1
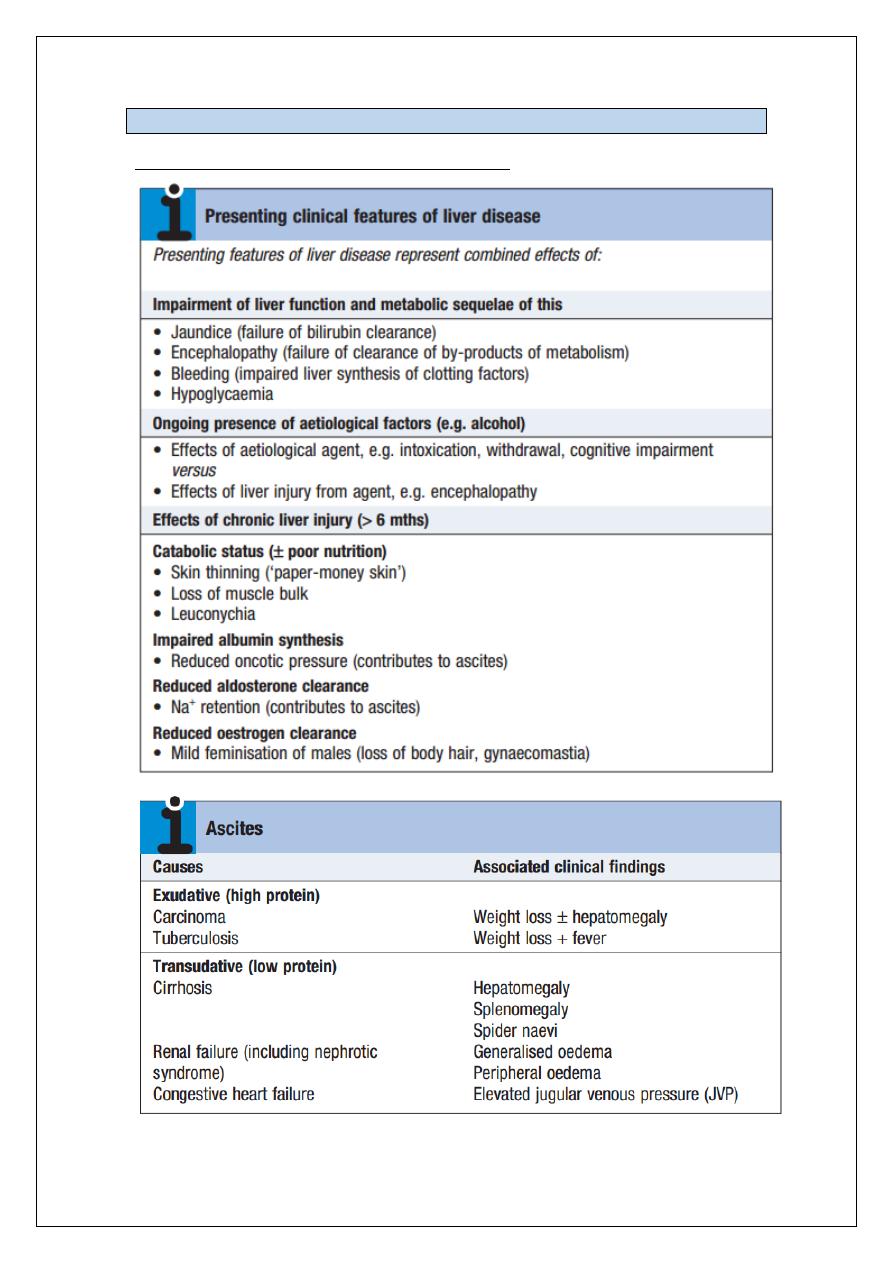
CLINICAL EXAMINATION OF THE ABDOMEN FOR LIVER AND BILIARY DISEASE
History and significance of abdominal signs
Applied anatomy
Normal liver structure and blood supply: The liver has multiple functions, including key roles
in metabolism, control of infection, elimination of toxins and by-products of metabolism.
2
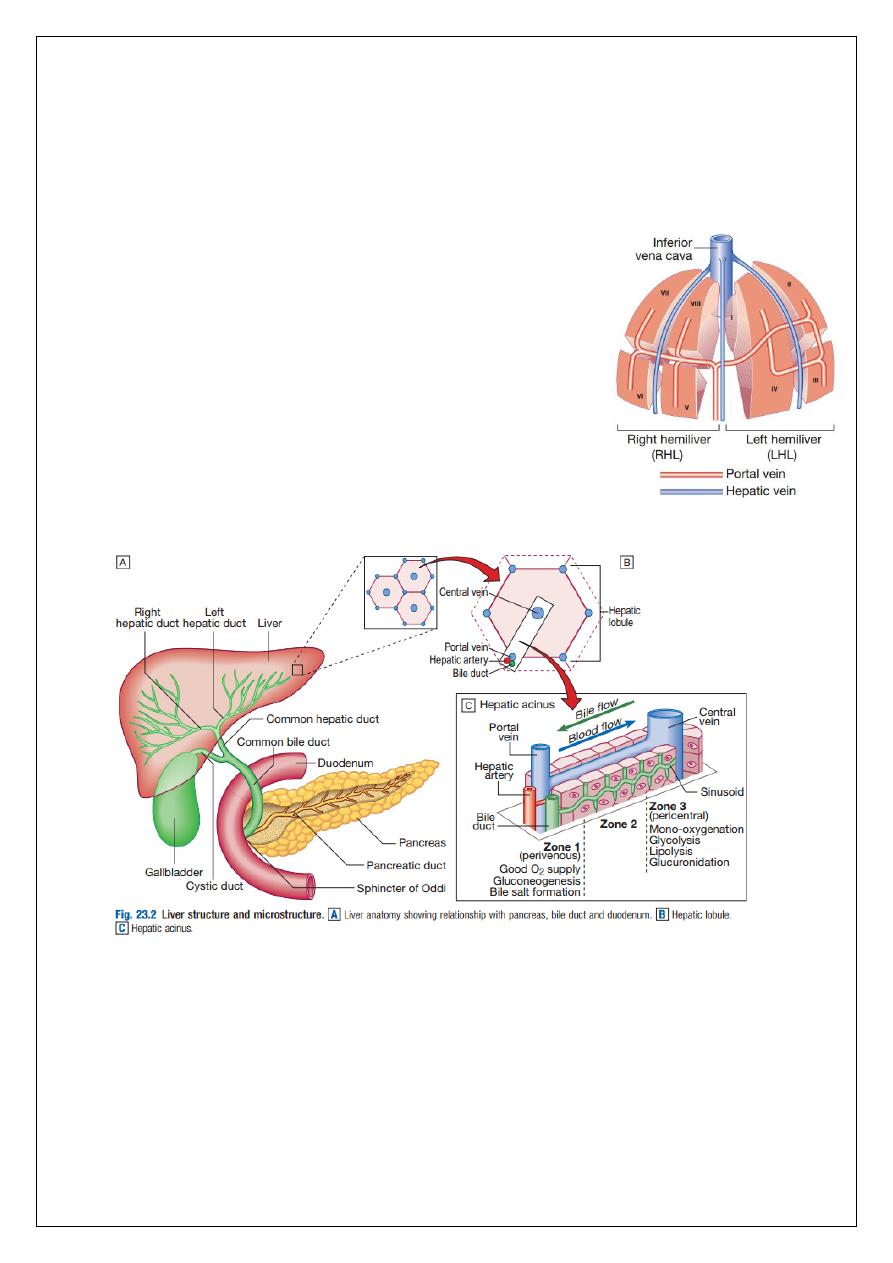
It is classically divided into left and right lobes by the falciform ligament, but a more useful
functional division is into the right and left hemilivers, based on blood supply. These are
further divided into eight segments according to subdivisions of the hepatic and portal veins.
Each segment has its own branch of the hepatic artery and biliary tree. The segmental
anatomy of the liver has an important influence on imaging and treatment of liver tumours,
given the increasing use of surgical resection.
A liver segment is made up of multiple smaller units known
as lobules, comprised of a central vein, radiating sinusoids
separated from each other by single liver cell (hepatocyte)
plates, and peripheral portal tracts. The functional unit of
the liver is the hepatic acinus. Blood flows into the acinus
via a single branch of the portal vein and hepatic artery
situated centrally in the portal tracts. Blood flows outwards
along the hepatic sinusoids into one of several tributaries of
the hepatic vein at the periphery of the acinus. Bile, formed
by active and passive excretion by hepatocytes into
channels called cholangioles which lie between them, flows
in the opposite direction from the periphery of the acinus.
The cholangioles converge in interlobular bile ducts in the portal tracts.
The hepatocytes in each acinus lie in three zones, depending on their position relative to the
portal tract. Those in zone 1 are closest to the terminal branches of the portal vein and
hepatic artery, and are richly supplied with oxygenated blood, and blood containing the
highest concentration of nutrients and toxins. Conversely, hepatocytes in zone 3 are furthest
from the portal tracts and closest to the hepatic veins, and are therefore relatively hypoxic
and exposed to lower concentrations of nutrients and toxins compared to zone 1. The
different perfusion and toxin exposure patterns, and thus vulnerability, of hepatocytes in the
different zones contribute to the often-patchy nature of liver injury.
3
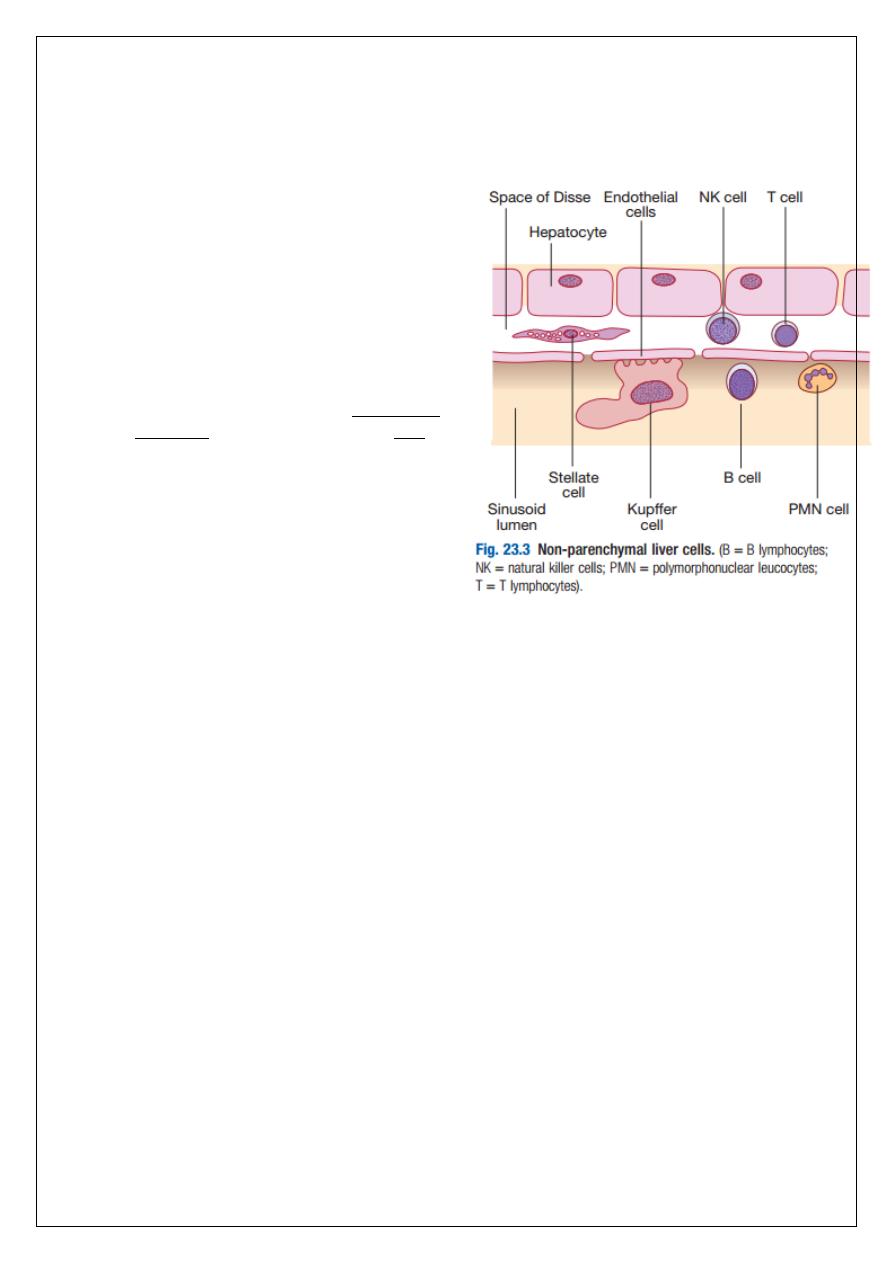
Liver cells:
Hepatocytes comprise 80% of liver cells. The
remaining 20% are the endothelial cells lining
the sinusoids, epithelial cells lining the
intrahepatic bile ducts, cells of the immune
system (including macrophages (Kupffer cells)
and unique populations of atypical
lymphocytes), and a key population of non-
parenchymal cells called stellate or Ito cells.
Endothelial cells line the sinusoids, a network of
capillary vessels that differ from other capillary
beds in the body in that there is no basement
membrane. The endothelial cells have gaps
between them (fenestrae) of about 0.1 micron
in diameter, allowing free flow of fluid and
particulate matter to the hepatocytes.
Individual hepatocytes are separated from the
leaky sinusoids by the space of Disse, which
contains stellate cells that store vitamin A and
play an important part in regulating liver blood
flow. They may also be immunologically active
and play a role in the liver’s contribution to defence against pathogens. The key role of
stellate cells in terms of pathology is in the development of hepatic fibrosis, the precursor of
cirrhosis. They undergo activation in response to cytokines produced following liver injury,
differentiating into myofibroblasts, which are the major producers of the collagen-rich
matrix that forms fibrous tissue.
Blood supply
The liver is unique as an organ as it has dual perfusion, receiving a majority of its supply via
the ❶portal vein, which drains blood from the gut via the splanchnic circulation and is the
principal route for nutrient trafficking to the liver, and a minority from the ❷hepatic artery.
The portal venous contribution is 50–90%. The dual perfusion system, and the variable
contribution from portal vein and hepatic artery, can have important effects on the clinical
expression of liver ischaemia (which typically exhibits a less dramatic pattern than ischaemia
in other organs, a fact that can sometimes lead to it being missed clinically), and can raise
practical challenges in liver transplant surgery.
Biliary system and gallbladder
Hepatocytes provide the driving force for bile flow by creating osmotic gradients of bile
acids, which form micelles in bile (bile acid-dependent bile flow), and of sodium (bile acid-
independent bile flow). Bile is secreted by hepatocytes and flows from cholangioles to the
biliary canaliculi. The canaliculi join to form larger intrahepatic bile ducts, which in turn
merge to form the right and left hepatic ducts. These ducts join as they emerge from the
liver to form the common hepatic duct, which becomes the common bile duct after joining
the cystic duct (see Fig. 23.2). The common bile duct is approximately 5 cm long and 4–6 mm

