromosomal disorders.
Types of chromosomal abnormalities
1- 95% of affected persons have trisomy 21 die to meiotic
nondisjunction, so their chromosome count is 47. The
parents of such children have a normal karyotype and are
normal in all respects.
Maternal age has a strong influence on the incidence of
Down syndrome. It occurs in 1 in 1550 live births in
women younger than 20 years, in contrast to 1 in 25 live
births in women older than 45 years.
2- 4% of all patients with trisomy 21, the extra chromosomal
material is present not as an extra chromosome but as a
translocation of the long arm of chromosome 21 to
chromosome 22 or 14. Such cases are frequently (but not
always) familial.
3- Approximately 1% of trisomy 21 patients are mosaics,
usually having a mixture of 46- and 47-chromosome cells.
Clinical features of Down syndrome.
1- Epicanthic folds and flat facial profile.
2- Trisomy 21 is a leading cause of mental retardation.
3- 40% of patients with trisomy 21 have cardiac
malformations.
4- Serious infections are another important cause of
morbidity and mortality.
5- The chromosomal imbalance, in some undefined manner,
also increases the person's risk of developing acute
leukemias, particularly acute megakaryocytic leukemia.
Prognosis
1- Improved remarkably in the recent past as a result of
better control of infections. Currently, the median age at
death is 47 years.
2- Most of those who survive into middle age develop
histologic, metabolic, and neurochemical changes of
Alzheimer disease. Many develop frank dementia.
Chromosome 22q11.2 Deletion Syndrome
Chromosome 22q11.2 syndrome encompasses a spectrum
of disorders that result from a small interstitial deletion of
band 11 on the long arm of chromosome 22.

Unit 6: Genetic and Pediatric Diseases
201
The clinical features of this deletion include
1- Congenital heart disease affecting the outflow tracts.
2- Abnormalities of the palate.
3- Facial dysmorphism.
4- Developmental delay.
5- Thymic hypoplasia with impaired T-cell immunity.
6- Parathyroid hypoplasia causing hypocalcemia.
Previously, these clinical features were believed to
represent two different disorders: DiGeorge syndrome
and velocardiofacial syndrome.
1. DiGeorge syndrome (thymic hypoplasia with
diminished T-cell immunity and parathyroid
hypoplasia with hypocalcemia) .
2. velocardiofacial syndrome (congenital heart disease
affecting outflow tracts, facial dysmorphism, and
developmental delay).
Patau syndrome (Trisomy 13)
Trisomy 13 type: 47,xx,+13
Translocation type: 46, xx,+13 (13;14)(q10;q10)
Mosaic 46 xx/47xx=13
Edward Syndrome (Trisomy 18)
Trisomy 18ype: 47,xx,+18
Mosaic 46, xx/47,xx,+18
II) Cytogenetic disorders involving sex chromosomes
Klinefelter Syndrome
This syndrome is best defined as male hypogonadism that
develops when there are at least two X chromosomes and
one or more Y chromosomes.
Abnormalities
1- Most patients are 47,XXY. This karyotype results from
nondisjunction of sex chromosomes during meiosis. The
extra X chromosome may be of either maternal or
paternal origin. Advanced maternal age and a history of
irradiation of either parent may contribute to the meiotic
error resulting in this condition.
2- Approximately 15% of patients show mosaic patterns,
including 46,XY/47,XXY, 47,XXY/48,XXXY, and
variations on this theme.
The presence of a 46,XY line in mosaics is usually
associated with a milder clinical condition.
Clinical manifestation
1- In some it may be expressed only as hypogonadism,
2- Most patients have a distinctive body habitus with an
increase in length between the soles and the pubic bone,
which creates the appearance of an elongated body. Also
characteristic is eunuchoid body habitus.
3- Reduced facial, body, and pubic hair and gynecomastia
are also frequently noted.
4- The testes are markedly reduced in size, sometimes to
only 2 cm in greatest dimension. Along with the testicular
atrophy, the serum testosterone levels are lower than
normal and urinary gonadotropin levels are elevated.
5- Rarely patients are fertile.
