
Unit 1: Principles of Drugs Therapy
4
Pharmacology
:
represents the scientific study of the
origin, nature, chemistry, effects, and uses of drugs.
5 branches of pharmacology (as we see in the figure below).
Pharmacodynamics: the study of the biochemical and
physical effect of drugs and the mechanisms of drug
actions in living organisms.
Pharmacokinetics: deals with handling of drugs by the
body (refers to the absorption, distribution, metabolism
and excretion of drug in a living organism.
Toxicology: represents the study of undesirable effects of
drugs in living organisms.
Pharmacotherapeutics: is the application of drugs for
treatment of disease.
Pharmacognosy: deals with natural drugs (that is-plants,
animals or minerals and their products.
Drug
:
small molecules that when introduced into the
body, it alters the body’s function by interaction with the
molecular of the cells.
-A prescription drug can only be used safely under the
supervision of a health care professional who is licensed
to prescribe or dispense drugs according to state law.
Drugs are used in three principal ways: to cure disease, to
suppress disease or to prevent disease.
Characteristic of drugs
:
3 essential points are important in drugs:
Molecular Shape: The drugs interact with specific sites
within the target tissue called receptors. The shape of the
receptor site decided what kind of drug molecule may
interact with it. As the optimal fit increased, the response
to the drug increased too.
Molecular size: The majority of drugs have molecular
weight range from 100 to 1000 which enables them for
convenient administration and efficient absorption and
distribution.
Chemical nature: drugs are either highly active or inert.
Many drugs are either weak acids or weak bases. The
electrical charge of molecules is an important issue to be
considered when we explain the tissue drug interaction.
Drug nomenclature
A drug has at least three names:
chemical, generic and trade. To avoid confusion we use
the generic name.
Chemical name Generic name Trade name
----------------------------------------------------------------------
6-chloro-2H-1,2,4-ben- Chlordiazepoxide Librium
zothiadiazine-7-sulfona-
mide 1,1-dioxide
7-chloro-1,3-dihydro-1- Diazepam Valium
methyl-5-phenyl-2H-1,4-
benzodiazepine-2-one
ethyl 1-methyl-4-phenyli- Meperidine Pethidine
sonipecotate hydrochlo-
ride
methyl 7-chloro-6,7,8-trideoxy-
6-(1-methyl-trans-4-propyl-L-2
-pyrrolidinecarboxamido)-1
-thio-L-threo-α-D-galacto
-octopyranoside-2-0-
dihydrogen phosphate. clindamycin Dalacin
Mechanism of drug action:
1) drug acts on cell membrane: by
a. Action on specific receptors: that the drugs changes the
functional macromolecular component of the cells, causes
the alteration of the cellular components. The part when
the drug acts is called the receptor. The living system
contains many types of receptors. For one drug there is
one receptor (specific receptor) or more than one type of
receptor e.g Histamine acts on two types of receptors H
1
and H
2
. Ach acts on muscarinic and nicotinic receptors.
b. Interference with selective passage of ions across
membrane e.g Ca+2 channel blocker as with Nifedipine,
Diltiazem and Verapamil ( anti-hypertension ) .

Unit 1: Principles of Drugs Therapy
5
c. Inhibition of membrane bound enzyme and pump.
Inhibition membrane bound ATP
ase
by cardiac glycoside
in treatment of heart failure (as by digoxin). Inhibition of
the pump as by tricyclic antidepressants
(chloropromazine).
d. Physiochemical effects. These types of drugs appear to
act on lipid, protein and water constituents of the nerve
cells as e.g. with general anesthetic agent (Halothane) and
local anesthetic agent (Lidocaine).
2) Drug acts on metabolic processes within the cell:
a. Enzyme inhibition. As with cholinesterase enzyme by
pyridostigmine, Xanthine oxidase enzyme by Allopurinol
(reduces the production of uric acid) and Monoamine
oxidase (inactivate the nor-epinepherine, serotonin and
dopamine) by phenalzine.
b. Through inhibition of transport processes across the
cells e.g. blocked of anion transport in the renal tubules
cells by Probencid, this can be used to delay excretion of
pencillin and to enhance elimination of urease.
c. Through incorporation into large molecules e.g 5-
Fluorouracil (anticancer) into
m
RNA in place of amino
acid Uracil.
d. In case of antimicrobial agent, by altering metabolic
processes of the m.o as e.g penicillin interfere with the
formation of bacterial cell wall.
3) Drug acts outside of the cells:
a. Chemical action. Some drugs react chemically with
known simple chemical equation e.g.
Antacids as aluminum hydroxide which neutralized
the acidity of stomach.
Dimercaprol (BAL) antagonise the effect of non-
organic substances such as Hg, Cd.
b. Physical action (through osmosis) e.g. 1.liquid paraffin
which acts as laxative. 2.MgSo
4
, Caster oil which act as
purgative.Mannitol which acts as diuretic.
Receptors:
Is a component of the cell, most of them are protein
macromolecules when agonist (drug that activate receptor)
binds to receptor will undergo a serious of alteration in
conformation which induce changes in system within the
cell that in turn bring the response to the drug.
e.g. activation of ß-adrenoceptors by a catecholamine as
adrenaline (the first messenger) increases the activity of
adenylate cyclase which raises the rat of formation of
c
AMP (second messenger) a modulator of the activity of
several enzyme that cause the cell to act.
Radioligand binding studies have shown that the receptor
number do not remain constant, when tissues are exposed
continuously to an agonist (is a drug that has affinity and
intrinsic activity or efficacy) the number of receptors
decreases (down-regulation) and this may be a cause of
tachyphylaxis which means (loss of response or efficacy)
due to frequent repeated doses e.g in asthmatic patients
(using adrenoceptors agonist which are bronchodilator).
In case the tissues when exposed continuously to
antagonist (is a drug that has affinity without efficacy)
this lead to formation of new receptors, as with ß-
adrenoceptor blockers agent (antihypertensive agent).
This is called up-regulation. Withdrawal of this agent may
cause angina pectoris.
Drug receptor interaction:
In classical receptor, there is a theory developed by Clark.
It was assumed that drug effect is proportional to the
fraction of receptor occupied by drug, and that maximal
effect results when all receptor are occupied.
At equilibrium the rate of forward and backward are equal.
When we have much quantities of receptors so the
binding of these quantities of receptors depend on the
concentration of drug. At high concentration of drug there
is a high binding and high response, if the concentration
of a drug decreases there is a decrease in the response.