4
wide. The distal portion of the duct passes through the head of the pancreas and usually
joins the pancreatic duct before entering the duodenum through the ampullary sphincter
(sphincter of Oddi). It should be noted, though, that the anatomy of the lower common bile
duct can vary widely. Common bile duct pressure is maintained by rhythmic contraction and
relaxation of the sphincter of Oddi; this pressure exceeds gallbladder pressure in the fasting
state, so that bile normally flows into the gallbladder, where it is concentrated tenfold by
resorption of water and electrolytes. The gallbladder is a pear-shaped sac typically lying
under the right hemiliver, with its fundus located anteriorly behind the tip of the 9th costal
cartilage. Anatomical variation is common and should be considered when assessing
patients clinically and radiologically. The function of the gallbladder is to concentrate, and
provide a reservoir for, bile. Gallbladder tone is maintained by vagal activity, and
cholecystokinin released from the duodenal mucosa during feeding causes gallbladder
contraction and reduces sphincter pressure, so that bile flows into the duodenum. The body
and neck of the gallbladder pass postero-medially towards the porta hepatis, and the cystic
duct then joins it to the common hepatic duct. The cystic duct mucosa has prominent
crescentic folds (valves of Heister), giving it a beaded appearance on cholangiography.
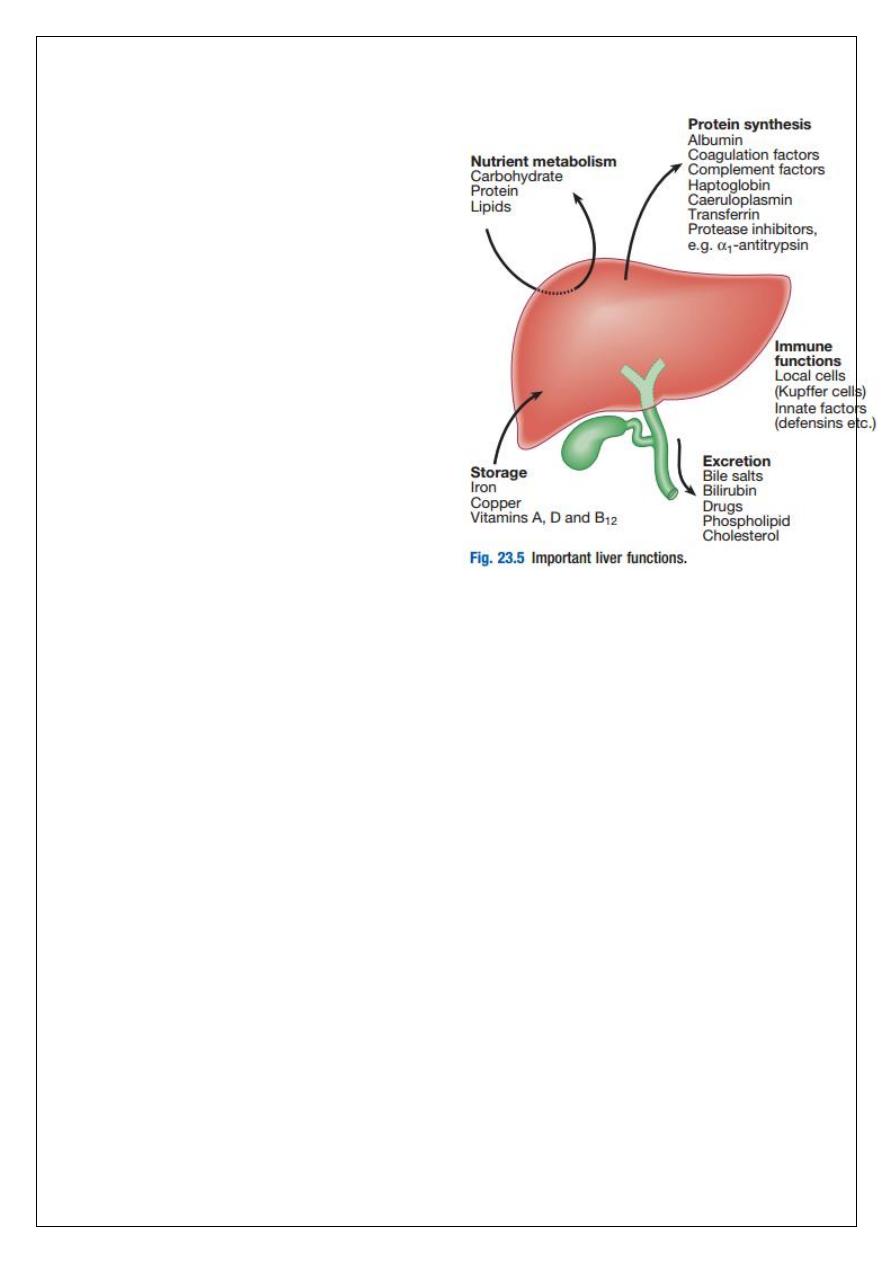
Hepatic function
Carbohydrate, amino acid and lipid metabolism:
The liver plays a central role in carbohydrate, lipid and amino acid metabolism, and is also
involved in metabolising drugs and environmental toxins. An important and increasingly
recognised role for the liver is in the integration of metabolic pathways, regulating the
response of the body to feeding and starvation.
Abnormality in metabolic pathways and their regulation can play an important role both in
liver disease (e.g. non-alcoholic fatty liver disease (NAFLD)) and in diseases that are not
conventionally regarded as diseases of the liver (such as type II diabetes mellitus and inborn
errors of metabolism).
Hepatocytes have specific pathways to handle each of the nutrients absorbed from the gut
and carried to the liver via the portal vein.
5
Amino acids from dietary proteins are
used for synthesis of plasma proteins,
including albumin. This plays a critical
role in maintaining oncotic pressure in
the vascular space and in the
transport of small molecules like
bilirubin, hormones and drugs
throughout the body. Amino acids
that are not required for the
production of new proteins are
broken down, with the amino group
being converted ultimately to urea.
Following a meal, more than half of
the glucose absorbed is taken up by
the liver and stored as glycogen or
converted to glycerol and fatty acids,
thus preventing hyperglycaemia.
During fasting, glycogen is broken
down to release glucose
(gluconeogenesis), thereby preventing
hypoglycaemia.
The liver plays a central role in lipid
metabolism, producing very low-density lipoproteins and further metabolising low-
and high-density lipoproteins. Dysregulation of lipid metabolism is thought to have a
critical role in the pathogenesis of NAFLD. Lipids are now recognised to play a key
part in the pathogenesis of hepatitis C, facilitating viral entry into hepatocytes.
Clotting factors:
The liver produces key proteins that are involved in the coagulation cascade. Many of these
coagulation factors (II, VII, IX and X) are post-translationally modified by vitamin K-
dependent enzymes, and their synthesis is impaired in vitamin K deficiency. Reduced clotting
factor synthesis is an important and easily accessible biomarker of liver function in the
setting of liver injury. Prothrombin time (PT; or the International Normalised Ratio, INR) is
therefore one of the most important clinical tools available for the assessment of
hepatocyte function.
Note that the deranged PT or INR seen in liver disease may not directly equate to increased
bleeding risk, as these tests do not capture the concurrent reduced synthesis of
anticoagulant factors, including protein C and protein S. In general, therefore, correction of
PT using blood products before minor invasive procedures should be guided by clinical risk
rather than the absolute value of the PT.
6
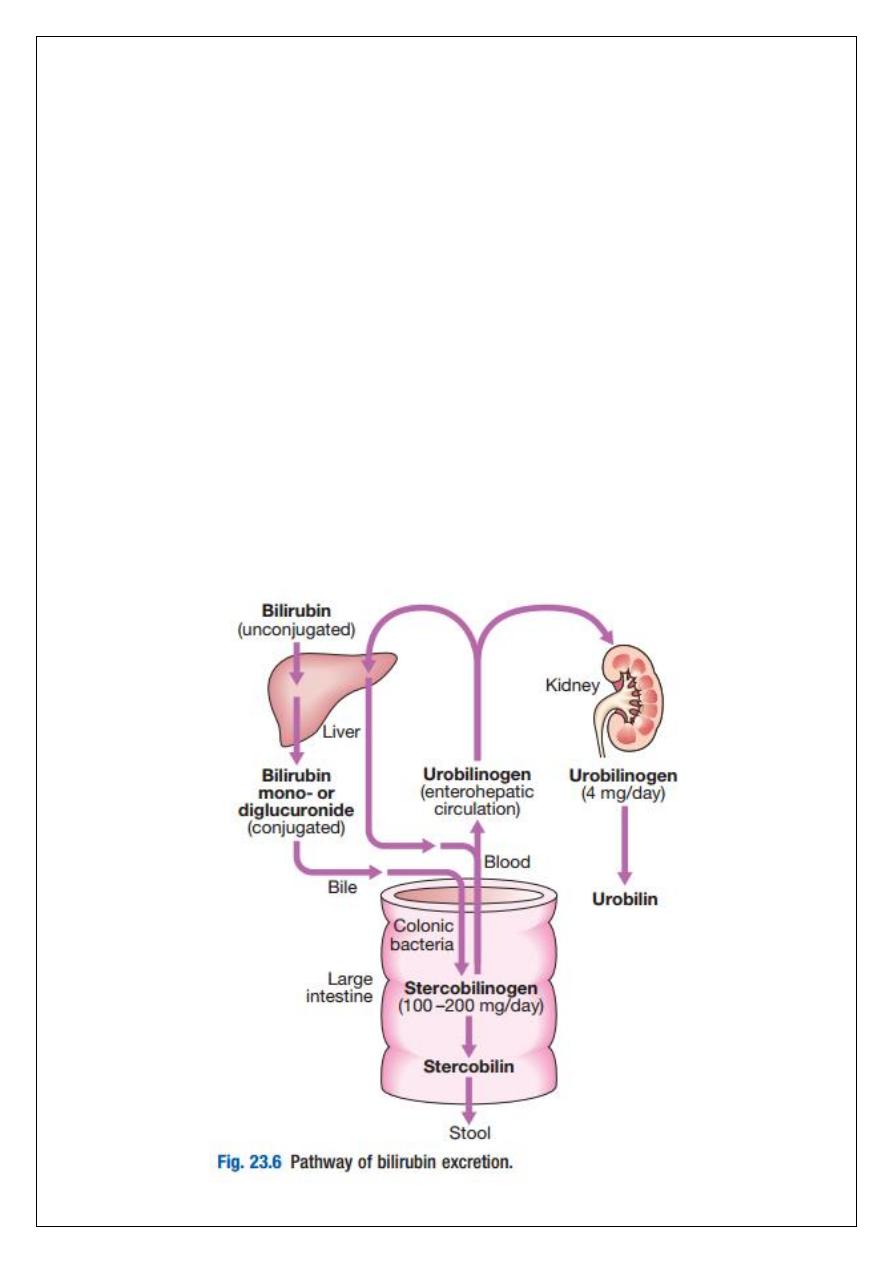
Bilirubin metabolism and bile
The liver plays a central role in the metabolism of bilirubin and is responsible for the
production of bile. Between 250–300 mg of unconjugated bilirubin is produced from the
catabolism of haem daily. Bilirubin in the blood is normally almost all unconjugated and,
because it is not water-soluble, is bound to albumin and does not pass into the urine.
Unconjugated bilirubin is taken up by hepatocytes at the sinusoidal membrane, where it is
conjugated in the endoplasmic reticulum by UDP-glucuronyl transferase, producing bilirubin
mono- and diglucuronide. Impaired conjugation by this enzyme is a cause of inherited
hyperbilirubinaemias. These bilirubin conjugates are water-soluble and are exported into the
bile canaliculi by specific carriers on the hepatocyte membranes. The conjugated bilirubin is
excreted in the bile and passes into the duodenal lumen. Once in the intestine, conjugated
bilirubin is metabolised by colonic bacteria to form stercobilinogen, which may be further
oxidised to stercobilin. Both stercobilinogen and stercobilin are then excreted in the stool,
contributing to its brown colour. Biliary obstruction results in reduced stercobilinogen in the
stool, and the stools become pale. A small amount of stercobilinogen (4 mg/day) is absorbed
from the bowel, passes through the liver, and is excreted in the urine, where it is known as
urobilinogen or, following further oxidisation, urobilin. The liver secretes 1–2 L of bile daily.
Bile contains bile acids (formed from cholesterol), phospholipids, bilirubin and cholesterol.
Several biliary transporter proteins have been identified. Mutations in genes encoding these
proteins have been identified in inherited intrahepatic biliary diseases presenting in
childhood, and in adult-onset disease such as intrahepatic cholestasis of pregnancy and
gallstone formation.
Storage of vitamins and minerals