6- Klinefelter syndrome may be associated with mental
retardation, the degree of intellectual impairment is
typically mild and in some cases is undetectable.
7- Patients with Klinefelter syndrome have several associated
disorders, such as breast cancer (20 times more common
than in normal males), extragonadal germ cell tumors and
autoimmune diseases such as systemic lupus erythematosus
Turner Syndrome
Turner syndrome, characterized by primary hypogonadism
in phenotypic females, results from partial or complete
monosomy of the short arm of the X chromosome.
Abnormalities
A- The entire X chromosome is missing in 57% of patients,
resulting in a 45,X karyotype. These patients are the most
severely affected, and the diagnosis can often be made at
birth or early in childhood.
Clinical features
1- Significant growth retardation, leading to abnormally
short stature (below third percentile);
2- Swelling of the nape of the neck due to distended
lymphatic channels (in infancy) that is seen as webbing of
the neck in older children;
3- Low posterior hairline;
4- Cubitus valgus (an increase in the carrying angle of the
arms);
5- Shieldlike chest with widely spaced nipples;
6- High-arched palate;
7- Lymphedema of the hands and feet; and a
8- Vriety of congenital malformations such as horseshoe
kidney, bicuspid aortic valve, and coarctation of the aorta.
9- Cardiovascular abnormalities are the most common cause
of death in childhood.
10- In adolescence, affected girls fail to develop normal
secondary sex characteristics; the genitalia remain
infantile, breast development is minimal, and little pubic
hair appears.
11- Most have primary amenorrhea,
12- The mental status of these patients is usually normal
13- Hypothyroidism caused by autoantibodies occurs
especially in women with isochromosome Xp. As many
as 50% of these develop clinical hypothyroidism.

Unit 6: Genetic and Pediatric Diseases
201
Note: In adult patients, a combination of short stature
and primary amenorrhea should prompt strong suspicion
of Turner syndrome. The diagnosis is established by
karyotyping.
B. Approximately 43% of patients with Turner syndrome
either are mosaics (one of the cell lines being 45,X) or
have structural abnormalities of the X chromosome.
The most common is deletion of the small arm.
In contrast to the patients with monosomy X, those who
are mosaics or have deletion variants may have an
almost normal appearance and may present only with
primary amenorrhea.
III- Single-gene disorders with
atypical patterns of inheritance
Three groups of diseases resulting from mutations
affecting single genes do not follow the mendelian rules
of inheritance:
1- Diseases caused by triplet-repeat mutations.
2- Diseases caused by mutations in mitochondrial genes.
3- Diseases associated with genomic imprinting.
1- Triplet-Repeat Mutations:
In all cases, gene functions are altered by an expansion of
the repeats, but the precise threshold at which
premutations are converted to full mutations differs with
each disorder.
The expansion in fragile X syndrome occurs during
oogenesis, in other disorders such as Huntington disease,
premutations are converted to full mutations during
spermatogenesis.
The expansion may involve any part of the gene and can
be grouped into two broad categories, (figure 2)
1- Those that affect untranslated regions (as in fragile X
syndrome) as the mutations affect noncoding regions,
there is "loss of function," since protein synthesis is
suppressed (e.g., FMRP).
2- Coding regions (as in Huntington disease). In which the
mutations involving translated parts of the gene give rise
to abnormal proteins that interfere with function of
normal proteins. Many of these so-called gain-of-function
mutations involve CAG repeats that encode
polyglutamine tracts, and the resultant diseases are
sometimes referred to as "polyglutamine diseases,"
affecting primarily the nervous system. Accumulation of
mutant proteins in aggregates within the cytoplasm is a
common feature of these diseases.
Fragile X Syndrome
Fragile X syndrome is the prototype of diseases in which
the mutation is characterized by a long repeating sequence
of 3 nucleotides.
Fragile X syndrome is characterized by
1- Mental retardation and an abnormality in the X
chromosome.
2- It is one of the most common causes of familial mental
retardation.
Clinical features
1- Affected males have moderate to severe mental
retardation.