Quantitative description of drug action:
As we see in the following equation at equilibrium
condition:
If K
d
is low that means K
2
is less; the drug with high
affinity. If half of receptors are bound to a drug the
concentration of R and DR is equal.
So the concentration of a drug necessary to bind 50% of
receptor equal the K
d

Unit 1: Principles of Drugs Therapy
6
Dose response curve:
It is plotted between the biological effect of the drug in
the vertical axis and the log dose on the horizontal axis as
in the following figure.
This figure shows typical dose response curve for drugs
showing differences in potency and efficacy.
Drug A is more potent than drug B, but both drugs have
the same maximum effect (efficacy).
Drug C shows lower potency & efficacy than drug A or B.
Types and location of receptors:
A. Ligand gated ion channel: they are responsible for
regulation the flow of ions across cell membrane. The
activity of these channels is regulated by the binding of a
ligand to the channel as e.g nicotinic acid. Stimulation of
the nicotinic receptors by acetylcholine results in Na
influx and that generates an action potential like
activation and contraction of skeletal muscle cells. Other
e.g group of benzodiazepine compounds as diazepam
(valium). It enhances the stimulation of GABA receptors,
this result in increasing Cl influx or hyperpolarization.
B. G protein coupled receptors: binding of appropriate
ligand to extracellular region of the receptor activates the
G protein which have three subunits (α,β andÝ). G protein
releases guanosin diphosphate then change to guanosin
triphosphate in α subunit, leads to activates adenyl
cyclase. When ligand (as hormone) is no longer present,
the receptor reverts to its resting state. GTP on the α
subunit is hydrolysed to GDP and adenyl cyclase is
deactivated.
C. Enzyme linked receptors: e.g of these receptors are
insulin receptors and others having a Tyrosin kinase
activity as a part of their structure. when the ligand binds
to the receptor subunit, the receptor undergoes
conformational changes (converting from its inactive
form into an active kinase form).
D. Intracellular receptors: differ from other three types of
receptor is entirely intracellular therefore, the ligand must
diffuse into the cell to interact with the receptor.
-the ligand must be sufficiently lipid soluble to move
across membrane easily attached to plasma albumin as
steroid hormones.
-the activated ligand-receptor complex migrates to the
nucleus, resulting in regulation of Gene expression.
Cellular responses are not observed until considerable
time elapsed (30 minutes or more) and duration of
response will be "hours to days"
Definitions:
Affinity
: is the ability of the drug to form a combination
with the receptor.
Efficacy
: is the ability of the drug to produce a response.
Agonists
: is a drug that has affinity and efficacy.
Antagonists:
is a drug that has affinity without efficacy.
Partial agonists
: is a drug that has affinity and some
efficacy and it has the ability to antagonise the action of
other drug that has higher efficacy (this type of drug has
both agonist and antagonist action).
Inverse agonists
: Some substances produce effects that
are specifically opposite to those of agonist. e.g action of
Diazepam on Benzodiazepine receptors in CNS produces
sedation, anxiolysis, muscle relexation and controls
convulsions, β-carbolines which also bind to this receptor
cause stimulation, anxiety, increase muscle tone and
convultion they are inverse agonists.
ED
50
:
the dose of the drug that produces 50% of
maximum response.
LD
50
:
the dose of the drug that kill 50% of experimented
animals.
TD
50
(medium toxic dose):
the dose which is required to
produce toxic effect in 50% of animals.
-so the dose we need for a particular effect should be
small and its named effective dose and if this dose
increase, it will show a toxic effect then it is called toxic
dose and if we increased the dose to a lethal level it will
cause death and then we name it lethal dose.
Potency:
The response that produced by a drug per unit weight.
Additive effect:
when the combined effect of two drugs
is the sum of individual action. Alcohol with ether.
Drug dependence:
is a state arising from repeated or
continuous administration of drugs.The subject feels a
desire to continue using the drug. Treatment of drug
dependence by using alternative drug as in case of heroin
we use methadone, in case of alcohol we use diazepam
Idiosyncrasy:
represents the qualitative abnormal
reaction to a drug due to genetic abnormality.

Unit 1: Principles of Drugs Therapy
7
Tolerance:
It develops when the dose is increased to get the effect of
a previously administrated smaller dose, e.g.
1) There is natural tolerance which is not induced by the
drug but it is due to inherent factors i.e pharmacogenetic
2) Acquired tolerance which is developed after a prolonged
time of using a drug. acquired tolerance is familiar
especially with opioids. It is due to pharmacological
efficacy at receptor site or down-regulation of receptor.
It is divided:
a) Reduced efficacy at receptor site as in opioids.
b) Enzyme induction (increased metabolism) as in
alcohol. The effect of drug is reduced so we need large
dose of drug to produce previous response.
c) Cross-tolerance happens between drugs of similar
structure or metabolized by the same enzyme and
sometimes between dissimilar drugs as with antibiotics.
Synergism or potentiation
:
occurs when 2 drugs with different sites or mechanism of
action produce greater effects when taken together than
either dose when taken alone.
Therapeutic index (TI)
: is the ratio of the LD
50
/ED
50
.
The largest the ratio the better drug safety while those
with lower index, it is more dangerous as e.g. therapeutic
index of morphine is 4 while diazepam is 500
Drug binding to receptor
: either:
Reversible is a weak bound. The drug receptor complex
can be separated easily. It is the type of most therapeutic
drugs. Irreversible when the bound is strong and the
complex is not separated easily.
Types of antagonism:
3 types of may involve in the interaction of drugs:
1) Pharmacological antagonism:
This type of drug compete with agonist for the same
receptor. It can be divided into:
a) Competitive reversible antagonism:
It can combine reversibly with receptor & it can be
overcome by increasing concentration of agonist
b) Noncompetitive irreversible antagonism:
It can combine irreversibly with receptor by a covalent
bond. The increasing dose of agonist will never overcome
the inhibition.
This antagonism decreases both efficacy & potency. The
log dose response curve is not only shifted to the right but
the maximal effect is also reduced.
2) Chemical antagonism
: by which one drug can
antagonize the action of second drug by chemical action.
e.g. protamine which is protein with +vely charged used
to counteract the effect of heparin which is –vely charged.
3) Physiological antagonism
:
By which one compound opposes the physiological action
of other e.g glucocorticoid hormone increases the blood
sugar while insulin decreases blood sugar.
Relation between receptor & clinical use of drug
1) The receptor determines the qualitative relationship
between dose of the drug & pharmacological activity.