7
Vitamins A, D and B12 are stored by the liver in large amounts, while others, such as vitamin
K and folate, are stored in smaller amounts and disappear rapidly if dietary intake is
reduced.
The liver is also able to metabolise vitamins to more active compounds, e.g. 7-
dehydrocholesterol to 25(OH) vitamin D. Vitamin K is a fat-soluble vitamin and so the
inability to absorb fat soluble vitamins, as occurs in biliary obstruction, results in a
coagulopathy. The liver also stores minerals such as iron, in ferritin and haemosiderin, and
copper, which is excreted in bile.
Immune regulation
Approximately 9% of the normal liver is composed of immune cells. Cells of the innate
immune system include Kupffer cells derived from blood monocytes, the liver macrophages
and natural killer (NK) cells, as well as ‘classical’ B and T cells of the adaptive immune
response.
Kupffer cells constitute the largest single mass of tissue-resident macrophages in the body
and account for 80% of the phagocytic capacity of this system. They remove aged and
damaged red blood cells, bacteria, viruses, antigen–antibody complexes and endotoxin.
They also produce a wide variety of inflammatory mediators that can act locally or may be
released into the systemic circulation.
INVESTIGATION OF LIVER AND HEPATOBILIARY DISEASE
Investigations play an important role in the management of liver disease in three settings:
identification of the presence of liver disease
establishing the aetiology
understanding disease severity (in particular, identification of cirrhosis with its
complications).
When planning investigations it is important to be clear as to which of these goals is being
addressed. Suspicion of the presence of liver disease is normally based on blood
biochemistry abnormality (‘liver function tests’, or ‘LFTs’), undertaken either as a result of
clinical suspicion or, increasingly, in the setting of health screening. Less commonly,
suspicion arises after a structural abnormality is identified on imaging.
Aetiology is typically established through a combination of history, specific blood tests and,
where appropriate, imaging and liver biopsy. Staging of disease (in essence, the
identification of cirrhosis) is largely histological, although there is increasing interest in non-
invasive approaches, including novel imaging modalities, serum markers of fibrosis and the
use of predictive scoring systems. The aims of investigation in patients with suspected liver
disease are shown below.