2- They express a characteristic physical phenotype that
includes a long face with a large mandible, large everted
ears, and large testicles (macro-orchidism). Although
characteristic of fragile X syndrome, these abnormalities
are not always present or may be quite subtle.
3- Sometimes, The only distinctive physical abnormality that
can be detected in at least 90% of postpubertal males with
fragile X syndrome is macro-orchidism.
Cytogenitic abnormalities
Fragile X syndrome results from a mutation in the FMR1
gene, which maps to Xq27.3.
Like all X-linked recessive disorders, this disease affects
males. However, unlike patients with other X-linked
recessive disorders, approximately 20% of males who
are known to carry the fragile X mutation may be
clinically and cytogenetically normal. These "carrier
males" can transmit the disease to their grandsons through
their phenotypically normal daughters. (figure 1)
Another peculiarity is the presence of mental retardation
in 50% of carrier females. These unusual features have
been related to the dynamic nature of the mutation .
In the normal population, the number of CGG repeats in
the FMR1 gene is small, averaging around 29, whereas
affected individuals have 200 to 4000 repeats. These so-
called full mutations are believed to arise through an
intermediate stage of premutations characterized by 52 to
200 CGG repeats. Carrier males and females have
premutations. During oogenesis (but not
spermatogenesis) the premutations can be converted to
full mutations by further amplification of the CGG
repeats, which can then be transmitted to both the sons
and the daughters of the carrier female. These
observations provide an explanation for why some carrier
males are unaffected (they have premutations), and
certain carrier females are affected (they inherit full
mutations). Recent studies indicate that premutations are
not so benign after all. Approximately 30% of females
Unit 6: Genetic and Pediatric Diseases
220
carrying the premutation have premature ovarian failure
(before the age of 40 years), and about one-third of
premutation-carrying males exhibit a progressive
neurodegenerative syndrome starting in their sixth
decade. This syndrome, referred to as fragile X-associated
tremor/ataxia, is characterized by intention tremors and
cerebellar ataxia and may progress to parkinsonism.
However it is clear that the abnormalities in permutation
carriers are milder and occur later in life.
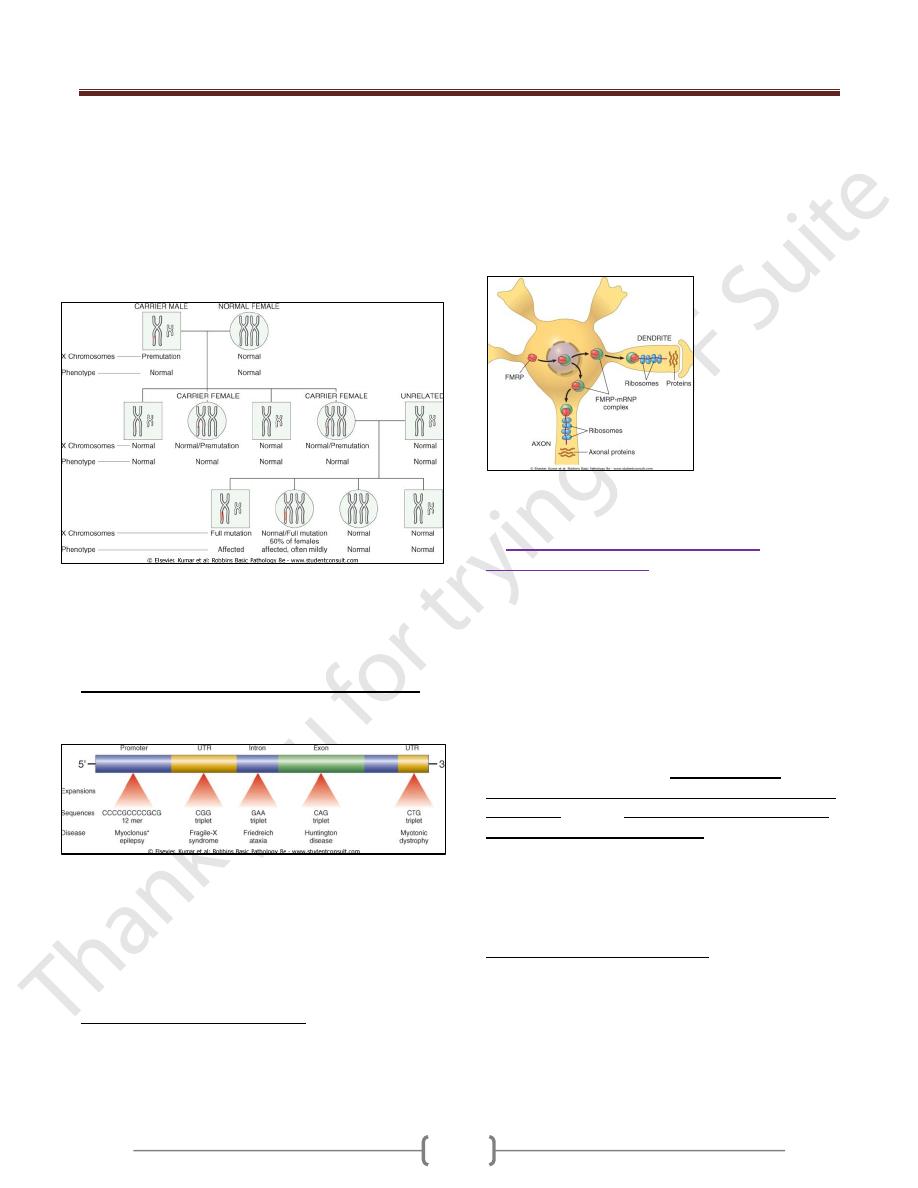
Figure 1: Fragile X pedigree. Note that in the first generation, all
sons are normal and all females are carriers. During oogenesis in