2) The receptor is responsible for selective of drug action
(agonist)
3) The receptor is also responsible for the action of
antagonists.
4) The receptor determines the quantitative dose of agonist
by using dose response curve.
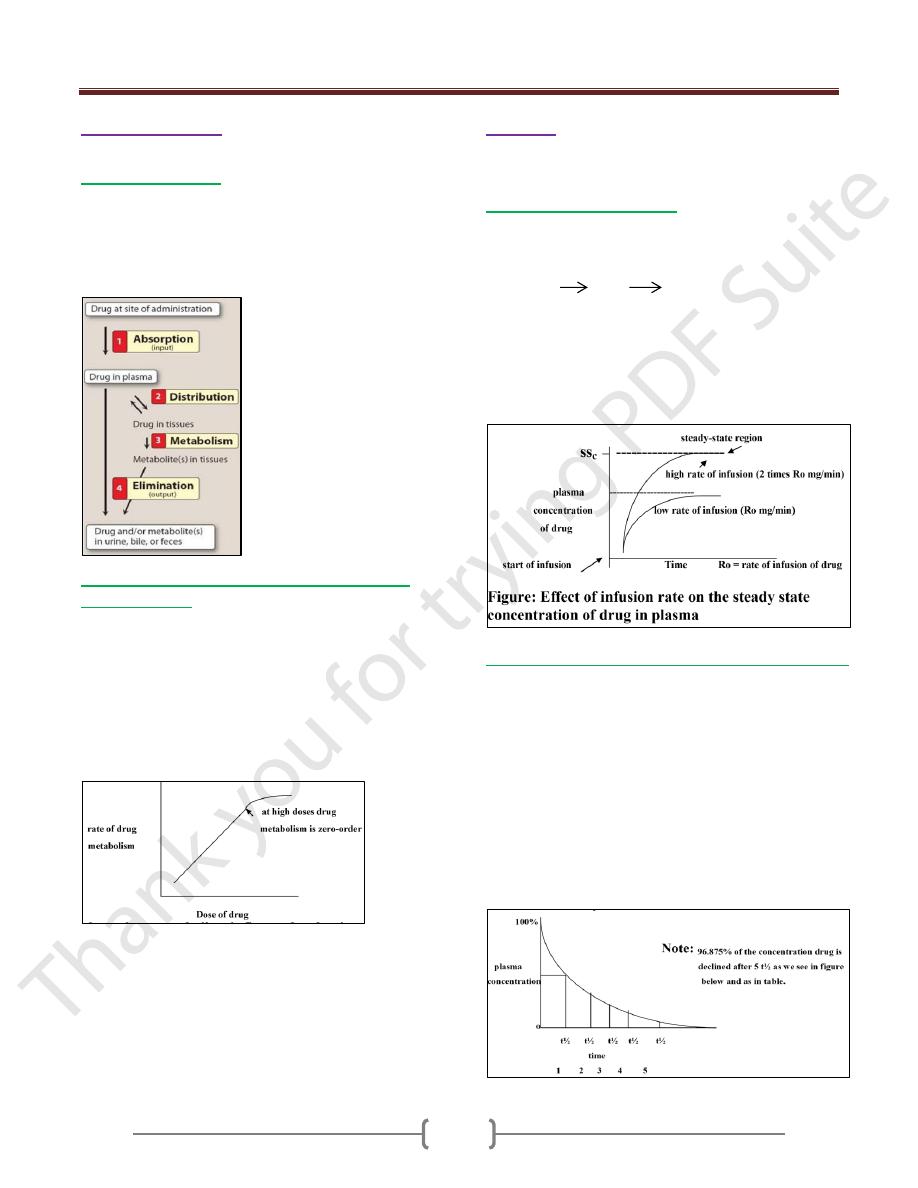
Unit 1: Principles of Drugs Therapy
8
Order of reaction:
There are 2 kinds of reaction
1)
First order reaction
: in this case drugs taken in the
body are subjected to absorption, distribution, metabolism
& elimination as in the figure below.
Generally, the rate of these process occur proportional to
the concentration of drug (by increase the concentration
of drug, these process will increase).
2) Zero order reaction (saturation kinetics) (Non-
linear kinetics):
As the amount of drug in the body raises these processes
(absorption, distribution, metabolism & excretion)
become saturated, that means the rate of these processes
reach maximum & no more increase in these process even
with increase the concentration of the drug. This may be
due to limited amount of enzyme. The enzyme is
saturated by a high free-drug concentration, and the rate
of metabolism remains constant.
At low doses, drug metabolism is first order that is
proportional to drug dose.
When first order is transformed into zero order, the
following changes will occur:
1) Metabolism rate remains fixed
2) The concentration of the drug will increase.
3) The half-life will increase.
Half-life:
Is the time needed for elimination of 50% of the drug
concentration in the plasma. It is constant for each drug.
Steady state concentration:
Means a state of equilibrium reached when drug doses are
given repeatedly after a period of time, the amount of
drug absorption equal drug elimination from body.
Drug in put body drug out put
At steady state, input (rate of infusion) equals output (rate
of elimination). That means the plasma concentration of
drug remains constant.
A faster rate of infusion does not change the time needed
to achieve steady state; only the steady-state
concentration (SS
c
) changes as in the following figure.
Plasma half-life (t1/2) & steady state concentration:
The steady state concentration is occurred when the drug
absorption is equal drug elimination from body.
I.V injection of a drug (single dose injection) to a body; it
will achieve the highest concentration in the plasma, then
there will be a sharp drop in the concentration from the
plasma due to the drug distribution around the body, this
is called distribution phase, which will be followed by a
steady state. Then the drug concentration decline as the
drug is being removed from the body by kidney or liver
this is kno as elimination phase. If the elimination of the
1
st
order kinetic, in this case the time taken for any
concentration to fall is always the same.
Schematic representation of drug absorption, distribution, metabolism, and elimination.
2. Zero order reaction (saturation kinetics).
As the amount of drug in the body raises these processes
(absorption, distribution, metabolism and excretion) become
saturated, that means the rate of these processes reach maximum
and no more increase in these process even with increase the
concentration of the drug. This may be due to limited amount of
enzyme. The enzyme is saturated by a high free-drug
concentration, and the rate of metabolism remains constant. This
is called Zero order kinetics or some times refereed as non-linear
kinetics as in the following figure.