8
Liver blood biochemistry
Liver blood biochemistry (LFTs) includes the measurement of serum bilirubin,
aminotransferases, alkaline phosphatase, gamma-glutamyl transferase and albumin.
Most analytes measured by LFTs are not truly ‘function’ tests but, given that they are
released by injured hepatocytes, instead provide biochemical evidence of liver cell damage.
Liver function per se is best assessed by the serum albumin, PT and bilirubin because of the
role played by the liver in synthesis of albumin and clotting factors and in clearance of
bilirubin.
Although LFT abnormalities are often non-specific, the patterns are frequently helpful in
directing further investigations. Also, levels of bilirubin and albumin and the PT are related
to clinical outcome in patients with severe liver disease, reflected by their use in several
prognostic scores: The Child–Pugh and MELD scores in cirrhosis, the Glasgow score in
alcoholic hepatitis and the King’s College Hospital criteria for liver transplantation in acute
liver failure.
Bilirubin and albumin
The degree of elevation of bilirubin can reflect the degree of liver damage. A raised bilirubin
often occurs earlier in the natural history of biliary disease (e.g. primary biliary cirrhosis)
than in disease of the liver parenchyma (e.g. cirrhosis) where the hepatocytes are primarily
involved.
Swelling of the liver within its capsule in inflammation can, however, sometimes impair bile
flow and cause an elevation of bilirubin level that is disproportionate to the degree of liver
injury.
Caution is therefore needed in interpreting the level of liver injury purely on the basis of
bilirubin elevation. Serum albumin levels are often low in patients with liver disease. This is
due to a change in the volume of distribution of albumin, and reduced synthesis. Since the
plasma half-life of albumin is about 2 weeks, albumin levels may be normal in acute liver
failure but are almost always reduced in chronic liver failure.
Alanine aminotransferase and aspartate aminotransferase
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are located in the
cytoplasm of the hepatocyte; AST is also located in the hepatocyte mitochondria. Although
both transaminase enzymes are widely distributed, expression of ALT outside the liver is
relatively low and this enzyme is therefore considered more specific for hepatocellular
damage. Large increases of aminotransferase activity favour hepatocellular damage, and this
pattern of LFT abnormality is known as ‘hepatitic’.
Alkaline phosphatase and gamma-glutamyl transferase
Alkaline phosphatase (ALP) is the collective name given to several different enzymes that
hydrolyse phosphate esters at alkaline pH. These enzymes are widely distributed in the

9
body, but the main sites of production are the liver, gastrointestinal tract, bone, placenta
and kidney. ALPs are post-translationally modified, resulting in the production of several
different isoenzymes, which differ in abundance in different tissues. ALP enzymes in the liver
are located in cell membranes of the hepatic sinusoids and the biliary canaliculi. Accordingly,
levels rise with intrahepatic and extrahepatic biliary obstruction and with sinusoidal
obstruction, as occurs in infiltrative liver disease.
Gamma-glutamyl transferase (GGT) is a microsomal enzyme found in many cells and tissues
of the body. The highest concentrations are located in the liver, where it is produced by
hepatocytes and by the epithelium lining small bile ducts. The function of GGT is to transfer
glutamyl groups from gamma-glutamyl peptides to other peptides and amino acids. The
pattern of a modest increase in aminotransferase activity and large increases in ALP and GGT
activity favours biliary obstruction and is commonly described as ‘cholestatic’ or
‘obstructive’. Isolated elevation of the serum GGT is relatively common, and may occur
during ingestion of microsomal enzyme-inducing drugs, including alcohol, but also in NAFLD.

10
Other biochemical tests
Other widely available biochemical tests may become altered in patients with liver disease:
• Hyponatraemia occurs in severe liver disease due to increased production of antidiuretic
hormone.
• Serum urea may be reduced in hepatic failure, whereas levels of urea may be increased
following gastrointestinal haemorrhage.
• When high levels of urea are accompanied by raised bilirubin, high serum creatinine and
low urinary sodium, this suggests hepatorenal failure, which carries a grave prognosis.
• Significantly elevated ferritin suggests haemochromatosis. Modest elevations can be seen
in inflammatory disease and alcohol excess.
Haematological tests
Blood count
The peripheral blood count is often abnormal and can give a clue to the underlying
diagnosis:
• A normochromic normocytic anaemia may reflect recent gastrointestinal
haemorrhage, whereas chronic blood loss is characterised by a hypochromic
microcytic anaemia secondary to iron deficiency. A high erythrocyte mean cell
volume (macrocytosis) is associated with alcohol misuse, but target cells in any
jaundiced patient also result in a macrocytosis. Macrocytosis can persist for a long
period of time after alcohol cessation, making it a poor marker of ongoing
consumption.
• Leucopenia may complicate portal hypertension and hypersplenism, whereas
leucocytosis may occur with cholangitis, alcoholic hepatitis and hepatic abscesses.
Atypical lymphocytes are seen in infectious mononucleosis, which may be
complicated by an acute hepatitis.
• Thrombocytopenia is common in cirrhosis and is due to reduced platelet production,
and increased breakdown because of hypersplenism. Thrombopoietin, required for
platelet production, is produced in the liver and levels fall with worsening liver
function. Thus platelet levels are usually more depressed than white cells and
haemoglobin in the presence of hypersplenism in patients with cirrhosis. A low
platelet count is often an indicator of chronic liver disease, particularly in the
context of hepatomegaly. Thrombocytosis is unusual in patients with liver disease
but may occur in those with active gastrointestinal haemorrhage and, rarely, in
hepatocellular carcinoma.
Coagulation tests

11
These are often abnormal in patients with liver disease. The normal half-lives of the vitamin
K-dependent coagulation factors in the blood are short (5–72 hours) and so changes in the
prothrombin time occur relatively quickly following liver damage; these changes provide
valuable prognostic information in patients with both acute and chronic liver failure. An
increased PT is evidence of severe liver damage in chronic liver disease. Vitamin K does not
reverse this deficiency if it is due to liver disease, but will correct the PT if the cause is
vitamin K deficiency, as may occur with biliary obstruction due to non-absorption of fat-
soluble vitamins.
Immunological tests
A variety of tests are available to evaluate the aetiology of hepatic disease (Boxes 23.4 and
23.5). The presence of liver-related autoantibodies can be suggestive of the presence of
autoimmune liver disease (although falsepositive results can occur in non-autoimmune
inflammatory disease such as NAFLD). Elevation in overall serum immunoglobulin levels can
also be suggestive of autoimmunity (immunoglobulin (Ig)G and IgM). Elevated serum IgA can
be seen, often in more advanced alcoholic liver disease and NAFLD, although the association
is not specifc.
Imaging
Several imaging techniques can be used to determine the site and general nature of
structural lesions in the liver and biliary tree. In general, however, imaging techniques are
unable to identify hepatic inflammation and have poor sensitivity for liver fbrosis unless
advanced cirrhosis with portal hypertension is present. Ultrasound Ultrasound is non-
invasive and most commonly used as a ‘frst-line’ test to identify gallstones, biliary
obstruction or thrombosis in the hepatic vasculature. Ultrasound is good for the
identifcation of splenomegaly and abnormalities in liver texture, but is less effective at
identifying diffuse parenchymal disease. Focal lesions, such as tumours, may not be detected
if they are below 2 cm in diameter and have echogenic characteristics similar to normal liver
tissue. Bubblebased contrast media are now routinely used and can enhance discriminant
12
capability. Doppler ultrasound allows blood flow in the hepatic artery, portal vein and
hepatic veins to be investigated. Endoscopic ultrasound provides high-resolution images of
the pancreas, biliary tree and liver.
Computed tomography and magnetic resonance imaging
Computed tomography (CT) detects smaller focal lesions in the liver, especially when
combined with contrast injection. Magnetic resonance imaging (MRI) can also be used to
localise and confrm the aetiology of focal liver lesions, particularly primary and secondary
tumours.