the carrier female, premutation expands to full mutation; hence,
in the next generation, all males who inherit the X with full
mutation are affected. However, only 50% of females who
inherit the full mutation are affected, and often only mildly.
The molecular basis of fragile X syndrome (Figure 2)
The CGG repeats are located in the 5' untranslated region
of the FMR1 gene.
Figure 2: Sites of expansion & the affected sequence in selected diseases
caused by nucleotide repeat mutations. UTR, untranslated region
In patients with this disease, the expanded CGG repeats
are hypermethylated. Methylation then extends up-stream
into the promoter region, resulting in transcriptional
silencing of the FMR1 gene.
Function of FMR protein (figure 3)
The product of the FMR1 gene, called FMR protein
(FMRP), is widely expressed in normal tissues, but higher
levels of transcripts are found in the brain and the testis.
Current evidence suggests that FMRP is an RNA-binding
protein that is transported from the cytoplasm to the
nucleus, where it binds specific mRNAs and transports
them to the axons and dendrites (Figure 3). It is in the
synapses that FMRP-mRNA complexes perform critical
roles in regulating the translation of specific mRNAs. The
absence of this finely coordinated "shuttle" function
seems to underlie the causation of fragile X syndrome.
Figure 3: A model for the action of familial mental retardation
protein (FMRP) in neurons.
2- Diseases Caused By Mutations in
Mitochondrial Genes
Mitochondria contain several genes that encode enzymes
involved in oxidative phosphorylation. Inheritance of
mitochondrial DNA differs from that of nuclear DNA in
that the former (mitochondrial DNA) is associated with
maternal inheritance.
The reason for this peculiarity is that ova contain
mitochondria within their abundant cytoplasm, whereas
spermatozoa contain few, if any, mitochondria. Hence, the
mitochondrial DNA complement of the zygote is derived
entirely from the ovum. Thus, mothers transmit
mitochondrial genes to all of their offspring, both male
and female; however, daughters but not sons transmit
the DNA further to their progeny.
Diseases caused by mutations in mitochondrial genes are
rare. Because mitochondrial DNA encodes enzymes
involved in oxidative phosphorylation, diseases caused by
mutations in such genes affect organs most dependent on
oxidative phosphorylation (skeletal muscle, heart, brain).
Leber hereditary optic neuropathy is the prototypical
disorder in this group. This neurodegenerative disease
manifests itself as progressive bilateral loss of central
vision that leads in due course to blindness.
