SINGLE GENE DISORDERS
AR Disorders
Dec. 14. 2015

Single Gene Disorders
• Cystic Fibrosis
• Phenylketonuria
• Galactosemia
• Glycogen storage disorders (Glycogenoses)
• Lysosomal Storage Diseases
• Neurofibromatosis
Multifactorial inheritance
Lect 3. Dec 2015

The three major categories of genetic
disorders:
(1) Single-gene disorders
(2) Multifactorial inheritance
(3) Chromosomal disorders.
(4) Single-gene disorders with nonclassic patterns of
inheritance.(Do not follow the Mendelian pattern
of inheritance).
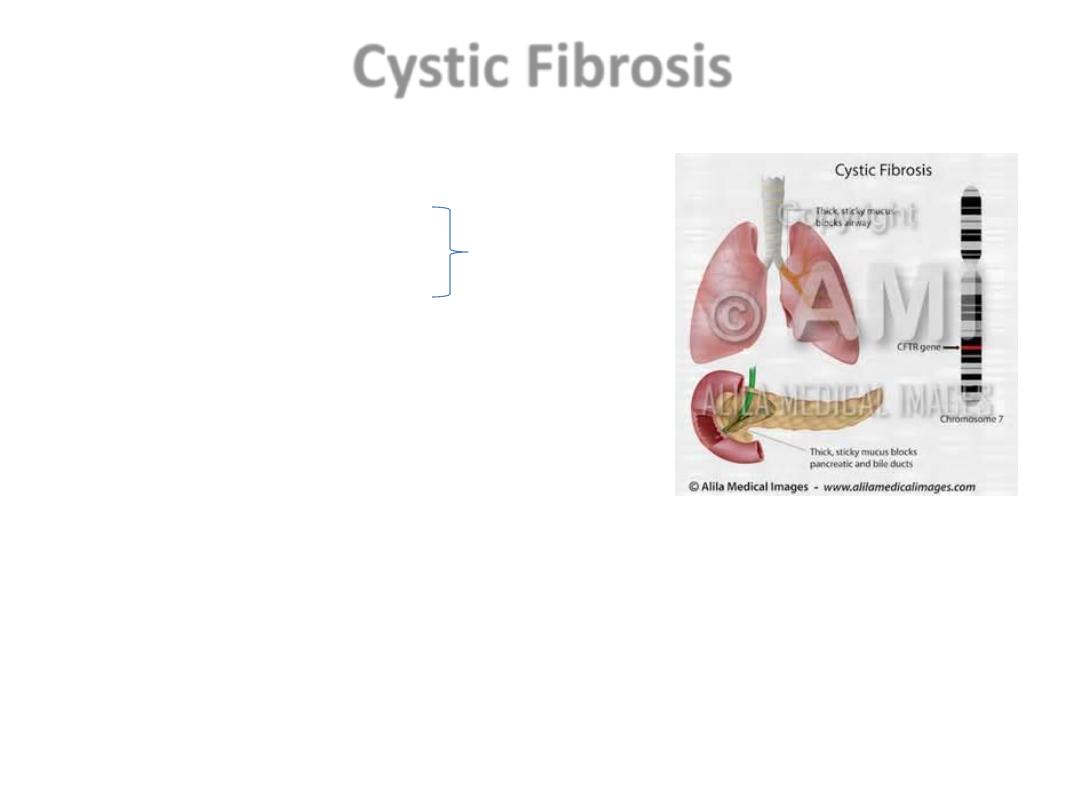
Cystic Fibrosis
AR
• Resp T., GIT., Reprod. sys.
• 1.Ch. Pulmonary infections
• 2. Pancreatic insufficiency (85 – 90%)
Dx: Hypertonic salty sweat.
• Malabsorption, Vit def, diarrhea,
Meconeum ileus
Causes of death;
• cardiopulmonary complications,
• Transplant complications,
• Liver failure.
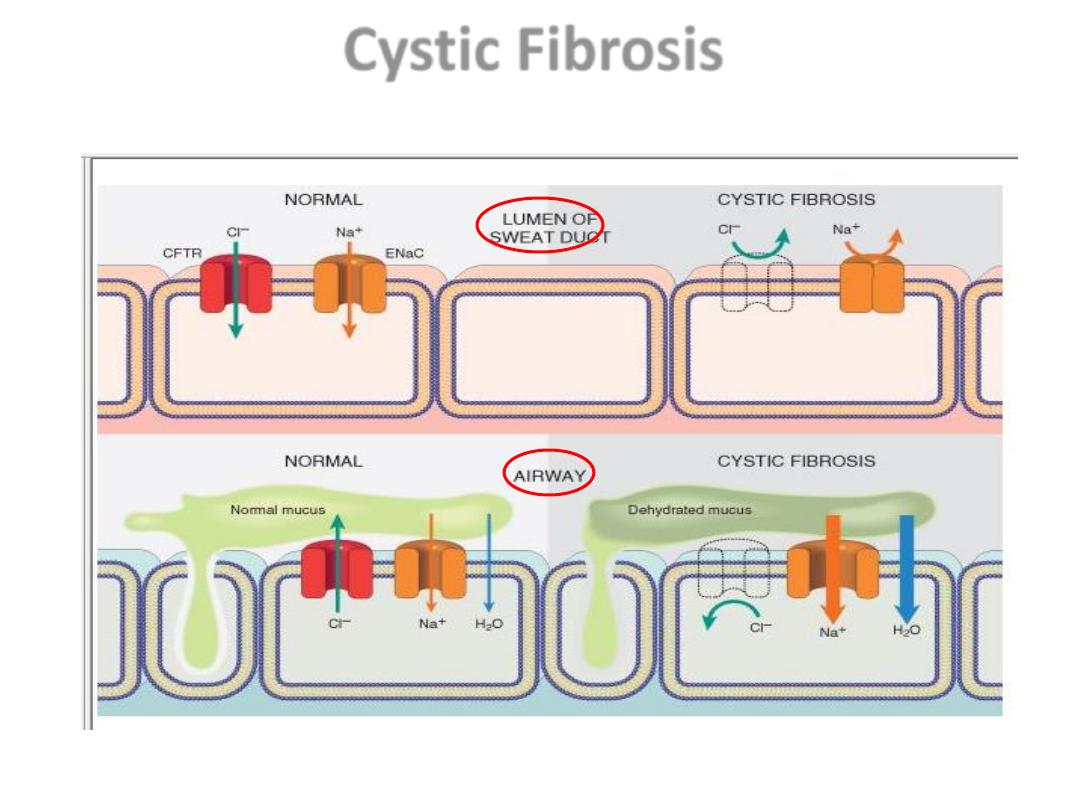
Cystic Fibrosis