Unit 1: Principles of Drugs Therapy
9
Factors affecting t
1/2
1) Rate of excretion (inversely proportional, short t
1/2
)
2) Rate of metabolism (inversely proportional, short t
1/2
)
3) Storage of drugs in tissues (longer t
1/2
)
4) Protein binding (longer t
1/2
)
Mechanism of drug movement
A) Passive diffusion
The most common mechanism, involve movement of a
drug from an area of high concentration to one of lower
concentration. For example, after oral administration, the
initial concentration of a drug is higher in the GIT than in
the blood. This promotes movement of the drug into the
bloodstream. When the drug is circulated, the
concentration is higher in the blood than in body cells, so
that the drug moves into the fluid surroundings the cells
or into the cells themselves. Passive diffusion continues
until a state of equilibrium is reached between the amount
of drug in the tissues and the amount in the blood.
The passive diffusion allows lipid soluble material to pass
through the membranes (which is made of lipid layers &
protein)
Water in the pores of various size are found on cell
membrane present in stomach & widely in renal proximal
tubules not allow the passage of the drug by passive diff.
According to the concentration gradient, no energy is
involved. Most drugs exhibit high or low lipid solubility
according to environmental PH.
Classification of drugs according to physio-
chemical properties (environmental pH)
Those are variable ionized according to environmental pH.
Those are incapable of becoming ionized (permanently
unionized)
Those are permanently ionized.
1) Variable ionized according to environmental pH.
Drugs are either weak organic acids or bases. The degree
of ionization depends on pH so the drug may be highly
ionized or highly unionized depending on whether it’s
organic acid or base.
Highly ionized drugs = water soluble & lipid insoluble.
Highly unionized drug = water insoluble & lipid soluble.
So:
Note: ionized groups in a drug molecule tend either to
loss hydrogen ion (acidic groups) or to added a hydrogen
ion (basic groups).
There is dissociation constant for each drug expressed as K
d
.
pH = K
d
when the conc. of ionized equal the conc. of
unionized for acidic & basic compounds.
The passive diffusion only occurs to drug in unionized
from which is lipid soluble.
e.g of drug shows the effect of pH on it.
Aspirin (is acidic substances).
Stomach pH = 1.5
Upper intestine pH = 6.8
Lower intestine pH = 7.6
pH elsewhere = 7.4
Aspirin in the stomach: it is unionized so it is considered
to be lipid soluble, as a result it diffuse to the cell.
When aspirin enters the gastric epithelial cells whose pH
is 7.4 it is ionized & becomes less diffusible.
Aspirin in stomach even it reaches to pH 3-4. Aspirin is
lipid soluble so it is absorbed easily.
In intestine pH is alkaline so Aspirin not absorbed easily.
In plasma pH 7.4, Aspirin is water soluble (not absorbed,
diffusion to the tissue is slow.
Increase excretion of Aspirin in urine with alkaline agent,
Aspirin becomes more water soluble.
2) Drugs which are incapable of ionization
As digoxin, chloramphenicol, do not affected by change in
pH due to absence of ionized group. So they are lipid soluble
and cross tissue boundaries easily (They are nonpolar)

Unit 1: Principles of Drugs Therapy
10
3) Drugs which are ionized (polar) at all value of pH.
They are either –ve charge as heparin or +ve charge as
tubocurarine. So heparin & tubocurarine are given
parenterally.
B. Facilitated diffusion
Is a similar process, except that drug molecules combine
with a carrier substance. The complex is then translocated
across the membrane at a much faster rate than that
associated with the diffusion of drug alone. This system
requires no cellular energy and is only slightly sensitive to
temperature changes as with vit B
12
absorption.
C. Active transport.
Some drugs move into or out of cells against a
concentration gradient i.e by active transport from high
concentration to low concentration and are more rapid
than transfer by diffusion. This process requires a carrier
substance and the release of cellular energy like with the
absorption of Iron by the gut, Levodopa across the BBB
and the secretion of many organic acid and bases by renal
tubular and bilary duct cells.
Note: Active transport is subjected to saturation and can
be inhibited.
Absorption:
Is the transfer of a drug from its site of administration to
the blood stream. The rate of absorption depends on the
method of drug administration.
Drug administration
: either
Enteral:
a.
By mouth:
Swallowed or sublingual (buccal absorption)
Swallowed: Giving a drug by mouth provides many
advantages to the patient; oral drugs are easily self-
administered and limit the number of systemic infections
that could complicate treatment. Moreover, toxicities or
overdose by the oral route may be overcome with
antidotes such as activated charcoal. On the other hand,
the pathways involved in drug absorption are the most
complicated, and the drug is exposed to harsh
gastrointestinal (GI) environments that may limit its
absorption. Some drugs are absorbed from the stomach;
however, the duodenum is a major site of entry to the
systemic circulation because of its larger absorptive
surface. Ingestion of drugs with food, or in combination
with other drugs, can influence absorption. The presence
of food in the stomach delays gastric emptying, so drugs
that are destroyed by acid (for example, penicillin)
become unavailable for absorption .
Note: Enteric coating of a drug protects it from the acidic
environment; the coating may prevent gastric irritation,
and depending on the formulation, the release of the drug
may be prolonged, producing a sustained-release effect.
S u b l i n g u a l : Placement under the tongue allows a drug
to diffuse into the capillary network and, therefore, to
enter the systemic circulation directly. Administration of
an agent, sublingually, has several advantages including
rapid absorption, convenience of administration, low
incidence of infection, avoidance of the harsh GI
environment, and avoidance of first-pass.
b. By rectum.
c. By vagina.
Parentral:
The parenteral route introduces drugs directly across
the body's barrier defenses into the systemic
circulation or other vascular tissue. Parenteral
administration is used for drugs that are poorly
absorbed from the GI tract (for example heparin) and
for agents that are unstable in the GI tract (for
example, insulin). Parenteral administration is also
used for treatment of unconscious patients and under
circumstances that require a rapid onset of action. In
addition, these routes have the highest bioavailability
and are not subject to first-pass metabolism or harsh
GI environments. Parenteral administration provides
the most control over the actual dose of drug
delivered to the body. However, these routes are
irreversible and may cause pain, fear, and infections.
The three major parenteral routes are intravascular by
subcutaneous (SC), Intramuscular (I.M), Intravenous
(IV) or infusion, Inhalation (for volatil liquid), topical
application for local (skin, eye, nose, ear)
Other routes
; e.g intrathecal, transdermal, intranasal,
intratracheal, intrapleural.