13
Hepatic angiography is seldom used nowadays as a diagnostic tool, since CT and MRI are
both able to provide images of hepatic vasculature, but it still has a therapeutic role in the
embolisation of vascular tumours such as hepatocellular carcinoma. Hepatic venography is
now rarely performed.
Cholangiography
Cholangiography can be undertaken by magnetic resonance cholangiopancreatography,
endoscopy (endoscopic retrograde cholangiopancreatography, ERCP) or the percutaneous
approach (percutaneous transhepatic cholangiography, PTC). The latter does not allow the
ampulla of Vater or pancreatic duct to be visualised. MRCP is as good as ERCP at providing
images of the biliary tree but has fewer complications and is the diagnostic test of choice.
Both endoscopic and percutaneous approaches allow therapeutic interventions, such as the
insertion of biliary stents across malignant bile duct strictures. The percutaneous approach is
only used if it is not possible to access the bile duct endoscopically.
Histological examination
An ultrasound-guided liver biopsy can confrm the severity of liver damage and provide
aetiological information. It is performed percutaneously with a Trucut or Menghini needle,
usually through an intercostal space under local anaesthesia, or radiologically using a
transjugular approach. Percutaneous liver biopsy is a relatively safe procedure but carries a
mortality of about 0.01%. The main complications are abdominal and/or shoulder pain,
bleeding and biliary peritonitis. Biliary peritonitis is rare and usually occurs when a biopsy is

14
performed in a patient with obstruction of a large bile duct. Liver biopsies can be carried out
in patients with defective haemostasis if:
the defect is corrected with fresh frozen plasma and platelet transfusion
the biopsy is obtained by the transjugular route, or
the procedure is conducted percutaneously under ultrasound control and the needle
track is then plugged with procoagulant material.
In patients with potentially resectable malignancy, biopsy should be avoided due to the
potential risk of tumour dissemination. Operative or laparoscopic liver biopsy may
sometimes be valuable. Although the pathological features of liver disease are complex, with
several features occurring together, liver disorders can be broadly classifed histologically
into fatty liver (steatosis), hepatitis (inflammation, ‘grade’) and cirrhosis (fbrosis, ‘stage’).
The use of special histological stains can help in determining aetiology. The clinical features
and prognosis of these changes are dependent on the underlying aetiology, and are
discussed in the relevant sections below
Non-invasive markers of hepatic fibrosis
Non-invasive markers of liver fbrosis have been developed and can reduce the need for liver
biopsy to assess the extent of fbrosis in some settings. Serological markers of hepatic fbrosis,
such as α2- macroglobulin, haptoglobin and routine clinical biochemistry tests, are used in
the Fibrotest®. The ELF® (Enhanced Liver Fibrosis) serological assay uses a combination of
hyaluronic acid, procollagen peptide III (PIIINP) and tissue inhibitor of metalloproteinase 1
(TIMP1). These tests are good at differentiating severe fbrosis from mild scarring, but are
limited in their ability to detect subtle changes. A number of noncommercial scores based on
standard biochemical and anthropometric indices have also been described that provide
similar levels of sensitivity and specifcity (e.g. the FIB4 Score). An alternative to serological
markers is transient elastography in which ultrasound-based shock waves are sent through
the liver to measure liver stiffness as a surrogate for hepatic fibrosis. Once again, this test is
good at differentiating severe fbrosis from mild scarring, but is limited in its ability to detect
subtle changes, and validity may be affected by obesity

15
PRESENTING PROBLEMS IN LIVER DISEASE
Liver injury may be either acute or chronic.
• Acute liver injury may present with non-specifc symptoms of fatigue and abnormal
LFTs, or with jaundice and acute liver failure.
• Chronic liver injury is defined as hepatic injury, inflammation and/or fibrosis
occurring in the liver for more than 6 months. In the early stages, patients can be
asymptomatic with fluctuating abnormal LFTs. With more severe liver damage,
however, the presentation can be with jaundice, portal hypertension or other signs
of cirrhosis and hepatic decompensation.
16
Acute liver failure
Acute liver failure is an uncommon but serious condition.
Definition: progressive deterioration in liver function and mental changes progressing
from confusion to coma occurring within 8 weeks of onset of the precipitating illness, in the
absence of evidence of preexisting liver disease.
This distinguishes it from instances in which hepatic encephalopathy represents a
deterioration in chronic liver disease. Liver failure occurs when there is insufficient metabolic
and synthetic function for the needs of the patient. Although the direct cause is usually
acute loss of functional hepatocytes, this can occur in different settings, which have
implications for outcome and treatment.
In a patient whose liver was previously normal (fulminant liver failure), the level of injury
needed to cause liver failure, and thus the patient risk, is very high. In a patient with pre-
existing chronic liver disease, the additional acute insult needed to precipitate liver failure is
much less. It is critical, therefore, to understand whether liver failure is a true acute event or
an acute deterioration on a background of pre-existing injury (which may itself not have
been diagnosed).
Although ultimately liver biopsy may be necessary, it is the presence or absence of the
clinical features suggesting chronicity that guides the clinician. More recently, newer
classifications have been developed to reflect differences in presentation and outcome of
acute liver failure. One such classification divides acute liver failure into hyperacute, acute
and subacute, according to the interval between onset of jaundice and encephalopathy.
Pathophysiology
Any cause of liver damage can produce acute liver failure, provided it is sufficiently severe
(Fig. 23.13). Acute viral hepatitis is the most common cause worldwide, whereas
paracetamol toxicity (p. 212) is the most frequent cause in the UK. Acute liver failure occurs
occasionally with other drugs, or from Amanita phalloides (mushroom) poisoning, in

17
pregnancy, in Wilson’s disease, following shock (p. 190) and, rarely, in extensive malignant
disease of the liver. In 10% of cases the cause of acute liver failure remains unknown and
these patients are often labelled as having ‘non-A–E viral hepatitis’ or ‘cryptogenic’ acute
liver failure.
Clinical assessment
Cerebral disturbance (hepatic encephalopathy and/or cerebral oedema) is the cardinal
manifestation of acute liver failure, but in the early stages this can be mild and episodic and
so its absence does not exclude a significant acute liver injury. The initial clinical features are
often subtle and include reduced alertness and poor concentration, progressing through
behavioural abnormalities such as restlessness and aggressive outbursts, to drowsiness and
coma (Box 23.9). Cerebral oedema may occur due to increased intracranial pressure, causing
unequal or abnormally reacting pupils, fixed pupils, hypertensive episodes, bradycardia,
hyperventilation, profuse sweating, local or general myoclonus, focal fits or decerebrate
posturing.
Papilloedema occurs rarely and is a late sign. More general symptoms include weakness,
nausea and vomiting.
Right hypochondrial discomfort is an occasional feature.
The patient may be jaundiced; however, this may not be present at the outset (e.g. in
paracetamol overdose) and there are a number of exceptions, including Reye’s syndrome, in
which jaundice is rare.
Occasionally, death may occur in fulminant cases of acute liver failure before jaundice
develops.
Fetor hepaticus can be present.
The liver is usually of normal size but later becomes smaller. Hepatomegaly is unusual and,
in the presence of a sudden onset of ascites, suggests venous outflow obstruction as the
cause.
Splenomegaly is uncommon and never prominent.
Ascites and oedema are late developments and may be a consequence of fluid therapy.
Other features are related to the development of complications .

18

19
Investigations
The patient should be investigated to determine the cause of the liver failure and the
prognosis (Boxes 23.10 and 23.11).
Hepatitis B core IgM antibody is the best screening test for acute hepatitis B
infection, as liver damage is due to the immunological response to the virus, which
has often been eliminated, and the test for HBsAg may be negative.
The PT rapidly becomes prolonged as coagulation factor synthesis fails; this is the
laboratory test of greatest prognostic value and should be carried out at least twice
daily. Its prognostic importance emphasises the necessity of avoiding the use of
fresh frozen plasma to correct raised PT in acute liver failure, except in the setting of
frank bleeding. Factor V levels can be used instead of the PT to assess the degree of
liver impairment.
The plasma bilirubin reflects the degree of jaundice.
Plasma aminotransferase activity is particularly high after paracetamol overdose,
reaching 100–500 times normal, but falls as liver damage progresses and is not
helpful in determining prognosis.
Plasma albumin remains normal unless the course is prolonged.
Percutaneous liver biopsy is contraindicated because of the severe coagulopathy,
but biopsy can be undertaken using the transjugular route if appropriate.