Unit 6: Genetic and Pediatric Diseases
222
3- Diseases Caused by Alterations of
Imprinted Regions (Genomic Imprinting)
All humans inherit two copies of each gene, carried on
homologous maternal and paternal chromosomes. It has
usually been assumed that there is no difference between
normal homologous genes derived from the mother or the
father. Indeed, this is true for many genes. However, it
has now been established that with respect to some genes,
functional differences exist between the paternal and
the maternal genes.
These differences arise from an epigenetic process called
genomic imprinting, whereby certain genes are differentially
"inactivated" during paternal & maternal gametogenesis.
Thus, maternal imprinting refers to transcriptional silencing
of the maternal allele, whereas paternal imprinting implies
that the paternal allele is inactivated. Imprinting occurs in
ovum or sperm and is then stably transmitted to all somatic
cells derived from the zygote.
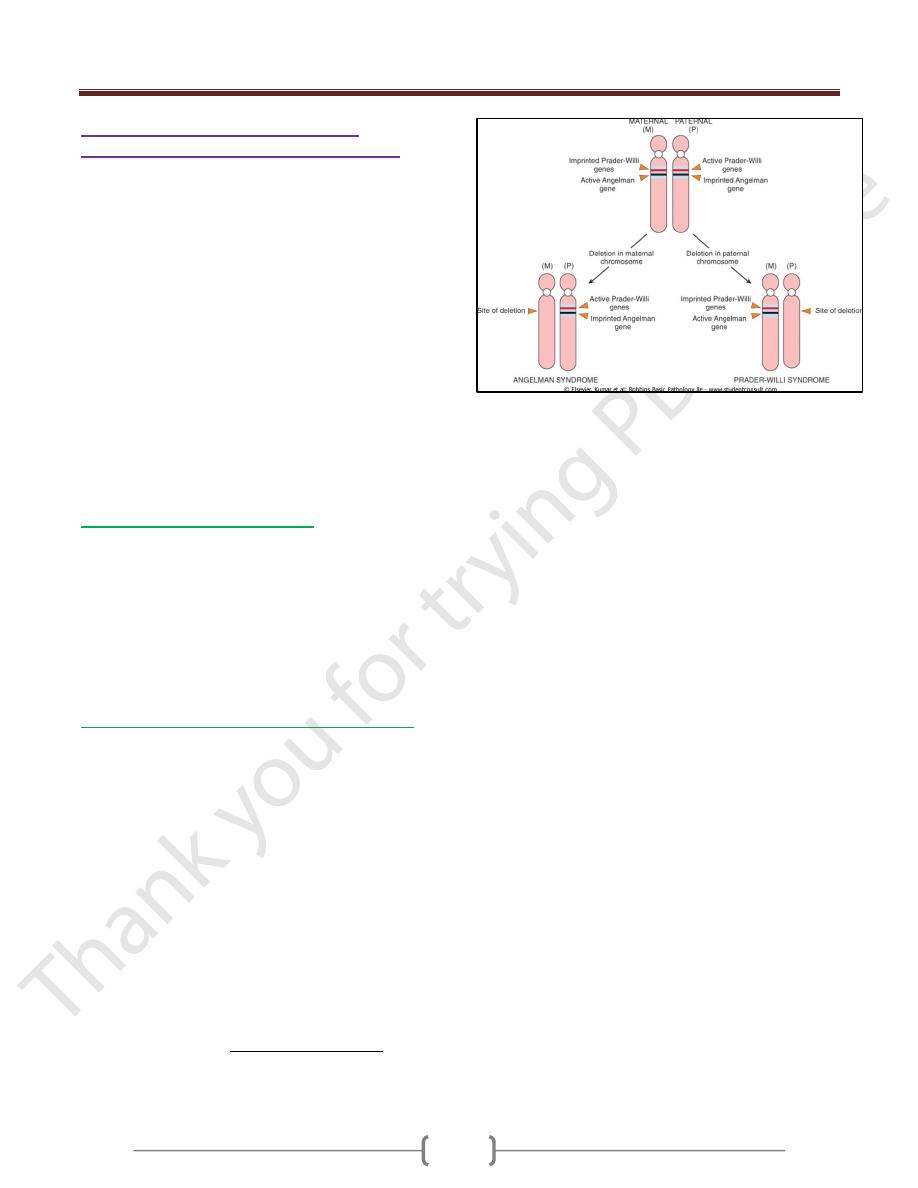
1- Prader-Willi syndrome (figure 4)
It is characterized by mental retardation, short stature,
hypotonia, obesity, small hands and feet, and
hypogonadism.