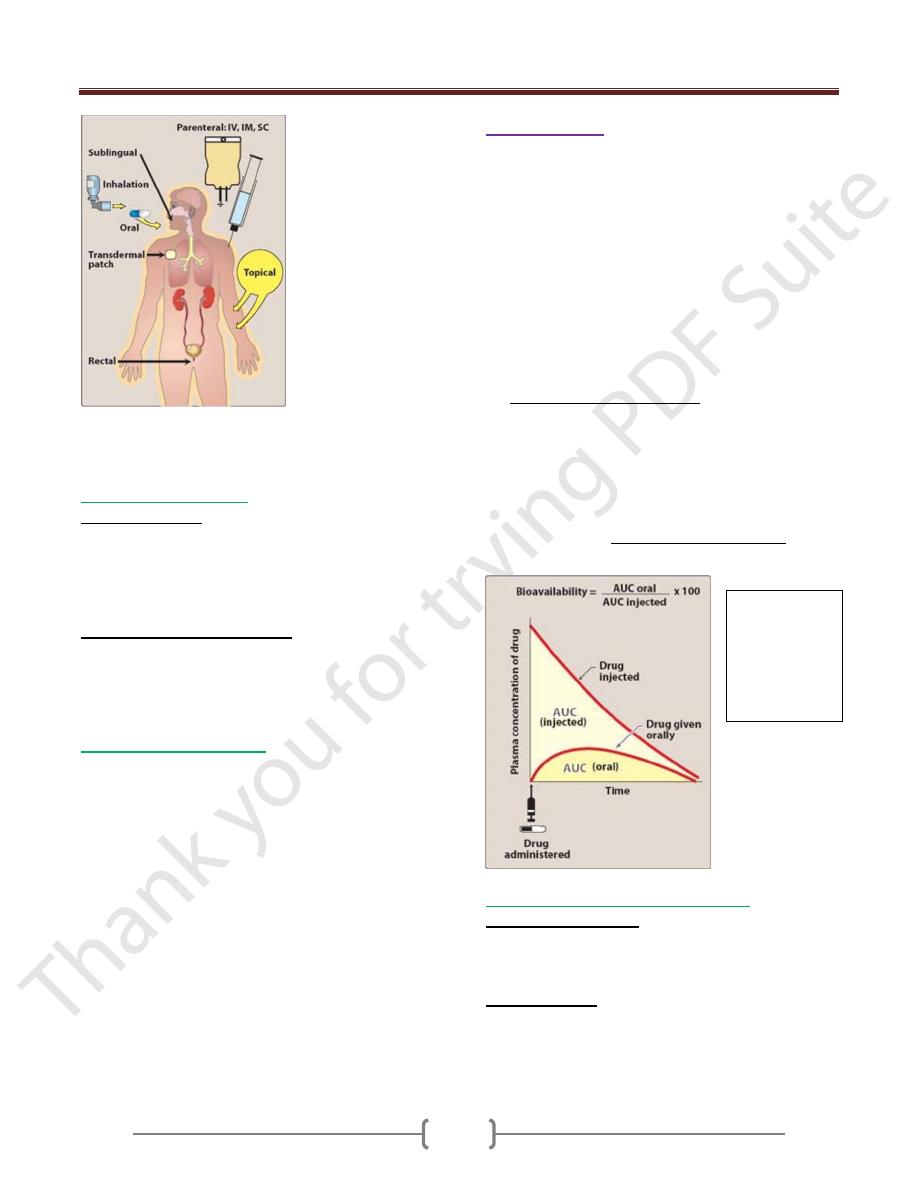
Unit 1: Principles of Drugs Therapy
11
Commonly used routes of drug administration. IV =
intravenous;
IM = intramuscular; SC = subcutaneous.
Absorption from G.I.T:
The small intestine is the main site for food and drug
absorption: it has the following advantages.
Wide surface area.
Wide range of pH.
Specialized epithelial cells for absorption.
Good blood supply.
The stomach is of less important.
It has highly acidic medium, so only acidic compound
are absorbed.
It is affected by gastric emptying.
It is with small surface area.
Enterohepatic circulation:
Bile salts circulate between the liver and intestine about 8
times during the day. A number of drugs form conjugates
with glucuronic acids in the liver and are excreted in the
bile. These glucuronides are too polar (ionized) to be
reabsorbed; they therefore remain in the gut where they
are hydrolyzed by enzymes and bacteria to release the
parent drug which is then reabsorbed and reconjugated in
the liver.
About 20% of total drug in the body is affected by
enterohepatic circulation which prolongs drug action as
e.g: with drug of sulindac (analgesic)
Pentaerythritol (prophylaxis angina)
Ethinyloestradiol (oral contraceptive)
Bioavailability:
Same dose of a drug if given i.v or orally will achieve a
different plasma concentration.
I.V administration of the drug, it enters the systemic
circulation. The drug is with 100% available.
If the same quantity of the drug is swallowed. It will
pass through portal circulation and then the systemic
blood, i.e. its availability is less than 100%. So
Bioavailability is the fraction of administered drug that
reaches the systemic circulation.
e.g: a 50% bioavailability of a tablet of 10 mg propranolol
would mean that a total of 5mg of propranolol would
reach the general circulation.
For determination of bioavailability of a drug. It is
determined by comparing plasma level of a drug after oral
administration with plasma drug level achieved by i.v
injection in which all of the agent enters the circulation.
When the drug is given orally, only part of the
administered dose appears in the plasma.
By plotting plasma concentration of the drug versus time ,
one can measure the area under the curve ( AUC). This
curve reflects the extent of absorption of the drug .
Factors that influence bioavailability:
1) Pharmaceutical factors:
The amount of drug that is released from a dosage form is
highly dependent on its particle size, adhesive substances,
salt form and excipients, alter the rate of absorption.
2) Biological factors: may alter the rate of Absorption as in case:
Disease of gut (diarrhea, vomiting…….etc).
Presence or absence of food.
Metabolism inside the gut wall.
Determination
of the
bioavailability
of a drug.
(AUC = area
under curve.)

Unit 1: Principles of Drugs Therapy
12
3) First-pass hepatic metabolism:
When a drug is absorbed across the GIT, it enters the
portal circulation before entering the systemic circulation.
As in figure.
First-pass metabolism can occur with orally administered
drugs
If the drug is rapidly metabolized by the liver, the amount
of unchanged drug reaches to the systemic circulation is
decreased. Many drug such as propranolol or lidocaine,
undergo significant biotransformation during a single
passage through the liver.