• CL channel defect in the sweat duct (top)
causes increased Cl & Na conc. in sweat.
• In the airway (bottom), decreased CL.
secretion and increased Na & H2O
reabsorption leading to dehydration of the
mucus layer coating epithelial cells, defective
mucociliary action, and mucus plugging of
airways. CFTR, Cystic fibrosis transmembrane
conductance regulator; ENaC, epithelial
sodium channel.
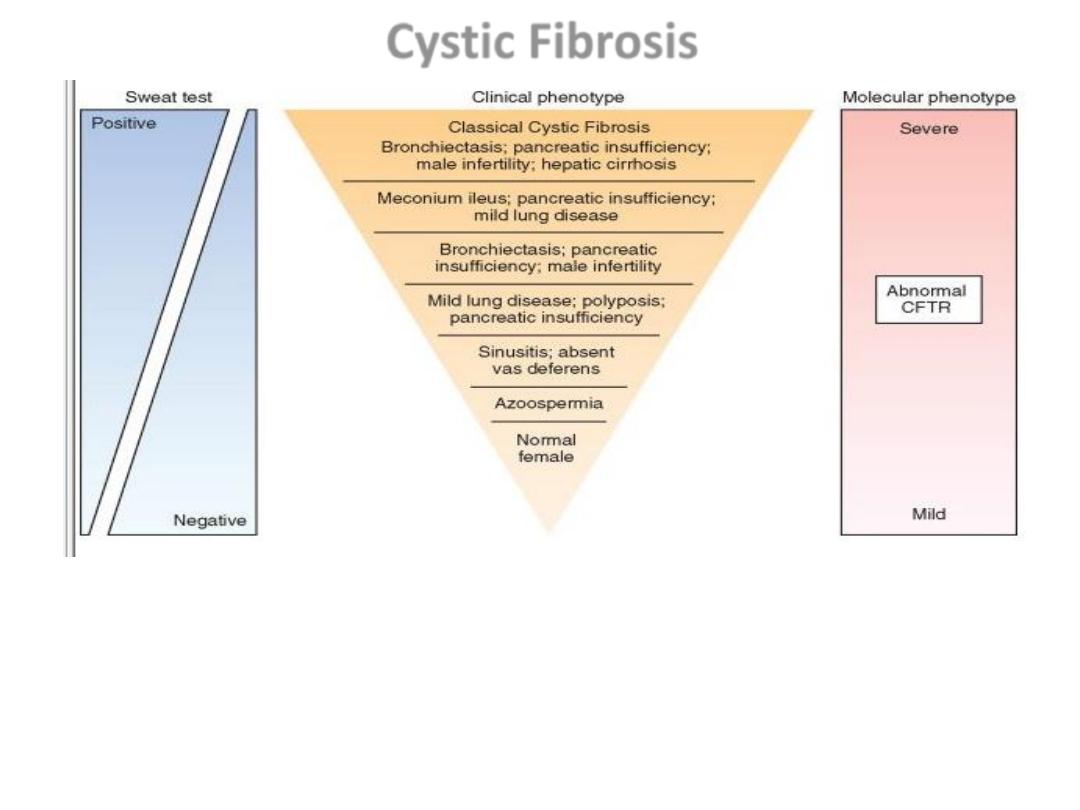
The many clinical manifestations of mutations in the cystic fibrosis gene, from
most severe to asymptomatic.
CFTR, Cystic fibrosis transmembrane conductance regulator
Cystic Fibrosis
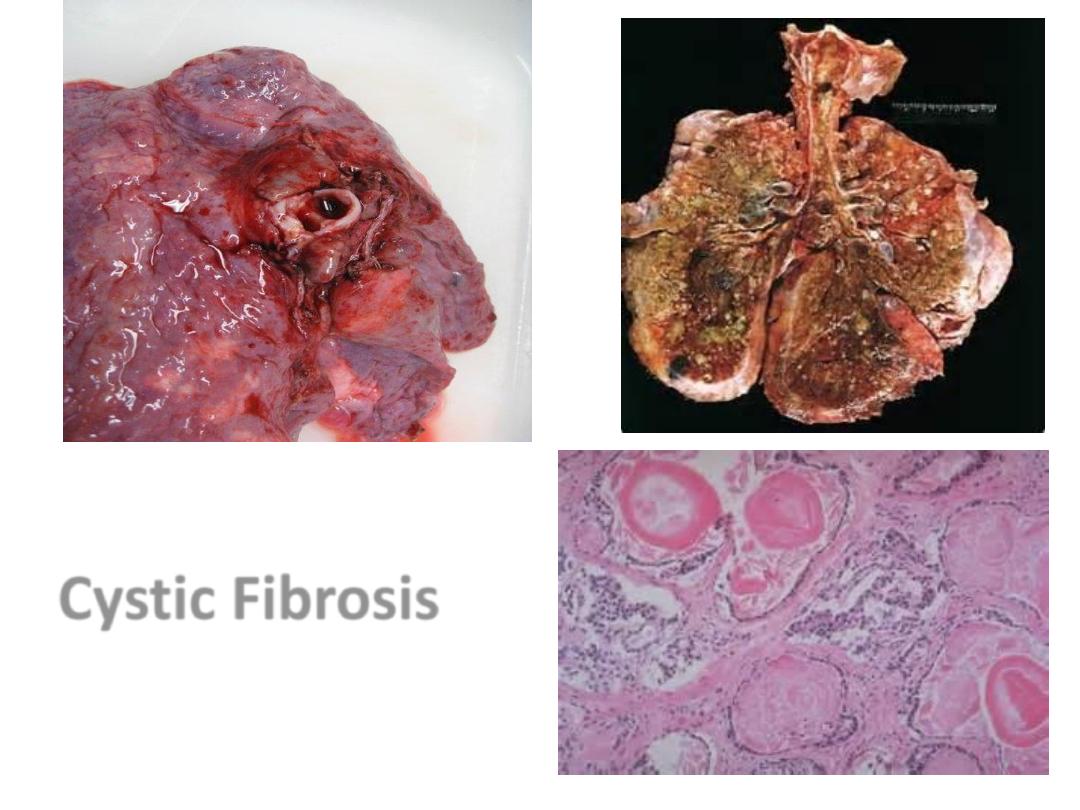
Pancreatiic CF
CF Lung
Cystic Fibrosis
Diseases Caused by Mutations in Genes
Encoding Enzyme Proteins
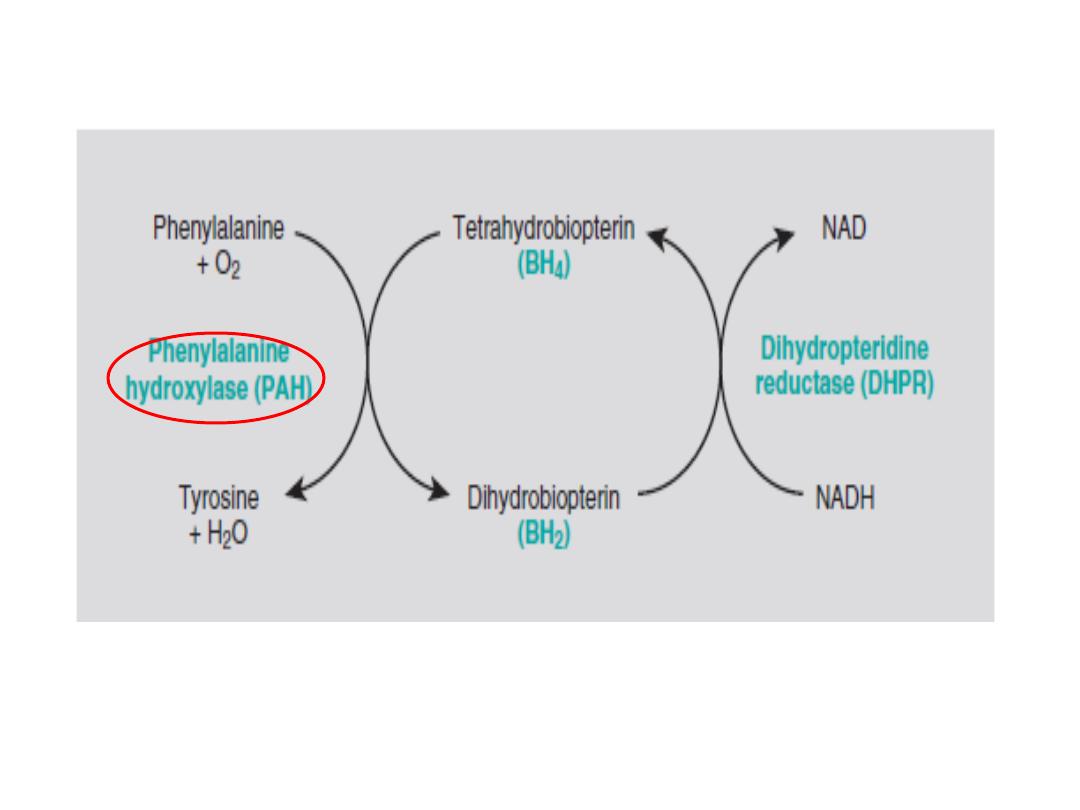
The phenylalanine hydroxylase system. NADH, nicotinamide adenine
dinucleotide, reduced form
Phenylketonuria
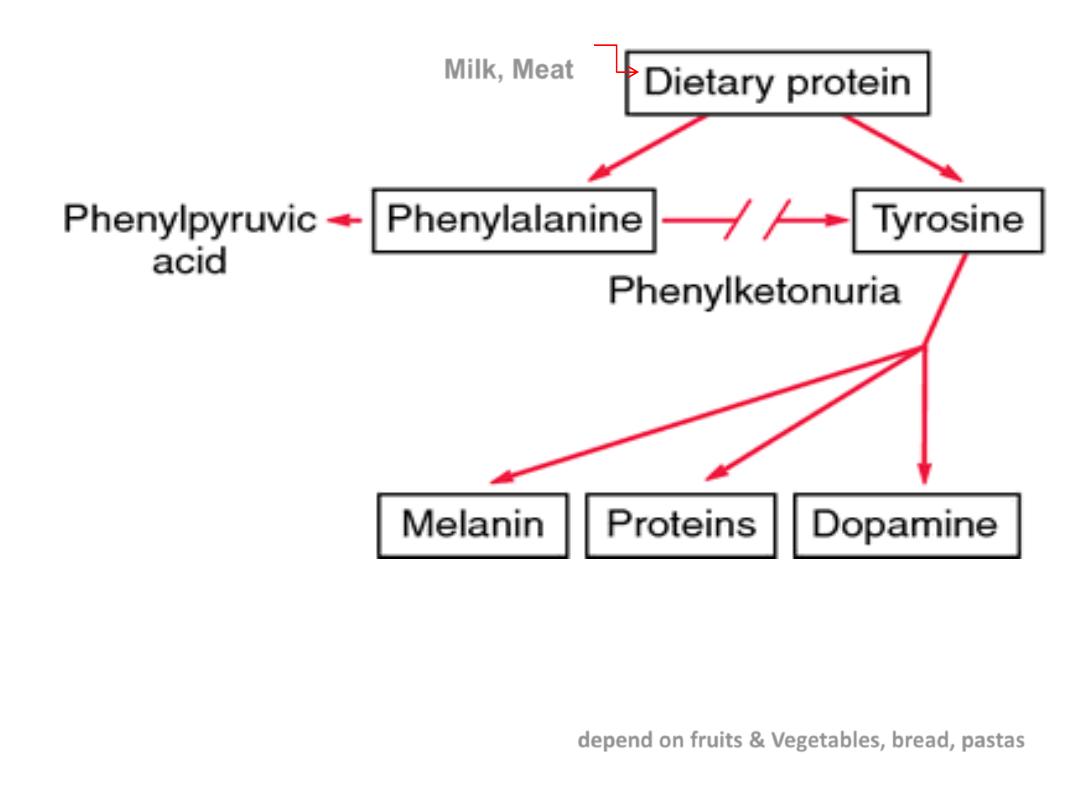
•
Deficiency of the enzyme---inability to convert phenylalanine into tyrosine, leads to intermediates
that excreted in sweat (odor), concomitant lack of tyrosine leads to lack of melanin.
•
Rx:
restriction of phenylalanine intake early in life.(
depend on fruits & Vegetables, bread, pastas
)
Milk, Meat

