THE IMMUNE SYSTEMLec. 2
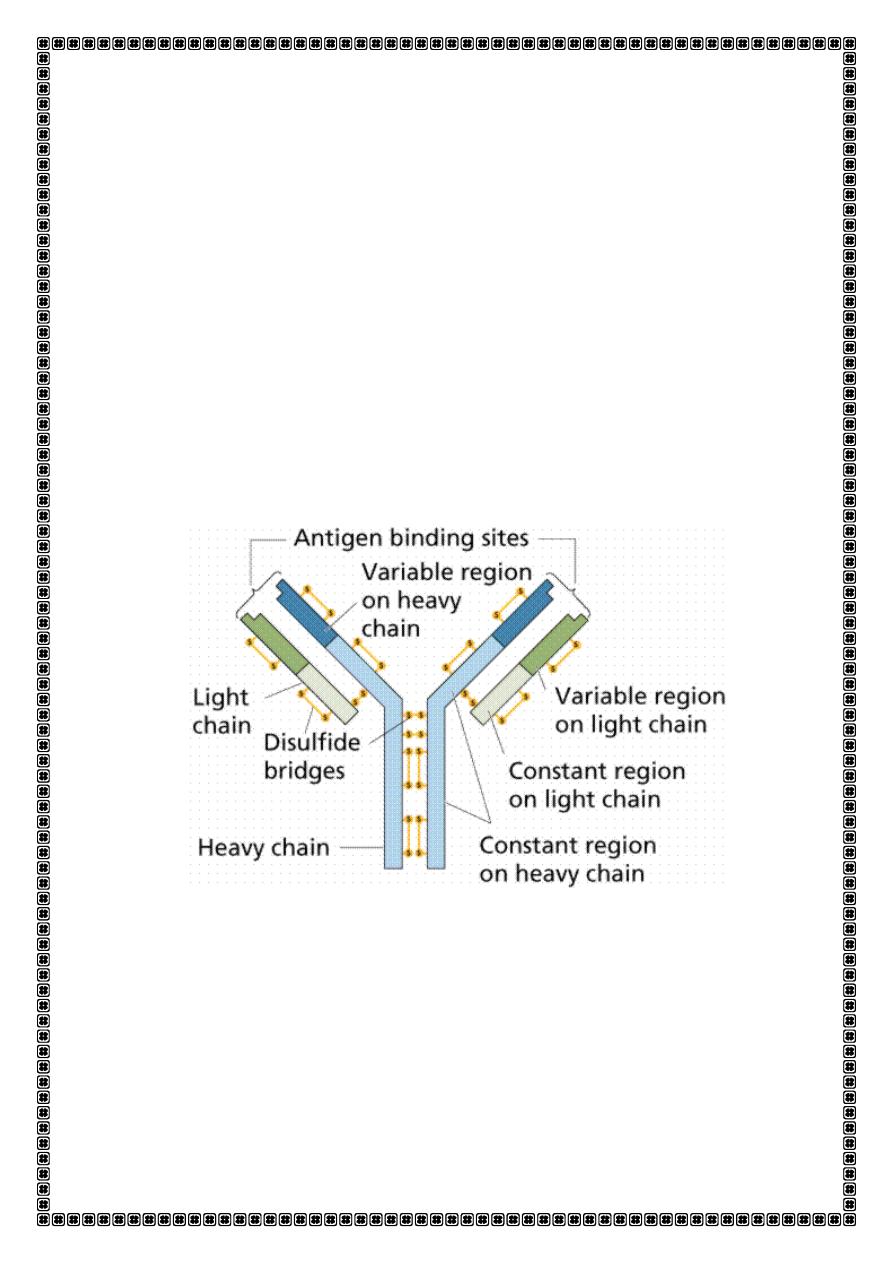
Antibody molecules (immunoglobulins)
Antibodies are glycoproteins. They consist of two heavy chains and two
light hai s eithe o polypeptides . The hea y hai dete i es
the antibody isotype or class, i.e. IgG, A, M, D or E.
.
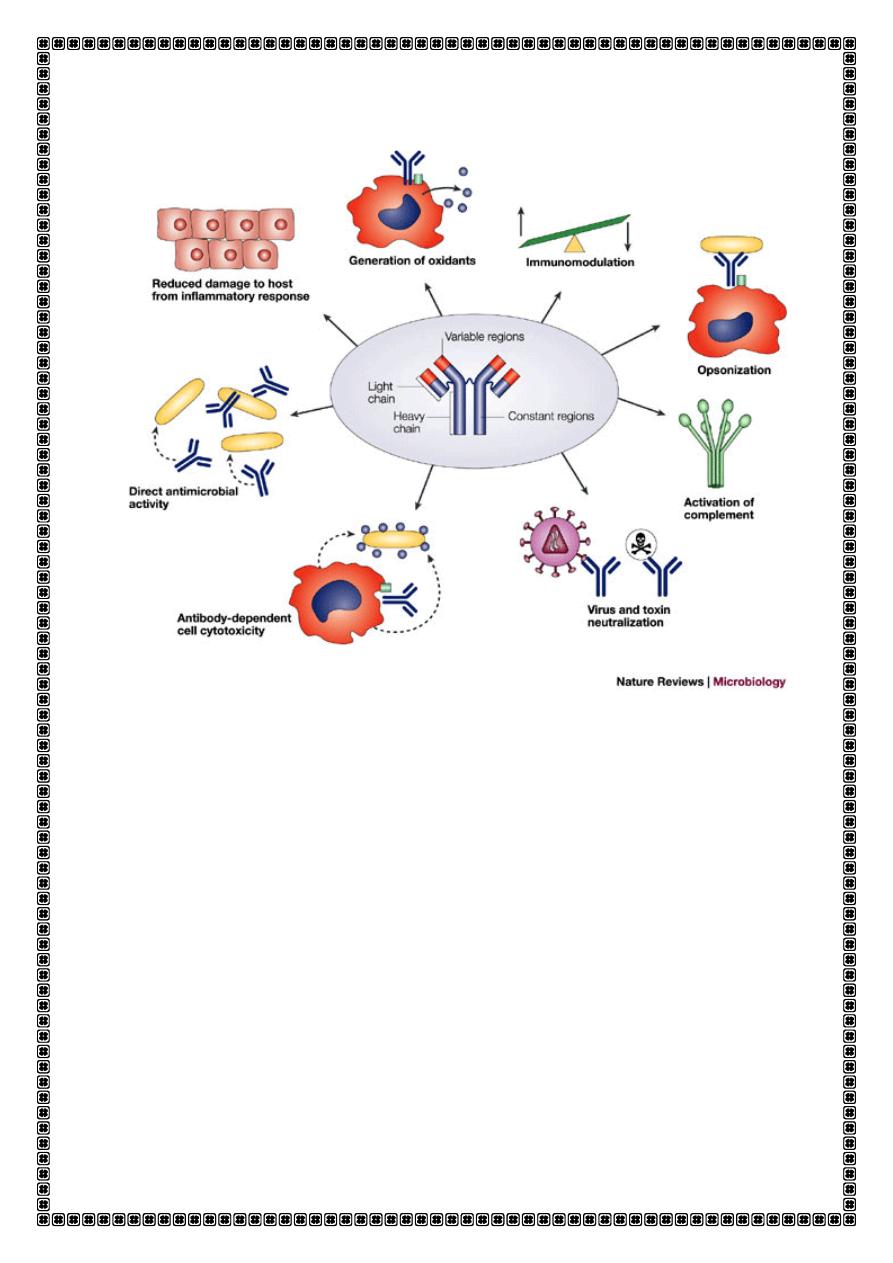
. The major functions of antibody are:,
Immune regulation
:,Ab acting as the antigen receptor on B cells and
presenting the antigen to helper T cells,
plays a part in antigen
presentation.
Antigen
is any substance capable of generating an immune respons,
Ag react with T.cells and B.cells to induce the formation of Ab, then Ag
react with these Ab and cells.
In primary immune respons when Ag first introduce in to body there is
lag phase of several days during which no Ab .detected ,then several
day later(5-10 days)IgM Ab appear.IN secondary immune respons A
group of B.cell called memory cell ,enhance immune response to
previously encountered Ag (the lag phase IS decrease)