20

21
Management
Patients with acute liver failure should be treated in a high-dependency or intensive care
unit as soon as progressive prolongation of the PT occurs or hepatic encephalopathy is
identified (Box 23.12), so that prompt treatment of complications can be initiated (Box
23.13). Conservative treatment aims to maintain life in the hope that hepatic regeneration
will occur, but early transfer to a specialised transplant unit should always be considered.
N-acetylcysteine therapy may improve outcome, particularly in patients with acute
liver failure due to paracetamol poisoning.
Liver transplantation is an increasingly important treatment option for acute liver
failure, and criteria have been developed to identify patients unlikely to survive
without a transplant (see Box 23.11). Patients should, wherever possible, be
transferred to a transplant centre before these criteria are met to allow time for
assessment and to maximise the time for a donor liver to become available. Survival
following liver transplantation for acute liver failure is improving, and 1-year survival
rates of about 60% can be expected.
22
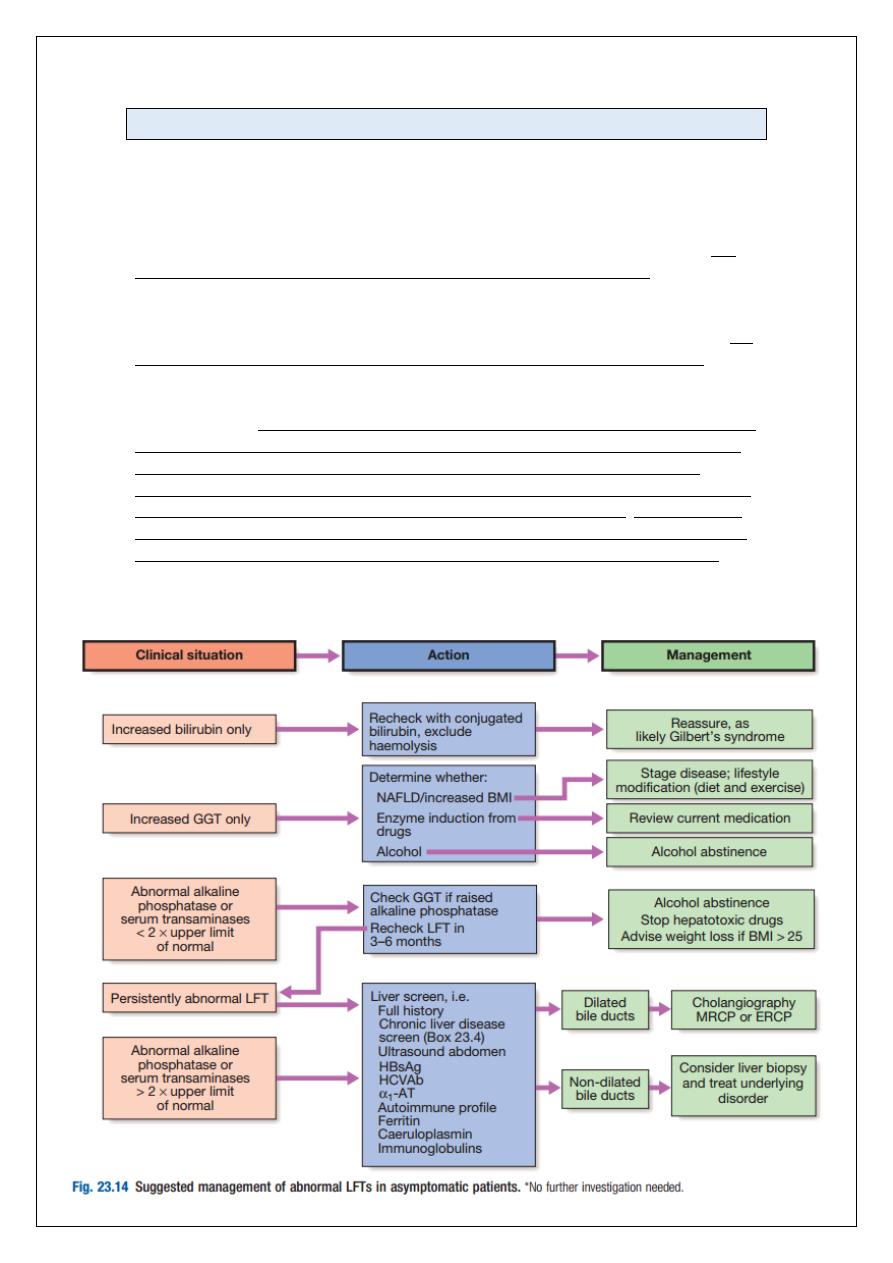
Abnormal liver function tests
Frequently, LFTs are requested in patients who have no symptoms or signs of liver disease,
as part of routine health checks, insurance medicals and drug monitoring. Many patients
with chronic liver disease are asymptomatic or have vague, non-specific symptoms.
Apparently asymptomatic abnormal LFTs are therefore a common occurrence. The
prevalence of abnormal LFTs has been reported to be as high as 10% in some studies. The
most common abnormalities are alcoholic or non-alcoholic fatty liver disease. Since effective
medical treatments are now available for many types of chronic liver disease, further
evaluation is usually warranted to make sure the patient does not have a treatable
condition. Although transient mild abnormalities in LFTs may not be clinically significant, the
majority of patients with persistently abnormal LFTs do have significant liver disease.
Biochemical abnormalities in chronic liver disease often fluctuate over time, and therefore
even mild abnormalities can indicate significant underlying disease and so warrant follow-up
and investigation. When abnormal LFTs are detected, a thorough history should be compiled
to determine the patient’s alcohol consumption, drug use (prescribed drugs or otherwise),
risk factors for viral hepatitis (e.g. blood transfusion, injecting drug use, tattoos), the
presence of autoimmune diseases, family history, neurological symptoms, and the presence
of features of the metabolic syndrome, including diabetes and/or obesity. The presence or
absence of stigmata of chronic liver disease does not reliably identify those individuals with
significant disease, and investigations are indicated, even in the absence of these signs.
Both the pattern of LFT abnormality, hepatitic or obstructive, and the degree of elevation
are helpful in determining the cause of underlying liver disease.