PKU
• AR
• Severe mental retardation,
seizures, and decreased
pigmentation of skin,
• can be avoided by restricting the
intake of phenylalanine in the
diet.
• Transplacental passage of
phenylalanine metabolites.
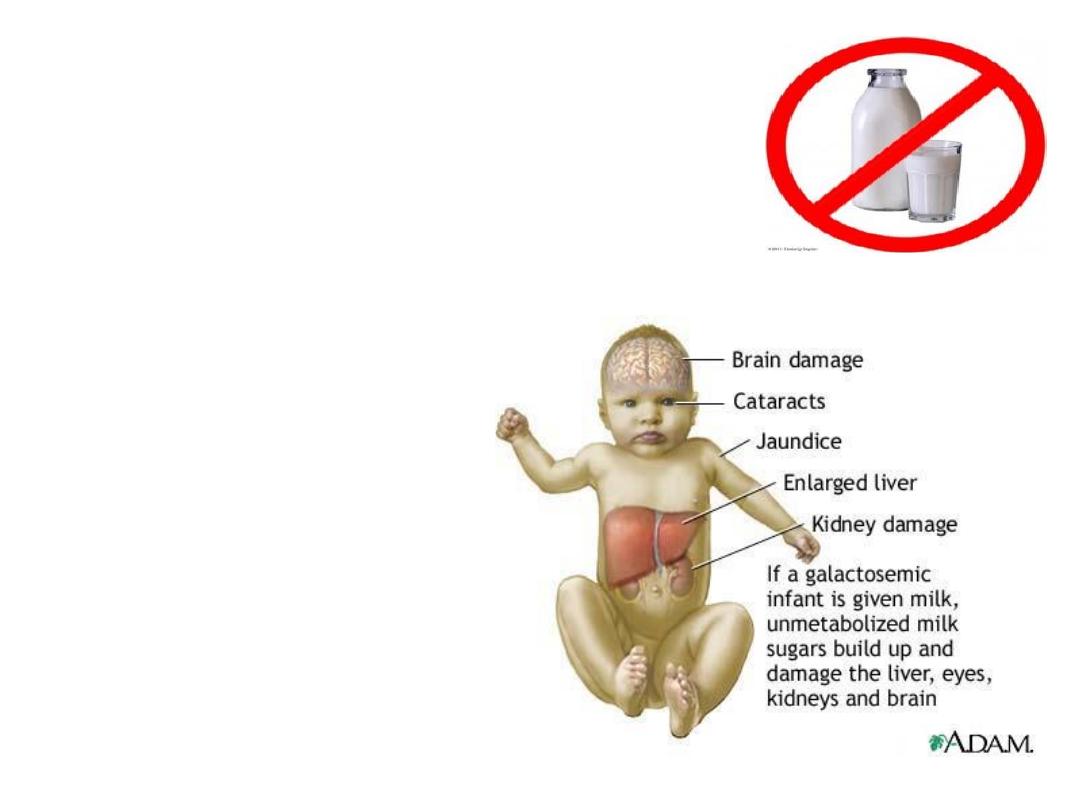
Galactosemia
• AR
• lack of the GALT enzyme,
• accumulation of
galactose-1-phosphate & its
metabolites in tissues.
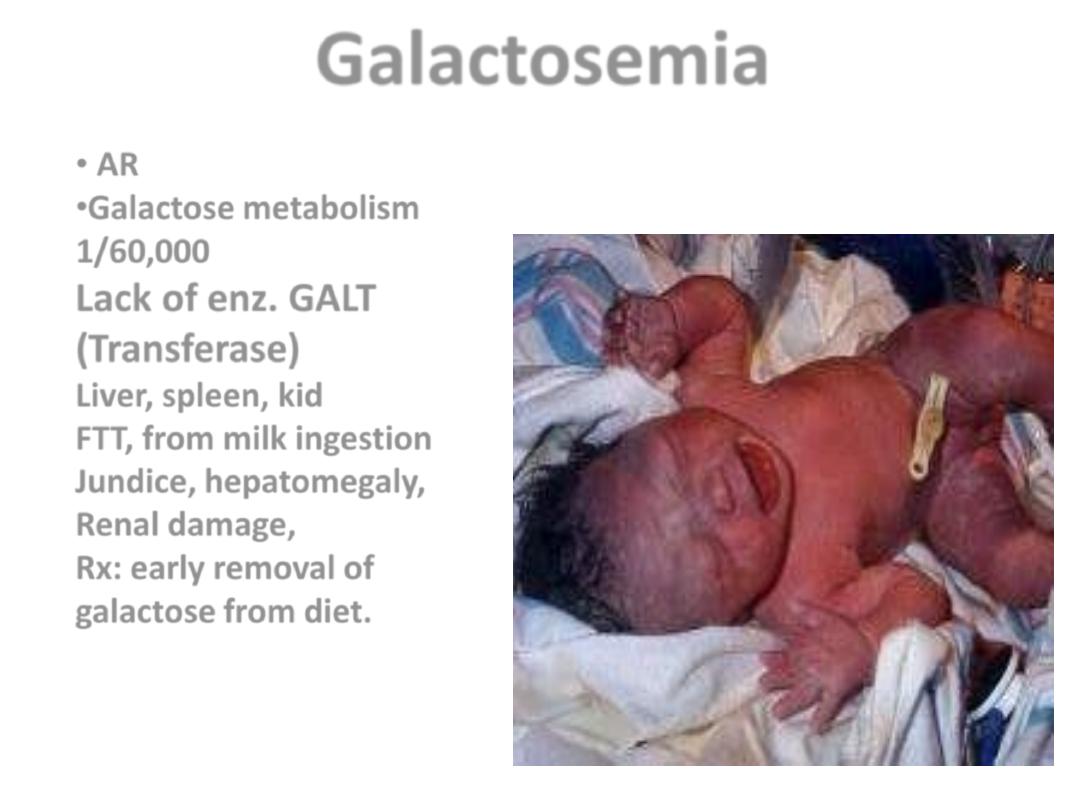
Galactosemia
• AR
•Galactose metabolism
1/60,000
Lack of enz. GALT
(Transferase)
Liver, spleen, kid
FTT, from milk ingestion
Jundice, hepatomegaly,
Renal damage,
Rx: early removal of
galactose from diet.