In 60% to 75% of cases, an interstitial deletion of band
q12 in the long arm of chromosome 15 [i.e.,
del(15)(q11;q13)] can be detected. It is striking that in all
cases the deletion affects the paternally derived
chromosome 15.
2- Angelman syndrome (happy puppet syndrome)
It is characterized by mental retardation, with ataxic gait,
seizures, and inappropriate laughter. Because of the
laughter and ataxia, this syndrome is also called the happy
puppet syndrome. A comparison of these two syndromes
clearly demonstrates the "parent-of-origin" effects on
gene function.
An interstitial deletion of band q12 in the long arm of
chromosome 15
It is striking that in all cases the deletion affects the
maternally derived chromosome 15.
The Angelman syndrome gene (imprinted on paternal
chromosome) is now known to encode a ligase that has a
role in the ubiquitin-proteasome proteolytic pathway .
This gene, called UBE3A, is expressed primarily from the
maternal allele in specific regions of the normal brain.
In Angelman syndrome, UBE3A is not expressed in
these areas of the brain-hence the neurologic disorder.
Figure 4: Genetics of Angelman and Prader-Willi syndromes.

Unit 6: Genetic and Pediatric Diseases
221
PEDIATRIC DISEASES
Tumors and tumor-like lesions
of infancy and childhood
Malignant neoplasms are the second most common cause
of death in children between the ages of 4 and 14 years
Benign tumors are even more common than are cancers.
Heterotopia or choristoma refers to microscopically
normal cells or tissues that are present in abnormal
locations. Examples include a pancreatic tissue "rest"
found in the wall of the stomach or small intestine, or a
small mass of adrenal cells found in the kidney, lungs,
ovaries, or elsewhere.
Hamartoma refers to an excessive but focal overgrowth
of cells and tissues native to the organ in which it occurs.
Although the cellular elements are mature and identical to
those found in the remainder of the organ, they do not
reproduce the normal architecture of the surrounding tissue.
Hamartomas can be thought of as the linkage between
malformations and neoplasms. Hemangiomas,
lymphangiomas, rhabdomyomas of the heart, and
adenomas of the liver are considered by some to be
hamartomas and by others to be true neoplasms.
Benign Tumors
1)
Hemangiomas
Is a benign and usually self-involuting tumor (swelling
or growth) of the endothelia
cells that line blood vessels
Are the most common tumors of infancy.
Both cavernous and capillary hemangiomas may be
encountered
In children most hemangiomas are located in the skin,
particularly on the face and scalp, where they produce flat
to elevated, irregular, red-blue masses; the flat, larger
lesions are referred to as port wine stains.
Hemangiomas may enlarge as the child gets older, but in
many instances they spontaneously regress.
The vast majority of superficial hemangiomas have no
more than a cosmetic significance; rarely, they may be
the manifestation of a hereditary disorder associated
with disease within internal organs, such as the von
Hippel-Lindau and Sturge-Weber syndromes.
2)
Lymphangiomas
Represent the lymphatic counterpart of hemangiomas.
They are characterized by cystic and cavernous spaces
lined by endothelial cells and surrounded by lymphoid
aggregates; the spaces usually contain pale fluid.
They may occur on the skin but, more importantly, are
also encountered in the deeper regions of the neck, axilla,
mediastinum, and retroperitoneum.
Though histologically benign, they tend to increase in size
after birth and may encroach on mediastinal structures or
nerve trunks in axilla.
3) Sacrococcygeal teratomas
Are the most common germ cell tumors of childhood,
accounting for 40% or more of cases.