Distribution:
Is the process by which a drug reversibly leaves the blood
stream and enters the extracellular fluid and/or the cells of
tissues. This depends on blood flow, capillary
permeability. The degree of drug binding to plasma
protein plays an important rule for drug distribution
through the cells.
Plasma protein binding:
-The bound fraction is pharmacologically inactive.
-The free fraction is pharmacologically active.
-The bound fraction acts as a reservoir for the drug.
-Free and bound fraction are at equilibrium.
-Free fraction is subjected to metabolism and excretion.
The main plasma proteins are:
Albumin
The binding of drug to albumin is reversible. It is the
main binding protein for many natural substances. It is
with low affinity to the basic but with good capacity (take
high amount of drug; a number of drug molecules binding
to a single albumin molecule) i.e. a lot is bounded but it is
readily released. Albumin is with high affinity to acidic
drug but with low capacity (one molecule of albumin with
one molecule of drug).
Competition for binding between drugs:
When two drugs are given, each with high affinity for
albumin, they compete for the available binding sites. The
drugs with high affinity for albumin can be divided into
two classes, depending on whether the dose of drug (the
amount of drug found in the body under conditions used
clinically) is greater than or less than the binding capacity
of albumin.
Class I drugs: If the dose of drug is less than the binding
capacity of albumin, then the dose/capacity ratio is low. The
binding sites are in excess of the available drug, and the
bound-drug fraction is high. This is the case for Class I drugs,
which included the majority of clinically useful agents.
Class II drugs: These drugs are given in doses that greatly
exceed the number of albumin binding sites. The
dose/capacity ratio is high, and a relatively high proportion
of the drug exists in the free state, not bound to albumin.
Clinical importance of drug displacement:
This assignment of drug classification assumes
importance when a patient taking a Class I drug, such as
warfarin, is given a Class II drug, such as a sulfonamide
antibiotic. Warfarin is highly bound to albumin, & only a
small fraction is free. This means that most of the drug is
sequestered on albumin and is inert in terms of exerting
pharmacologic actions. If a sulfonamide is administered,
it displaces warfarin from albumin, leading to a rapid
increase in the concentration of free warfarin in plasma,
because almost 100 percent is now free, compared with
the initial small percentage. [Note: The increase in
warfarin concentration may lead to increased therapeutic
effects, as well as increased toxic effects, such as bleeding

Unit 1: Principles of Drugs Therapy
13
Other binding proteins in the blood include
lipoprotein
&
α
1
-acid glycoprotein
, both of which are good for basic
drugs such as quindine, chloropromazine & imipramine
Other binding proteins in the blood include,
globulines
are good for the binding with thyroxine & sex hormones.
Note: Disease may modify the protein binding of the
drugs as in renal failure and chronic liver disease. e.g in
liver cirrhosis, plasma proteins are high, while in
pregnancy, they are low.
Note: For a drug to be effective therapeutically, it has to
achieve adequate plasma levels of the unbound form.
Drugs vary in the period required to reach equilibrium
between the body fluid and tissues.
When a drug is extensively bound to tissues. It has a large
apparent volume of distribution (v
d
).
For e.g, if 25mg of a drug (D=25mg) is administered and
the plasma concentration is 1.0mg/L, then the
v
d
= 25mg / 1.0mg /L=25L.
Metabolism:
In the body, the drugs are treated as foreign substances.
So
Drugs are subjected to various mechanisms to
get rid of them
1) Reducing lipid solubility.
Metabolic reactions are intended to reduce lipid solubility
and increase water solubility and then filtered by the
kidney without reabsorption.
2) Reducing biological activity.
Many possibilities:
a. A drug may be changed from active to inactive
metabolites, most drugs undergo such conversion.
b. A drug may change from active to pharmacological
active metabolites.
Diazepam (hypnotic) Oxazepam
(active) (active)
Amitriptyline (antidepressant) Nortriptyline
(active) (active)
c. A drug may be changed from inactive to
pharmacological active metabolites.
Inactive Active
Levodopa (Antiparkinsonism) Dopamine
Benorylate (analgesic) Salicylicacid + paracetamol
Enalapril (ACE
I
)
Enalaprilate
Chloral hydrate (hypnotic) Trichloroethanol
Metabolic processes:
The liver is the most important drug metabolizing organ,
other organ: kidney, gut, mucosa, lung and skin are also
important for metabolizing of some drugs.
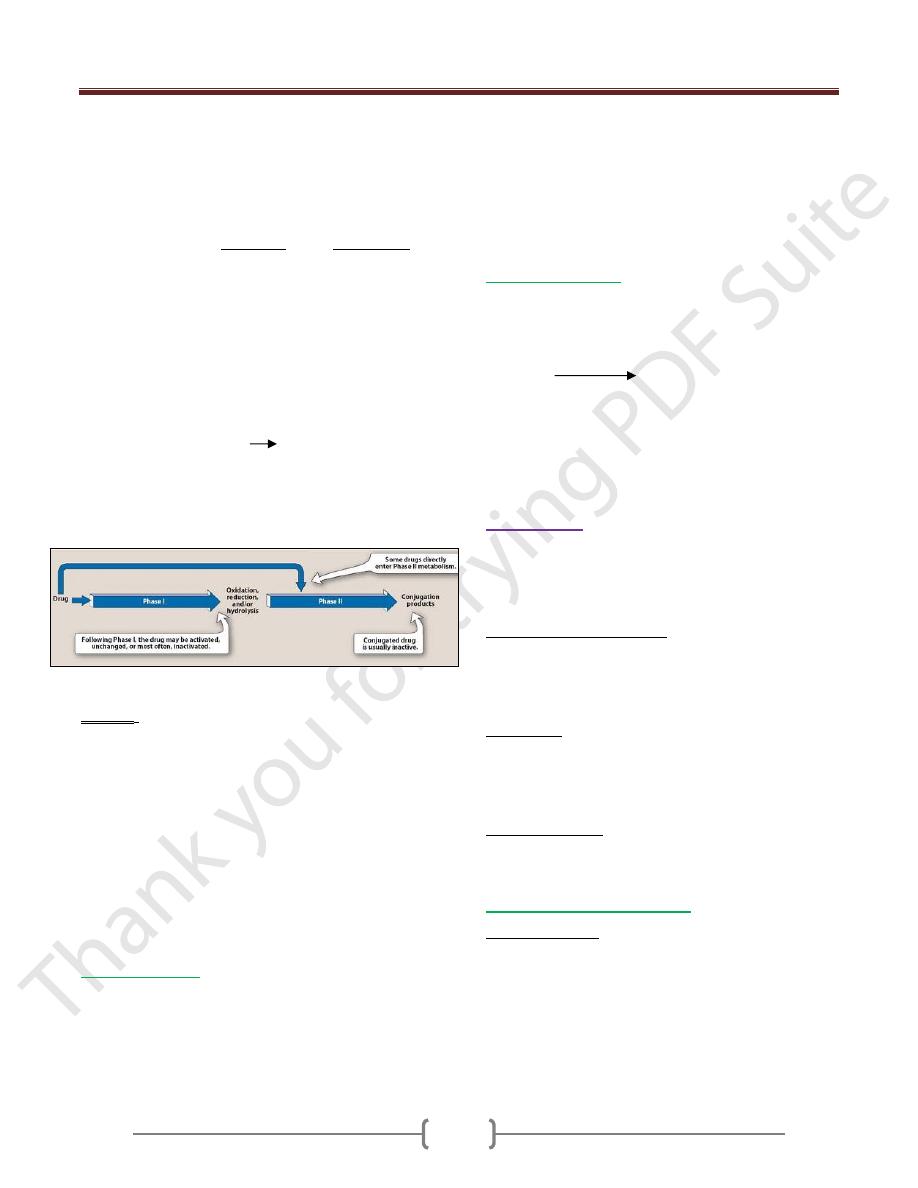
Metabolism involves in two broad phases:
Phase I: The change in drug molecule is by oxidation,
reduction or/and hydrolysis. The new metabolites may
have some pharmacological activity or without
pharmacological activity, but these new metabolites differ
from the parent drug molecules.