23

24
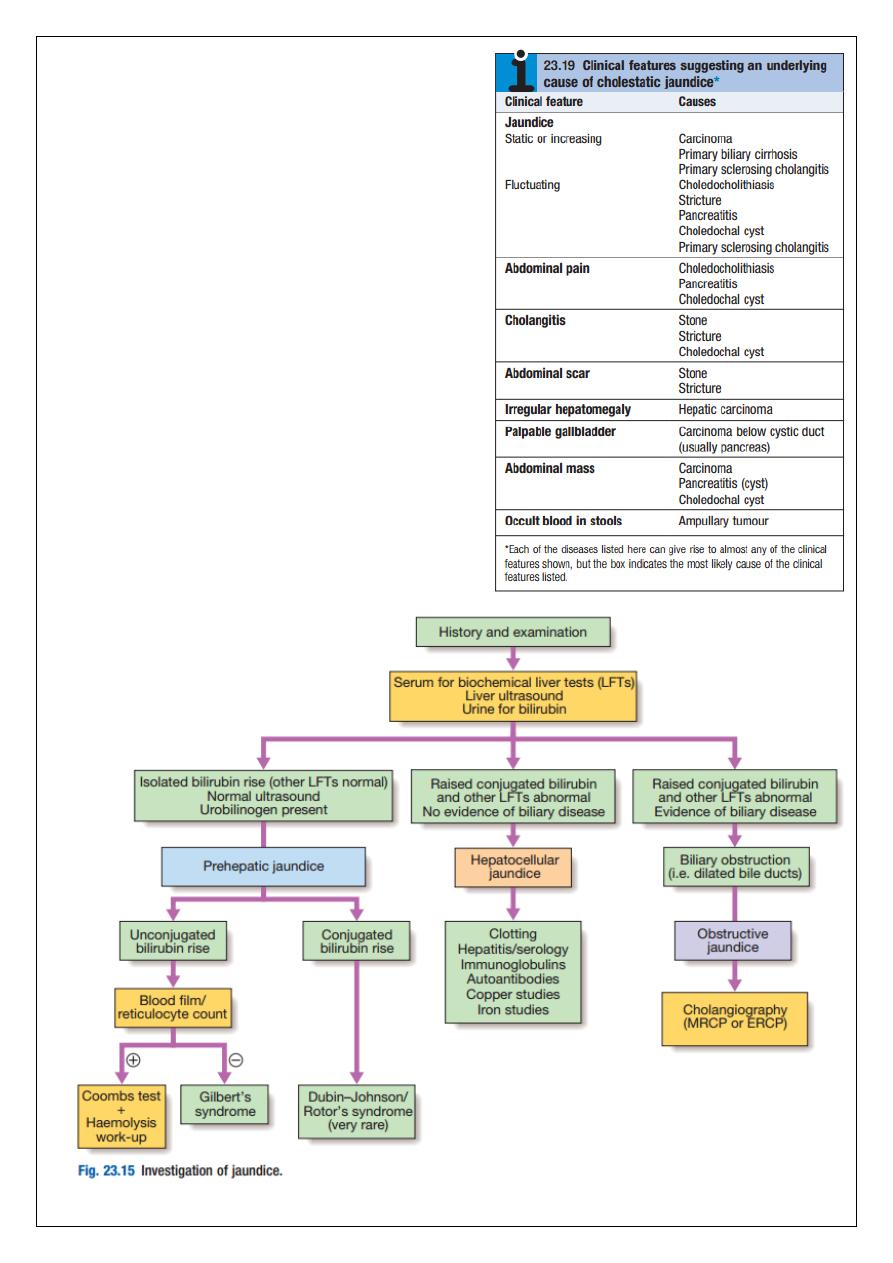
Jaundice
Jaundice is usually detectable clinically when the plasma bilirubin exceeds 2.5 mg/dL. The
causes of jaundice overlap with the causes of abnormal LFTs discussed above. In a patient
with jaundice it is useful to consider whether the cause might be pre-hepatic, hepatic or
post-hepatic and there are often important clues in the history.
Pre-hepatic jaundice
This is caused either by haemolysis or by congenital hyperbilirubinaemia, and is
characterised by an isolated raised bilirubin level.
In haemolysis, destruction of red blood cells or their marrow precursors causes increased
bilirubin production. Jaundice due to haemolysis is usually mild because a healthy liver can
excrete a bilirubin load six times greater than normal before unconjugated bilirubin
accumulates in the plasma. This does not apply to newborns, who have less capacity to
metabolise bilirubin.
The most common form of non-haemolytic hyperbilirubinaemia is Gilbert’s syndrome, an
inherited disorder of bilirubin metabolism. Other inherited disorders of bilirubin metabolism
are very rare.
Hepatocellular jaundice
Hepatocellular jaundice results from an inability of the liver to transport bilirubin into the
bile, occurring as a consequence of parenchymal disease. Bilirubin transport across the
hepatocytes may be impaired at any point between uptake of unconjugated bilirubin into
the cells and transport of conjugated bilirubin into the canaliculi. In addition, swelling of cells
and oedema resulting from the disease itself may cause obstruction of the biliary canaliculi.
In hepatocellular jaundice the concentrations of both unconjugated and conjugated bilirubin
in the blood increase. Hepatocellular jaundice can be due to acute or chronic injury, and
clinical features of acute or chronic liver disease may be detected clinically.
Characteristically, jaundice due to parenchymal liver disease is associated with increases in
transaminases (AST, ALT), but increases in other LFTs, including cholestatic enzymes (GGT,
ALP) may occur, and suggest specific aetiologies. Acute jaundice in the presence of an ALT of
greater than 1000 U/L is highly suggestive of an infectious cause (e.g. hepatitis A, B), drugs
(e.g. paracetamol) or hepatic ischaemia. Imaging is essential, in particular to identify
features suggestive of cirrhosis, to define the patency of the hepatic vasculature, and to
obtain evidence of portal hypertension. Liver biopsy has an important role in defining the
aetiology of hepatocellular jaundice and the extent of liver injury.
Obstructive (cholestatic) jaundice
Cholestatic jaundice may be caused by:
failure of hepatocytes to initiate bile flow
obstruction of the bile ducts or portal tracts
obstruction of bile flow in the extrahepatic bile ducts between the porta hepatis and
the papilla of Vater. In the absence of treatment, cholestatic jaundice tends to
become progressively more severe because conjugated bilirubin is unable to enter
the bile canaliculi and passes back into the blood, and also because there is a failure

25
of clearance of unconjugated bilirubin arriving at the liver cells. Cholestasis may
result from defects at more than one of these levels. Those confined to the
extrahepatic bile ducts may be amenable to surgical or endoscopic correction.
Clinical features comprise those due to cholestasis itself, those due to secondary
infection (cholangitis) and those of the underlying condition. Obstruction of the bile
duct drainage due to blockage of the extrahepatic biliary tree is characteristically
associated with pale stools and dark urine. Pruritus may be a dominant feature and
can be accompanied by skin excoriations. Peripheral stigmata of chronic liver disease
are absent. If the gallbladder is palpable, the jaundice is unlikely to be caused by
biliary obstruction due to gallstones, probably because a chronically inflamed stone-
containing gallbladder cannot readily dilate. This is Courvoisier’s Law, and suggests
that jaundice is due to a malignant biliary obstruction (e.g. pancreatic cancer).
Cholangitis is characterised by ‘Charcot’s triad’ of jaundice, right upper quadrant
pain and fever. Cholestatic jaundice is characterised by a relatively greater elevation
of ALP and GGT than the aminotransferases. Ultrasound is indicated to determine
whether there is evidence of mechanical obstruction and dilatation of the biliary
tree.
26

27
Hepatomegaly
Hepatomegaly may occur as the result of a general enlargement of the liver or because of
primary or secondary liver tumour. The most common liver tumour in Western countries is
liver metastasis, whereas primary liver cancer complicating chronic viral hepatitis is more
common in the Far East. Unlike metastases, neuro-endocrine tumours typically cause
massive hepatomegaly but without significant weight loss. Cirrhosis can be associated with
either hepatomegaly or reduced liver size in advanced disease. Although all causes of
cirrhosis can involve hepatomegaly, it is much more common in alcoholic liver disease and
haemochromatosis. Hepatomegaly may resolve in patients with alcoholic cirrhosis when
they stop drinking.
Ascites
Ascites is present when there is accumulation of free fluid in the peritoneal cavity. Small
amounts of ascites are asymptomatic, but with larger accumulations of fluid (> 1 L) there is
abdominal distension, fullness in the flanks, shifting dullness on percussion and, when the
ascites is marked, a fluid thrill. Other features include eversion of the umbilicus, herniae,
abdominal striae, divarication of the recti and scrotal oedema. Dilated superficial abdominal
veins may be seen if the ascites is due to portal hypertension.
Pathophysiology
Ascites has numerous causes, the most common of which are malignant disease, cirrhosis
and heart failure. Many primary disorders of the peritoneum and visceral organs can also
cause ascites, and these need to be considered even in a patient with chronic liver disease.
Splanchnic vasodilatation is thought to be the main factor leading to ascites in cirrhosis. This
is mediated by vasodilators (mainly nitric oxide) that are released when portal hypertension
causes shunting of blood into the systemic circulation. Systemic arterial pressure falls due to
pronounced splanchnic vasodilatation as cirrhosis advances. This leads to activation of the
renin–angiotensin system with secondary aldosteronism, increased sympathetic nervous
activity, increased atrial natriuretic hormone secretion and altered activity of the kallikrein–
kinin system. These systems tend to normalise arterial pressure but produce salt and water
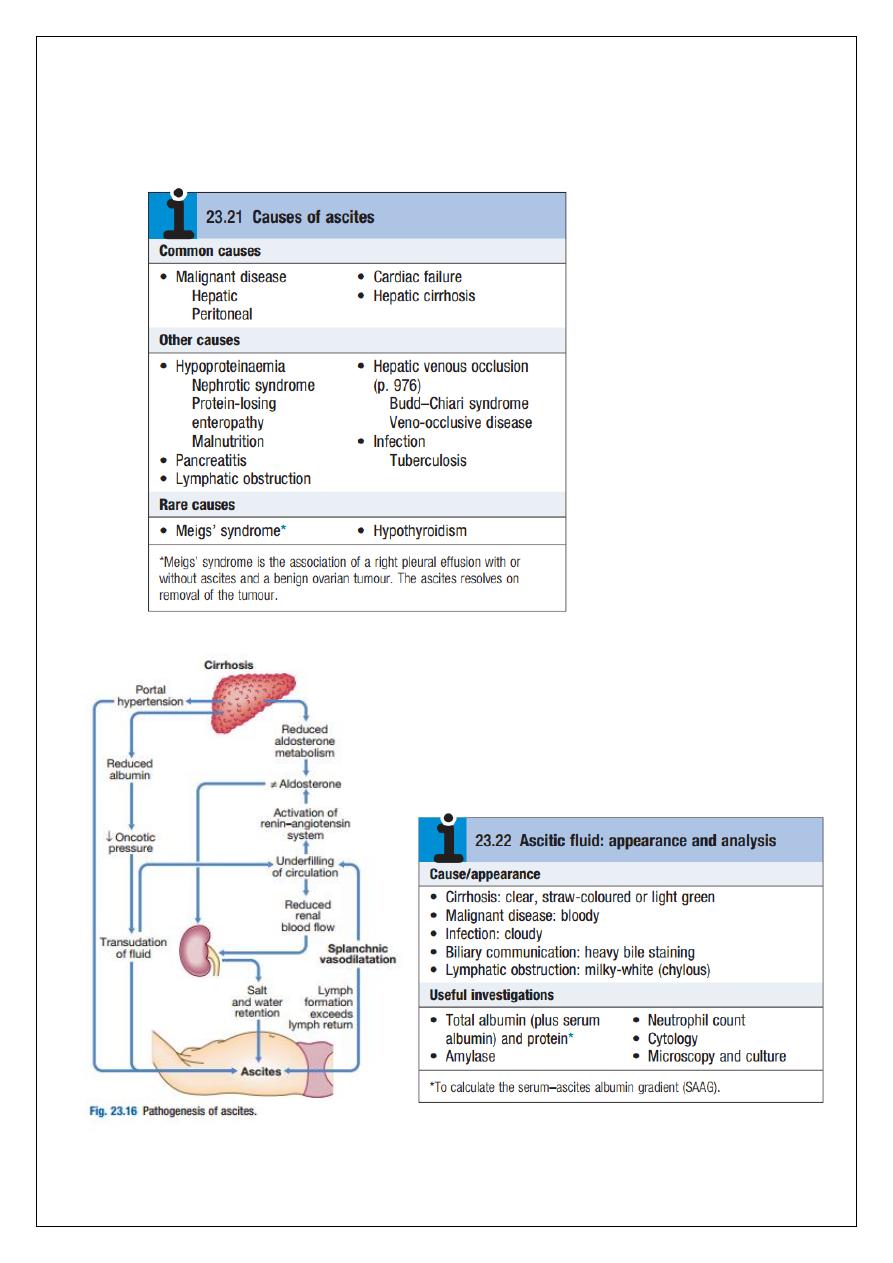
28
retention. In this setting the combination of splanchnic arterial vasodilatation and portal
hypertension alters intestinal capillary permeability, promoting accumulation of fluid within
the peritoneum.