Approximately 10% of sacrococcygeal teratomas are
associated with congenital anomalies, primarily defects of
the hindgut and cloacal region and other midline defects
(e.g., meningocele, spina bifida) not believed to result
from local effects of the tumor. Approximately 75% of
these tumors are histologically mature with a benign
course, and about 12% are unmistakably malignant and
lethal. The remainder is designated immature teratomas,
and their malignant potential correlates with the amount
of immature tissue elements present. Most of the benign
teratomas are encountered in younger infants (<4
months), whereas children with malignant lesions tend to
be somewhat older.
Malignant Tumors
The organ systems involved most commonly by
malignant neoplasms in infancy and childhood include the
hematopoietic system, neural tissue, and soft.
Malignant tumors of infancy and childhood differ
biologically and histologically from those in adults. The
main differences are as follows:
1) Relatively frequent demonstration of a close relationship
between abnormal development (teratogenesis) and tumor
induction (oncogenesis).
2) Prevalence of constitutional genetic abnormalities or
syndromes that predispose to cancerTendency of fetal and
neonatal malignancies to spontaneously regress or undergo
"differentiation" into mature elements Improved survival or
cure of many childhood tumors, so that more attention is
now being paid to minimizing the adverse delayed effects
of chemotherapy and radiotherapy in survivors, including
the development of second malignancies.
Neuroblastoma
The term "neuroblastic tumor" includes tumors of the
sympathetic ganglia and adrenal medulla that are derived

Unit 6: Genetic and Pediatric Diseases
221
from primordial neural crest cells populating these sites;
neuroblastoma is the most important member of this
family.
It is the second most common solid malignancy of
childhood after brain tumors, accounting for 7% to 10%
of all pediatric neoplasms, and as many as 50% of
malignancies diagnosed in infancy.
Neuroblastomas demonstrate several unique features in
their natural history, including spontaneous regression
and spontaneous- or therapy-induced maturation.
Most occur sporadically, the neoplasms may involve both
of the adrenals or multiple primary autonomic sites.
Few are familial with autosomal dominant transmission.
Factors influence prognosis
1) The stage of the tumor. Example, stage 4S (S means
special), because the outlook for these patients is
excellent, despite the spread of disease.
2) The age of the patient, children younger than 1 year have
a much more favorable outlook than do older children at a
comparable stage of disease.
3) Morphology is an independent prognostic variable in
neuroblastic tumors; evidence of schwannian stroma and
gangliocytic differentiation is indicative of a "favorable"
histology.
4) Amplification of the MYCN oncogene in neuroblastomas
is a molecular event that has profound impact on
prognosis. MYCN amplification is present in about 25% to
30% of primary tumors, most in advanced-stage disease;
the greater the number of copies, the worse the prognosis.
5) Deletion of the distal short arm of chromosome 1, gain of
the distal long arm of chromosome 17 and overexpression
of telomerase are all adverse prognostic factors.
6) Expression of TrkA, a high-affinity receptor for nerve
growth factor that is indicative of differentiation toward
sympathetic ganglia lineage, is associated with favorable
prognosis.
o The most important factors for the prognosis are the
age of the patient and stage of the tumor
Clinical Course
o Children younger than 2 years with neuroblastomas
generally present with protuberant abdomen resulting
from an abdominal mass, fever, and weight loss.
In older children the neuroblastomas may remain
unnoticed until metastases cause hepatomegaly, ascites,
and bone pain.
o Neuroblastomas may metastasize widely through the
hematogenous and lymphatic systems, particularly to
liver, lungs, and bones, in addition to the bone marrow.
o In neonates, disseminated neuroblastomas may present
with multiple cutaneous metastases with deep blue
discoloration to the skin (earning the rather unfortunate
moniker of "blueberry muffin baby").
o About 90% of neuroblastomas, regardless of location,
produce catecholamines (similar to the catecholamines
associated with pheochromocytomas), which are an
important diagnostic feature (i.e., elevated blood levels
of catecholamines and elevated urine levels of
catecholamine metabolites such as vanillylmandelic acid
[VMA] and homovanillic acid [HVA]). Despite the
elaboration of catecholamines, hypertension is much less
frequent with these neoplasms than with
pheochromocytomas.