Glycogen storage disorders
(Glycogenoses):
• AR
• deficiency of enzymes involved in glycogen synthesis or
degradation,
• result in excessive accumulation of glycogen or abnormal
form of glycogen in various tissues
.

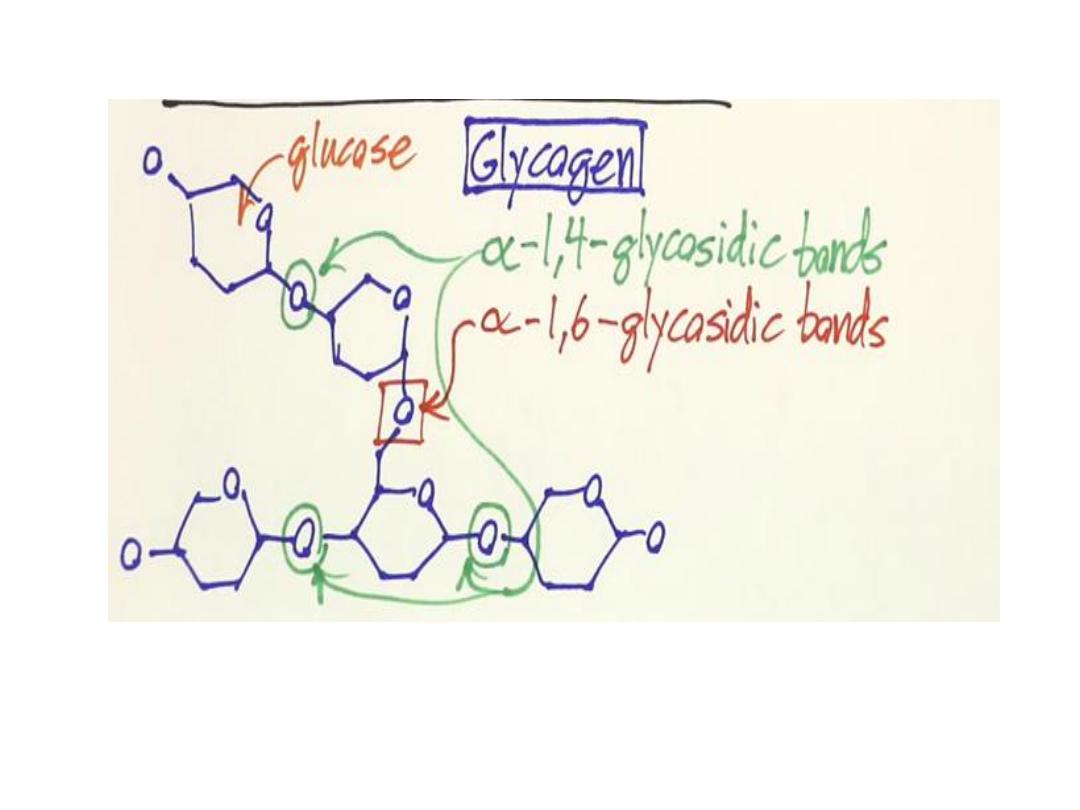


Glycogen storage disorders
1. Hepatic form:
• due to Glucose 6 phosphatase enzyme
deficiency called Von Gierke disease.