Binding of
Class I and
Class II drugs to
albumin when
drugs are
administered
alone (A, B) or
together (C).
Unit 1: Principles of Drugs Therapy
14
Phase I oxidation of some drugs results in the formation
of epoxides, which are short lived and highly reactive
metabolites. Epoxides bind to cell constituents
irreversibly through covalent bound. Such binding may
lead to toxicity. Gluathione in the liver helps the reduction
of epoxides toxicity.
e.g with the toxicity of Halothane or with paracetamol.
Glutathione is a part of an important defense mechanism
against hepatic damage by these two drugs. In case of
high toxicity with paracetamol the Glutathione quantities
are not sufficient to remove the oxidizing paracetamol. So
that means this quantity of paracetamol is toxic.
Phase I reaction utilized the P450 system: the Phase I
reactions most frequently are catalyzed by the cytochrome
P450 system (oxygen is introduced through a reduction step
coupled to NADPH:cytochrome P450 oxidoreduactase).
Drug + O
2
+ NADPH + H+ Drug
modified
+ H
2
O +
NADP+
The P450 system is important for metabolism of many
endogenous compounds (steroid, lipids, etc.) and for
biotransformation of exogenous substances.
The biotransformation of drugs
Phase II: The change in the drug molecule is by
conjugation. The conjugation is by glucuronic acid
sulphate or acetylate which is a polar molecule. The
kidney readily eliminates the conjugation compound,
results in polar usually more water-soluble compounds
that are most often therapeutically inactive.
e.g morphine, paracetamol and salicylate form
conjugation with sulfates.
Note: Not all drugs undergo Phase I and II reactions in
that order. For example, isoniazid is first acetylated (a
phase II reaction) and then hydrolyzed to isonicotinic acid
(a phase I reaction).
Enzyme induction:
Number of drugs such as rifampicine, ethanol or
Carbamazepine increase in the amount and activity of
microsomal enzymes and conjugating systems when
administered repeatedly.
The enzyme induction may use to treat certain disease as
e.g In neonatal jaundice, phenobarbitone is used to
increase the enzyme activity by induction leading to
decrease bilirubin.
Enzyme induction may cause a disease as e.g:
Treatment of patient by Phenytoin may cause increase in
vitamin D metabolism and causes rickets or osteomalacia.
Enzyme inhibition:
Some drugs may inhibit the enzyme. Such inhibition may
be of clinical application such as metabolism of
endogenous substance.
Xanthine oxidase
Xanthine Uric acid
Allopurinol inhibits the Xanthine oxidase for treatment of
gout. Other e.g Acetazolamide inhibits enzyme carbonic
anhydrase for treatment of glaucoma.
Angiotensin converting-enzyme inhibitors (ACEI
s
) as by
captopril to treat hypertension.
Elimination:
Drugs are eliminated from the body after being partly or
wholly converted to water-soluble metabolites or in some
cases without being metabolized. Accumulation of the
drug in the body, this may lead to toxic effects.
The causes of accumulation:
1. Frequent dosing more than supposed to be taken.
2. Renal failure this will reduce the elimination of a drug.
3. Hepatic failure or liver disease in this case the drug is
not metabolized by the liver.
Side effects:
They are unwanted effects occurring during therapy.
Nausea and vomiting may occure in the case of morphine
therapy. Diarrhea may occur due ampicillin
administration.
Secondary effects: They are the indirect results occurring
during drug therapy. As with tetracycline (as antibiotic)
may cause intestinal flora.