Retinoblastoma
Retinoblastoma is believed to arise from a cell of
neuroepithelial origin, usually in the posterior retina
1) Retinoblastoma is the most common malignant eye tumor
of childhood.
2) Retinoblastoma frequently occurs as a congenital tumor,
3) It can be multifocal and bilateral,
4) It undergoes spontaneous regression, and
5) Patients have a high incidence of second primary tumors.
6) The incidence decreases with age, most cases being
diagnosed before the age of 4 years.
Retinoblastomas occur in both familial and sporadic
patterns. Familial cases typically develop multiple
tumors that are bilateral, although they may be unifocal
and unilateral. Patients with familial retinoblastoma are
also at increased risk for developing osteosarcoma and
other soft tissue tumors. All sporadic non heritable tumors
are unilateral and unifocal.
Approximately 60% to 70% of the tumors are associated
with a germline mutation in the RB1 gene and are hence
heritable.
30% to 40% of the tumors develop sporadically, and these
have somatic RB1 gene mutations.
Clinical Features
The median age at presentation is 2 years, although the
tumor may be present at birth. The presenting findings
include poor vision, strabismus, a whitish hue to the pupil
("cat's eye reflex"), and pain and tenderness in the eye.
Prognosis
Untreated, the tumors are usually fatal, but after early
treatment with enucleation, chemotherapy, and
radiotherapy, survival is the rule.
Some tumors spontaneously regress.

Unit 6: Genetic and Pediatric Diseases
221
Wilms' Tumor
Wilms' tumor, or nephroblastoma, is the most common
primary tumor of the kidney in children.
Most cases occur in children between 2 & 5 years of age.
This tumor illustrates several important concepts of
childhood tumors:
1) The relationship between congenital malformation and
increased risk of tumors,
2) The histologic similarity between tumor and
developing organ, and finally,
3) The remarkable success in the treatment of childhood
tumors.
Three groups of congenital malformations are
associated with an increased risk of developing
Wilms' tumor;
1) Patients with the WAGR syndrome, characterized by
aniridia, genital abnormalities, and mental retardation,
have a 33% chance of developing Wilms' tumor.
2) Another group of patients, those with the so-called Denys-
Drash syndrome (DDS) also has an extremely high risk
(
∼90%) of developing Wilms' tumor. This syndrome is
characterized by gonadal dysgenesis and renal
abnormalities. Both of these conditions (WAGR,DDS)
are associated with abnormalities of the Wilms' tumor 1
(WT1) gene, located on chromosome 11p13.
o The nature of genetic aberration differs, however. Patients
with WAGR syndrome demonstrate loss of genetic
material (i.e., deletions) of WT1, and individuals with
DDS harbor a dominant negative inactivating mutation
in a critical region of the gene. (A dominant negative
mutation interferes with the function of the remaining
wild-type allele.)
o The WT1 gene is critical to normal renal and gonadal
development; inactivation of one copy of this gene results
in genitourinary abnormalities in humans.
3) A third group of patients, those with the Beckwith-
Wiedemann syndrome (BWS), also has an increased risk
of developing Wilms' tumor. These patients have
enlargement of individual body organs (e.g., tongue,
kidneys, or liver) or entire body segments
(hemihypertrophy); enlargement of adrenal cortical cells
(adrenal cytomegaly) is a characteristic microscopic
feature.
BWS is an example of a disorder of genomic imprinting.
The genetic locus that is involved in these patients is in
band p15.5 of chromosome 11 distal to the WT1 locus.
This region contains at least 10 genes that are normally
expressed from only one of the two parental alleles, with
transcriptional silencing of the other parental homologue .
One of the candidate genes in this region-insulin-like
growth factor-2 (IGF2) is normally expressed solely from
the paternal allele, while the maternal allele is imprinted
(i.e., silenced). In some Wilms' tumors, loss of imprinting
(i.e., re-expression of IGF2 by the maternal allele) can be
demonstrated, leading to overexpression of the IGF2
protein, which is postulated to result in both organ
enlargement and tumorigenesis.