Glycogen storage disorders
2- Myopathic form:
• deficiency of muscle phosphorylase enz.
• (
McArdle disease
)

Glycogen storage disorders
• 3- Two other forms of glycolysis;
• Pompe; deficiency of lysosomal acid maltase,
deposition of glycogen in every organ but
cardiomegaly is prominent .
• Brancher glycogenoses, effect on the liver,
heart, muscles…
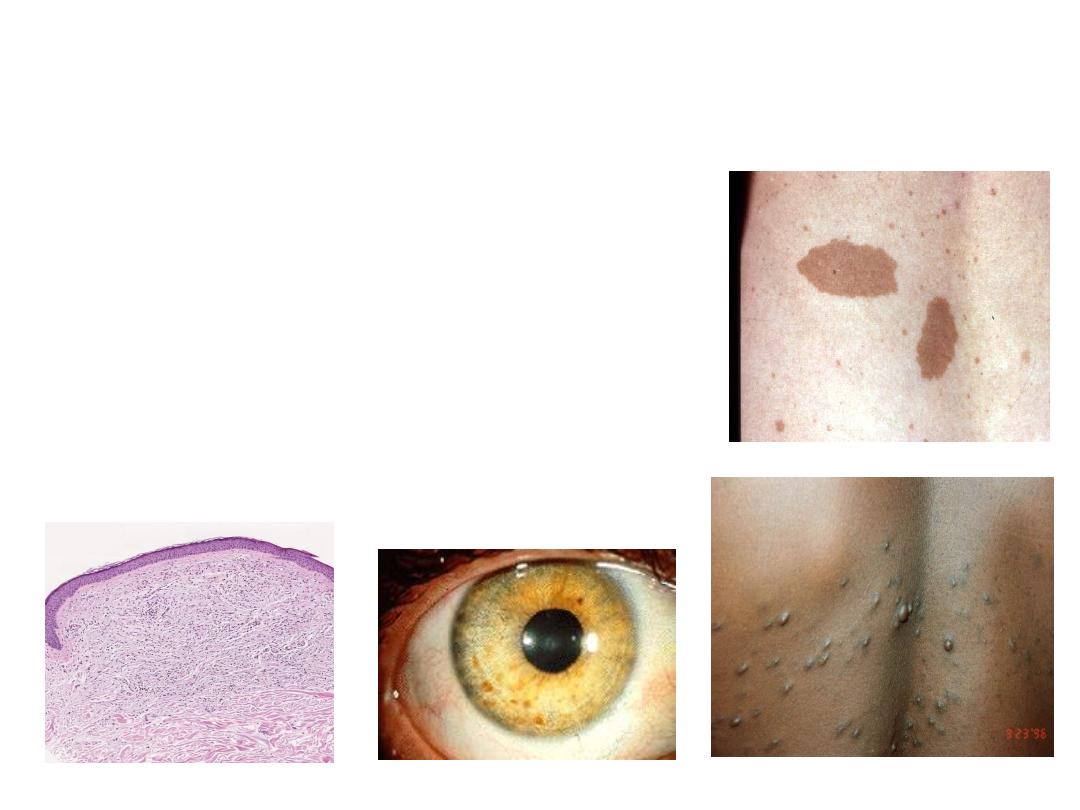
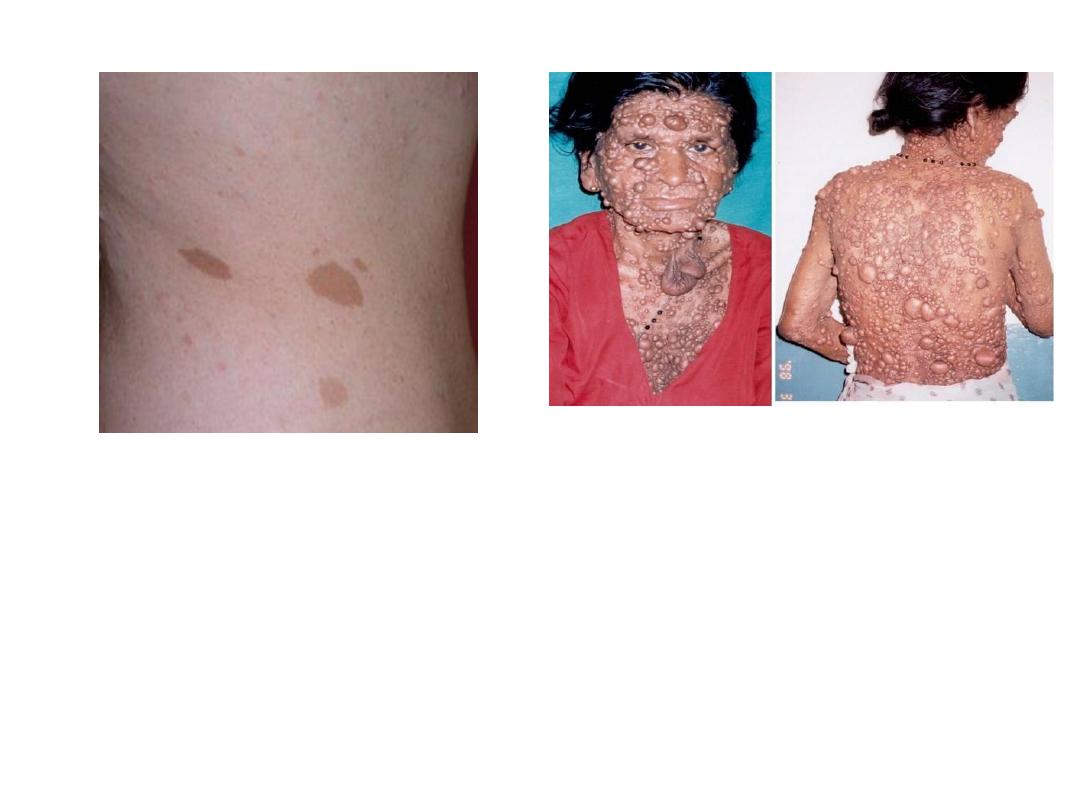
NEUROFIBROMATOSIS
1 and 2
• Diseases caused by mutation in
protein that regulate cell growth
• 1-von Recklinghausen
Neurofibromas, café-au-lait, Lisch
nodules
• 2- “acoustic” neurofibromatosis