Organs for drug elimination:
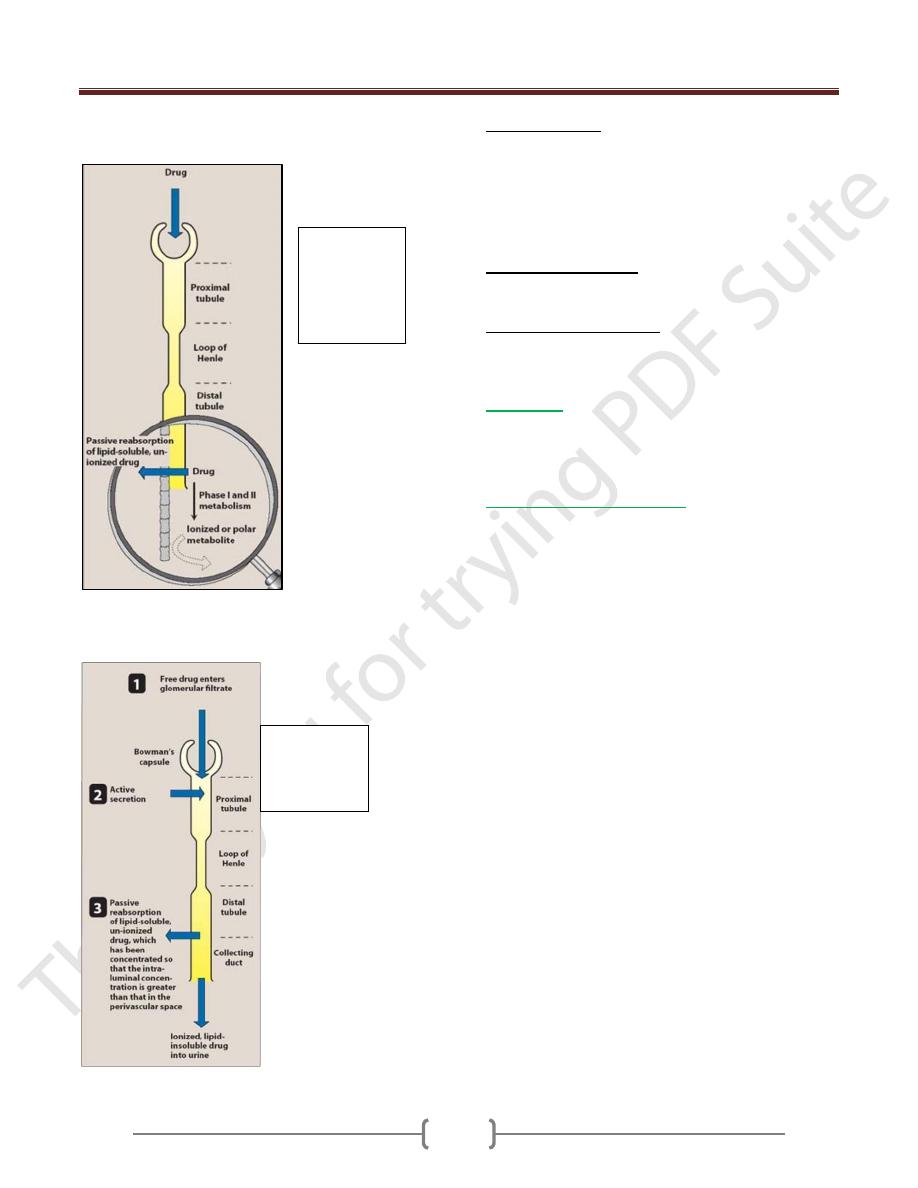
1) Renal elimination: it is the major organ of drug
elimination. The following mechanism are involved:
a. Glomerular filtration: it depends on the concentration of
free drug in plasma and its M.wt. Substances less than
1000 M.wt (including most drug) pass easily through the
pores of Glomerular membrane.
b. Renal tubular excretion: Strongly charged (ionized)
molecules are excreted by this system.
Unit 1: Principles of Drugs Therapy
15
c. Renal tubular reabsorption: if the fluid in the renal
tubules becomes more alkaline.
Acidic drug becomes less lipid soluble & its reabsorption
diminishes (ionized). Basic drug becomes unionized. It is
more lipids soluble. So its reabsorption increases.
2) Fecal elimination:
When a drug intended for systemic effect is taken by
mouth a proportion may remain in the bowel and be
excreted in the feces.
Or drug diffused from blood into the intestinal lumen and
is excreted in the feces.
Or bilary drug excretion that finally excreted in the feces.
3) Pulmonary elimination: only this route eliminates gases
and volatile liquids. Inhalation of general anesthetic agent
such as halothane is mainly eliminated through the lungs.
4) Other routes of elimination: sweat glands, salivary and
milk glands. The amount of drug excreted in these routes
is very small.
Clearance:
Clearance value can provide useful information about the
biological fate of a drug. It is the rate of elimination of
drug in urine relative to plasma concentration.
Prolongation of drug action:
1. Using large dose that means with increase of the
dose, but there is increased in toxic effect. This is not
always correct, other mechanisms are used.
2. Using vasoconstrictor agent will reduce local blood
flow so that distribution of drug away from an
injection site is retarded e.g local anaesthetic action is
prolonged by combination with adrenaline.
3. By slowing metabolism of drug as Carbidopa is used
in combination with Levodopa for Parkinsonism.
4. By delaying drug excretion as the use of probenecid
to block the renal excretion of penicillin when the
letter is used in single dose to treat gonorrhea.
5. By altering of molecular structure as with group of
hypnotic agents (benzodiazepine).
6. Pharmaceutical formulation as with sustained release
preparations.
Effect of drug
metabolism on
reabsorption in
the distal
tubule.
Drug
elimination
by the kidney

Unit 1: Principles of Drugs Therapy
16
Inherited influences (Pharmacogenetics):
Is concerned with drug response. Some individuals might
expected to respond to a fixed dose of drug, some would
show less than the usual response, most would show the usual
response & some would show more than usual response.
Heritable conditions causing increased of drugs
responses:
a. Acetylator status: Acetylation is an important rate of
metabolism for many drugs. As in case of slow
Acetylator of Isoniazide may cause peripheral
neuropathy, in case of rapid Acetylator causes
hepatocellular necrosis due to hepatotoxic
metabolites.
b. Defective carbon oxidation: it causes special risk of
adverse effects. As with standard doses of drugs that
include Bufuralol, metoprolol, timolol (increased
beta-blocked), haloperidol (excessive sedation).
c. Glucose-6-phosphate dehydrogenase (G-6PD)
deficiency: it is an important enzyme for the integrity
of RBCs. Individuals who are G-6PD deficient may
suffer acute hamolysis. Drugs that carry definite risk
of hamolysis e.g with Dapson, nitrofurantoin,
Primaquine, Quinolones and with less extent with
aspirin.
d. Pseudocholinesterase deficiency: it is non-specific
cholinesterase in tissue and plasma. The
neuromuscular blocking action of Suxamethonium is
terminated by plasma Pseudocholinesterase. In
deficiency state the metabolism of suxamethonium is
highly reduced.
Heritable conditions causing decreased drug
responses:
a. Resistance to coumarin (anticoagulant). Patients
require 20 times or more of the usual dose to obtain an
adequate clinical response.
b. Resistance to Heparin. Patients with Antithrombin III
deficiency require large doses of heparin for anticoagulant
effect.
c. Resistance to Suxamethonium: is characterised by
increased Pseudocholinesterase activity and failure of
normal doses of Suxamethonium to cause muscular
relaxation.
d. Resistance to vitamin D: individuals develop rickets
which responds only to large doses of vit D i.e. x1000 the
standard dose.