29
Investigations
Ultrasonography is the best means of detecting ascites, particularly in the obese and those
with small volumes of fluid. Paracentesis (if necessary under ultrasonic guidance) can be
used to obtain ascitic fluid for analysis. The appearance of ascitic fluid may point to the
underlying cause. Pleural effusions are found in about 10% of patients, usually on the right
side (hepatic hydrothorax); most are small and only identified on chest X-ray, but
occasionally a massive hydrothorax occurs. Pleural effusions, particularly those on the left
side, should not be assumed to be due to the ascites. Measurement of the protein
concentration and the serum–ascites albumin gradient (SAAG) are used to distinguish a
transudate from an exudate. Cirrhotic patients typically develop a transudate with a total
protein concentration below 25 g/L and relatively few cells. However, in up to 30% of
patients, the total protein concentration is more than 30 g/L. In these cases, it is useful to
calculate the SAAG by subtracting the concentration of the ascites fluid albumin from the
serum albumin. A gradient of more than 11 g/L is 96% predictive that ascites is due to portal
hypertension. Venous outflow obstruction due to cardiac failure or hepatic venous outflow
obstruction can also cause a transudative ascites, as indicated by an albumin gradient above
11 g/L but, unlike in cirrhosis, the total protein content is usually above 25 g/L.
Exudative ascites (ascites protein concentration > 25 g/L or a SAAG < 11 g/L) raises the
possibility of infection (especially tuberculosis), malignancy, hepatic venous obstruction,
pancreatic ascites or, rarely, hypothyroidism. Ascites amylase activity above 1000 U/L
identifies pancreatic ascites, and low ascites glucose concentrations suggest malignant
disease or tuberculosis. Cytological examination may reveal malignant cells (one-third of
cirrhotic patients with a bloody tap have a hepatocellular carcinoma). Polymorphonuclear
leucocyte counts above 250 × 10
6
/L strongly suggest infection (spontaneous bacterial
peritonitis, see below). Laparoscopy can be valuable in detecting peritoneal disease.
Management
Successful treatment relieves discomfort but does not prolong life; if over-vigorous, it can
produce serious disorders of fluid and electrolyte balance and precipitate hepatic
encephalopathy. Treatment of transudative ascites is based on restricting sodium and water
intake, promoting urine output with diuretics and, if necessary, removing ascites directly by
paracentesis. Exudative ascites due to malignancy is treated with paracentesis, but fluid
replacement is generally not required. During management of ascites, the patient should be
weighed regularly. Diuretics should be titrated to remove no more than 1 L of fluid daily, so
body weight should not fall by more than 1 kg daily to avoid excessive fluid depletion.
Sodium and water restriction
Restriction of dietary sodium intake is essential to achieve negative sodium balance, and a
few patients can be managed satisfactorily by this alone. Restriction of sodium intake to 100
mmol/day (‘no added salt diet’) is usually adequate. Drugs containing relatively large
amounts of sodium, and those promoting sodium retention such as non-steroidal anti-
inflammatory drugs (NSAIDs), must be avoided. Restriction of water intake to 1.0–1.5 L/day
is necessary only if the plasma sodium falls below 125 mmol/L.

30
Diuretics
Most patients require diuretics in addition to sodium restriction. Spironolactone (100–400
mg/day) is the first-line drug because it is a powerful aldosterone antagonist; it can cause
painful gynaecomastia and hyperkalaemia, in which case amiloride (5–10 mg/day) can be
substituted. Some patients also require loop diuretics, such as furosemide, but these can
cause fluid and electrolyte imbalance and renal dysfunction. Diuresis may be improved if
patients are rested in bed, perhaps because renal blood flow increases in the horizontal
position. Patients who do not respond to doses of 400 mg spironolactone and 160 mg
furosemide, or who are unable to tolerate these doses due to hyponatraemia or renal
impairment, are considered to have refractory or diuretic-resistant ascites and should be
treated by other measures.
Paracentesis
First-line treatment of refractory ascites is large-volume paracentesis. Paracentesis to
dryness is safe, provided the circulation is supported with an intravenous colloid such as
human albumin (6–8 g per litre of ascites removed, usually as 100 mL of 20% human albumin
solution (HAS) for every 1.5–2 L of ascites drained) or another plasma expander.
Paracentesis can be used as an initial therapy or when other treatments fail.
Transjugular intrahepatic portosystemic stent shunt
A transjugular intrahepatic portosystemic stent shunt (TIPSS) can relieve resistant ascites but
does not prolong life; it may be an option where the only alternative is frequent, large-
volume paracentesis. It can be used in patients awaiting liver transplantation or in those
with reasonable liver function, but can aggravate encephalopathy in those with poor
function.
Peritoneo-venous shunt
The peritoneo-venous shunt is a long tube with a non-return valve running subcutaneously
from the peritoneum to the internal jugular vein in the neck; it allows ascitic fluid to pass
directly into the systemic circulation. Complications, including infection, superior vena caval

31
thrombosis, pulmonary oedema, bleeding from oesophageal varices and disseminated
intravascular coagulopathy, limit its use and insertion of these shunts is now rare.
Complications
Renal failure
Renal failure can occur in patients with ascites. It can be pre-renal due to vasodilatation
from sepsis and/or diuretic therapy, or due to hepatorenal syndrome.
Hepatorenal syndrome
This occurs in 10% of patients with advanced cirrhosis complicated by ascites. There are two
clinical types; both are mediated by renal vasoconstriction due to underfilling of the arterial
circulation.
Type 1 hepatorenal syndrome is characterised by progressive oliguria, a rapid rise of
the serum creatinine and a very poor prognosis (without treatment, median survival
is less than 1 month). There is usually no proteinuria, a urine sodium excretion of
less than 10 mmol/day and a urine/ plasma osmolarity ratio of more than 1.5. Other
non-functional causes of renal failure must be excluded before the diagnosis is
made. Treatment consists of albumin infusions in combination with terlipressin and
is effective in about two-thirds of patients. Haemodialysis should not be used
routinely because it does not improve the outcome.
Patients who survive should be considered for liver transplantation.
Type 2 hepatorenal syndrome usually occurs in patients with refractory ascites, is
characterised by a moderate and stable increase in serum creatinine, and has a
better prognosis.
Spontaneous bacterial peritonitis
Spontaneous bacterial peritonitis (SBP) may present with abdominal pain, rebound
tenderness, absent bowel sounds and fever in a patient with obvious features of cirrhosis
and ascites. Abdominal signs are mild or absent in about one-third of patients, and in these
patients hepatic encephalopathy and fever are the main features. Diagnostic paracentesis
may show cloudy fluid, and an ascites neutrophil count above 250 × 10
6
/L almost invariably
indicates infection. The source of infection cannot usually be determined, but most
organisms isolated are of enteric origin and Escherichia coli is most frequently found. Ascitic
culture in blood culture bottles gives the highest yield of organisms. SBP needs to be
differentiated from other intra-abdominal emergencies, and the finding of multiple
organisms on culture should arouse suspicion of a perforated viscus. Treatment should be
started immediately with broadspectrum antibiotics, such as cefotaxime or piperacillin/
tazobactam). Recurrence of SBP is common but may be reduced with prophylactic
quinolones such as norfloxacin or ciprofloxacin.
Prognosis
Only 10–20% of patients survive 5 years from the first appearance of ascites due to cirrhosis.
The outlook is not universally poor, however, and is best in those with well-maintained liver

32
function and a good response to therapy. The prognosis is also better when a treatable
cause for the underlying cirrhosis is present or when a precipitating cause for ascites, such as
excess salt intake, is found. The mortality at 1 year is 50% following the first episode of
bacterial peritonitis.