• Neurofibromatosis (commonly
abbreviated NF) is a AD disease

Penetrance and Expressivity
.
• Incomplete penetrance ; Some individuals inherit
the mutant gene but are phenotypically normal.
• 50% penetrance indicates that 50% of those who
carry the gene express the trait.
• Variable expressivity; If a trait is seen in all
individuals carrying the mutant gene but is
expressed differently among individuals.
• Because of other genes or environmental factors
that modify the phenotypic expression of the
mutant allele

• Highly penetrant, meaning that the presence
of the mutation is associated with the
disease in a large proportion of individuals

Complex Multigenic disorders.
• Most common
• Multi-”FACTORIAL”, not just multi-GENIC
• No single gene is sufficient to produce the
disease. i.e. Multigenic or Polygenic
• Since environmental interactions are
important.
i.e. Multifactorial disorders.
• low penetrance

“MULTIFACTORIAL” DISORDERS
• Atherosclerosis, diabetes mellitus, hypertension,
and autoimmune diseases
• Cleft lip, palate
• Congenital heart disease
• Coronary heart disease
• Gout
• Pyloric stenosis
• Schizophrenia
• Psychological depression
• MANY, MANY, MANY, MANY MORE

MULTIFACTORIAL INHERITANCE
• Multi-”FACTORIAL”, not just multi-GENIC
• Common phenotypic expressions governed
by “multifactorial” inheritance
– Hair color
– Eye color
– Skin color
– Height
– Intelligence
– Diabetes, type II

• We are now moving the discussion up from
ONE gene MULTI-genes Parts of
chromosomes WHOLE chromosomes.

LYSOSOMAL STORAGE DISEASES
• Lysosomes; Digestive sys of the cell.
• Rich in hydrolase enz.
• Involve in breakdown of Sphingolipid &
Mucopolysaccharide.
• Enz. Def. Accumulation of – in cells.
• Hepatospleenomegaly
• CNS involvement.

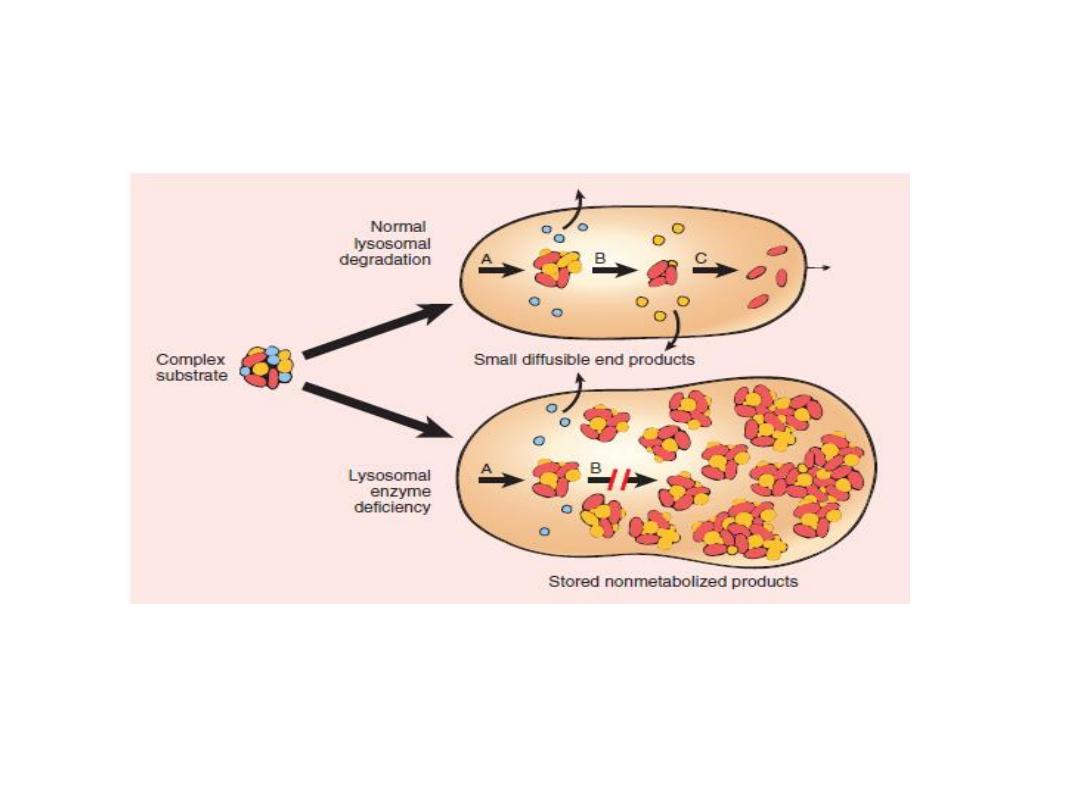
Lysosomal Storage Diseases
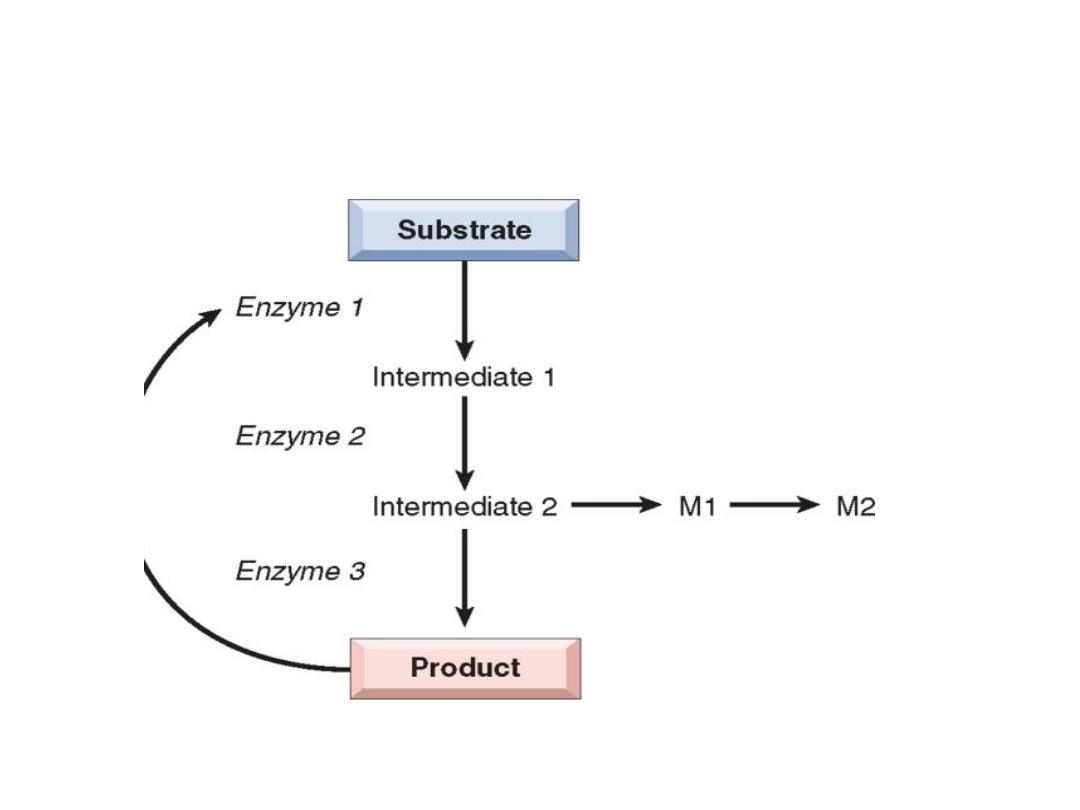
•
Pathogenesis of lysosomal storage diseases, a complex substrate is normally degraded by a series of
lysosomal enzymes(A, B, and C) into soluble end products
•
. If there is a deficiency or malfunction of one of the enzymes (e.g., B), catabolism is incomplete, and
insoluble ntermediates accumulate in the lysosomes.

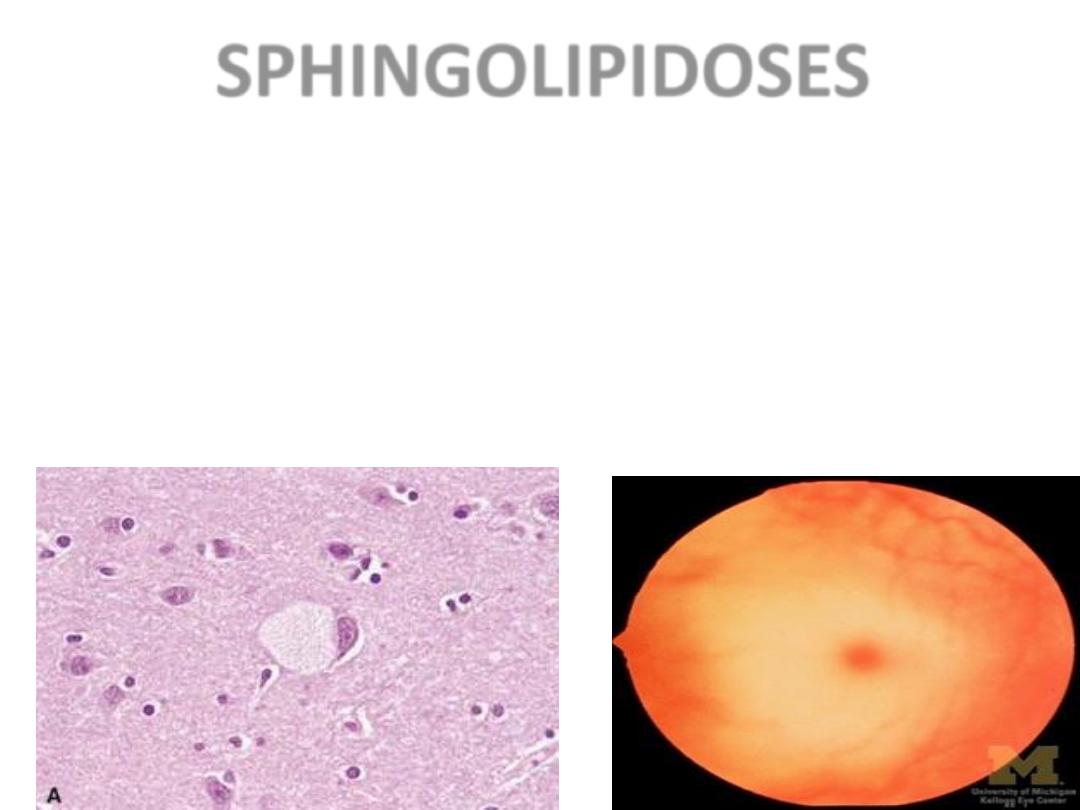
SPHINGOLIPIDOSES
• MANY types, Tay-Sachs most often referred to
– GANGLIOSIDES are ACCUMULATED
– Ashkenazi Jews (1/30 are carriers)
– CNS neurons a site of accumulation
– CHERRY RED spot in Macula
• Tay-Sachs disease (abbreviated TSD, also
known as
deficiency) is an
. In its most common variant known as
infantile Tay-Sachs disease it presents with a
relentless deterioration of mental and physical
abilities which commences at 6 months of age
and usually results in death by the age of four.
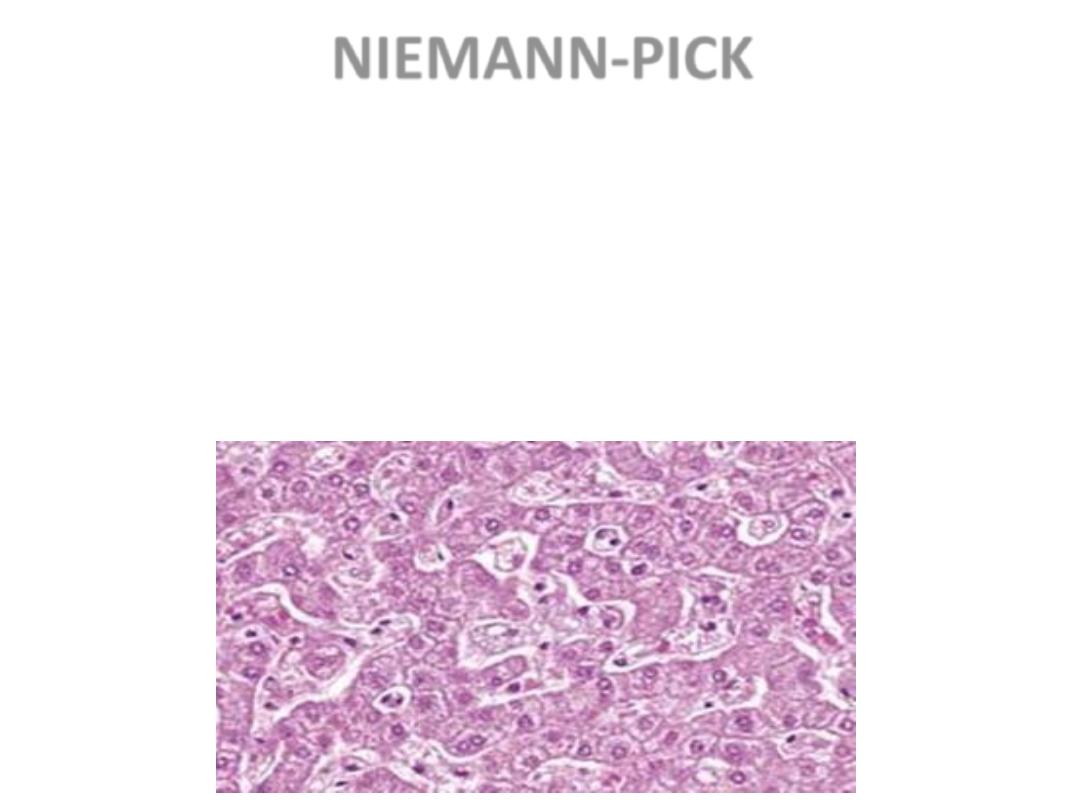
NIEMANN-PICK
• TYPES A, B, C
• SPHINGOMYELIN BUILDUP
• MASSIVE SPLENOMEGALY
• ALSO in ASHKANAZI JEWS
• OFTEN FATAL in EARLY LIFE, CNS,
ORGANOMEGALY
• Sphingomyelin (SPH), (sphin-go-my-e-lin (sfi
found
in animal
, especially in the
membranous
which surrounds
some
. It usually consists
and
. In humans
SPH represents ~85% of all sphingolipids.
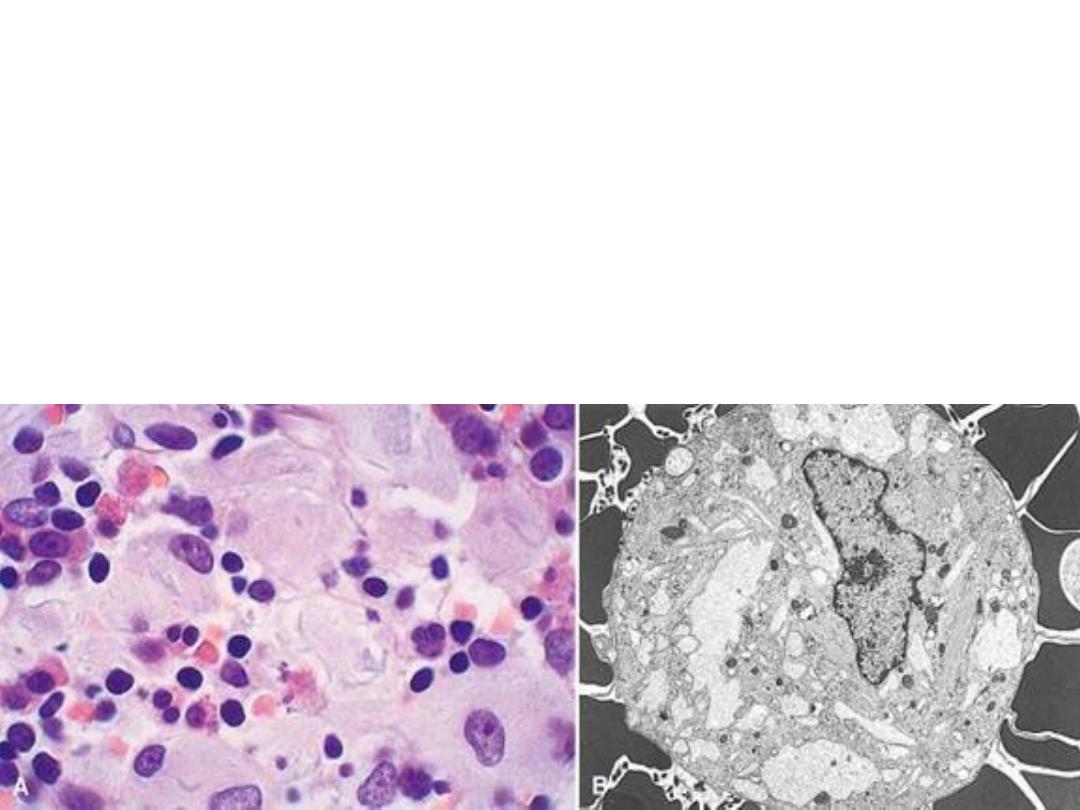
GAUCHER DISEASE
• GLUCOCEREBROSIDE BUILDUP
• 99% are type I, NO CNS involvement
• ALL MACROPHAGES, liv, spl, nodes, marrow

• Cerebrosides are
which are important
is the most well known cerebroside.
Glucocerebroside (also called glucosylceramide) is any of
the
is
• Gaucher's disease is the most common of the
. It is caused by a hereditary deficiency of the
glucosidase). The enzyme acts on a fatty
substance
(also known as
).
When the enzyme is defective, the substance accumulates,
particularly in cells of the mononuclear cell lineage.

MUCOPOLYSACCHARIDOSES
• HURLER/HUNTER, for I and II, respectively
• DERMATAN sulfate, HEPARAN sulfate
buildup
– coarse facial features
– clouding of the cornea
– joint stiffness
– mental retardation
– URINARY EXCRETION of SULFATES COMMON

Biochemical and Molecular Basis of Some Mendelian Disorders
Protein
Type/Function
Example
Molecular Lesion Disease
ENZYME
Phenylalanine
hydroxylase
Splice-site
mutation: reduced
amount
Phenylketonuria
Hexosaminidase
Splice-site
mutation or
frameshift
mutation with stop
codon: reduced
amount
Tay-Sachs disease
Adenosine
deaminase
Point mutations:
abnormal protein
with reduced
activity
Severe combined
immunodeficiency

ENZYME INHIBITOR
α
1
-Antitrypsin
Missense mutations:
impaired secretion from
liver to serum
Emphysema and liver
disease
RECEPTOR
Low-density lipoprotein
receptor
Deletions, point
mutations: reduction of
synthesis, transport to
cell surface, or binding
to low-density
lipoprotein
Familial
hypercholesterolemia
Vitamin D receptor
Point mutations: failure
of normal signaling
Vitamin D–resistant
rickets
TRANSPORT
Oxygen
Hemoglobin
Deletions: reduced
amount
α-Thalassemia
Defective mRNA
processing: reduced
amount
β-Thalassemia
Point mutations:
abnormal structure
Sickle cell anemia
Ions
Cystic fibrosis
transmembrane
conductance regulator
Deletions and other
mutations:
nonfunctional or
misfolded proteins
Cystic fibrosis

STRUCTURAL
Extracellular
Collagen
Deletions or point
mutations cause
reduced amount of
normal collagen or
normal amounts of
defective collagen
Osteogenesis
imperfecta;
Ehlers-Danlos
syndromes
Fibrillin
Missense mutations Marfan syndrome
Cell membrane
Dystrophin
Deletion with
reduced synthesis
Duchenne/Becker
muscular dystrophy
Spectrin, ankyrin, or
protein 4.1
Heterogeneous
Hereditary
spherocytosis
HEMOSTASIS
Factor VIII
Deletions, insertions,
nonsense mutations,
and others: reduced
synthesis or
abnormal factor VIII
Hemophilia A
GROWTH
REGULATION
Rb protein
Deletions
Hereditary
retinoblastoma
Neurofibromin
Heterogeneous
Neurofibromatosis
type 1

• Diseases caused by mutation in protein that
regulate cell growth:
• 2 classes of genes that regulate cell growth,
proto-oncogenes & cancer suppressor genes,
mutation affecting these genes lead to tumor.
• 5% of all CA, there is mutation affecting
certain tumor suppressor genes are present in
all cells of the body including germ cells---
transmitted to the offspring.

Move fast & break things

• Gene expression and mendelian traits are
usually described as dominant or recessive,
• in some cases both of the alleles of a gene
pair contribute to the phenotype—a
condition called codominance.
• Histocompatibility and blood group antigens
are good examples of codominant
inheritance

• With every AD disorder, some proportion
of patients do not have affected parents.
Such patients owe their disorder to new
mutations involving either the egg or the
sperm from which they were derived