,
the first antibody to be produced is IgM, which appears in the serum
after 5-10 days., other antibody classes (IgG, IgA and IgE) are produced
3-7 days later. If, some time later, a memory B cell is re-exposed to
antigen, the lag time between antigen exposure and the production of
antibody is decreased (to 2-3 days
HUMAN LEUCOCYTE ANTIGENS
Antigen presentation
. The immune system has the ability to recognize between 'self' and
'non-self' antigens. This process is facilitated by a
recognition system
called the major histocompatibility complex (MHC) which dictates the
way that antigen is recognized as foreign. In man, the products of MHC
are termed human leucocyte antigens (HLA).
The HLA system
: is cluster of genes is located on the short arm of
chromosome 6. The system comprises six genetic loci - HLA-A, -B, -C, -D,
-DR and
–DQ.The gene encodes
the HLA molecules
(cell surface antigen presenting proteins)which are
distributed throughout the body tissues and it is through differences in
HLA that cells are classified as self or non-self. The possibility of two
different individuals having the same combination of HLA molecules is
very remote.. The HLA genes code for cell-surface glycoproteins that
extend from the plasma membrane to the cytoplasm and are known as
class I and class II molecules. These glycoproteins consist of two chains

of u e ual size α a d β hai s . The hai s fo a g oo e i hi h a
antigenic peptide sits ready for presentation to T cells.
Class I HLA molecules
Class I (HLA-A, -B and -C) are expressed on all cell types except
erythrocytes and trophoblasts.. Class I molecules interact with CD8 T
cells during antigen presentation and therefore are involved in driving
mainly cytotoxic reactions.
Class II HLA molecules
Class II (HLA-D and -DR, D-related) are expressed only on professional
antigen-presenting cells (B cells, monocytes/macrophages, Langerhans'
cells, dendritic cells) and activated T cells. Class II antigens link with CD4
molecules during antigen presentation and the reaction induced by
cells bearing this molecule is therefore of the helper type.
T cells respond to protein antigens, but they cannot recognise these in
their native form. Instead, intact protein must be processed into
component peptides which can bind to the cell surface HLA . This
process is known as antigen processing and presentation, and it is
the
peptide/HLA complex
which is recognised by individual T cells.
T lymphocytes are classified into
1-Helper/inducer cells
Bear CD4 cell surface molecule ( cytokine-secreting cells), making up
about 75% of peripheral blood T cells) and the ability to recognize
antigen only when the Ag expressed with HLA class II on antigen-
presenting cells.

2-Cytotoxic/suppressor cells
Bear CD8cell surface molecule (mainly cytotoxic suppressor cells),
which account for the remainder.able to recognize antigen only when
presented with HLA class I molecules
These cell types are indistinguishable morphologically, but can be
separated by the presence of cell-surface moleculesCD(specific target
molecule on a cell that is recognized by one or more antibodies).
Components of the immune response.
Antigen is presented to T-helper cells (Th cells) by an antigen-
presenting cell. Th cells secrete lymphokines(cytokine), which
1-activate cytotoxic T cells (Tc cells) that are involved in antiviral and
anti-tumour activity.
2-activate NK cells and macrophage, which are involved inantitumor
activity.
3-induction of antibody responses by B cells
Helper T cells are unable to destroy pathogens or cells
directly, but through cytokine production are able to
activate macrophages to kill organisms within them and
further activate cytotoxic T cells and NK cells.
Some diseases show a close association with HLA type.
HLA - associations with disease
A1, B8, DR3
Polymyositis and dermatomyositis
A3, B14
Hereditary haemochromatosis
A28
Schizophrenia
B5
Behçet's syndrome

Polycystic kidney disease
Ulcerative colitis
B8, DR3, DR7, DQ2 Coeliac disease
B18
Hodgkin's disease
B27
Acute anterior uveitis
Ankylosing spondylitis
Psoriatic arthropathy
Reiter's syndrome
Juvenile arthritis
IMMUNE DEFICIENCY
Immune deficiency may arise through
1-primary ,intrinsic defects in immune function,
2- but is much more commonly due to secondary causes
The consequences(complications) of deficiencies of the immune system
include
1- recurrent infections,
2- autoimmunity and
3- susceptibility to malignancy.

Presenting problems in immune deficiency is
Recurrent infections
. Frequent, severe infections or infections caused by unusual organisms
or at unusual sites are the most useful indicator.
Warning signs of immune deficiency
≥ 8 ea i fe tio s ithi yea
≥ se ious si us i fe tio s ithi yea
≥ o ths o a ti ioti s ith little effe t
≥ p eu o ias ithi yea
Failure of an infant to gain weight or grow normally
Recurrent deep skin or organ abscesses
Persistent thrush in mouth or elsewhere on skin
after infancy
Need for intravenous antibiotics to clear infections
≥ deep-seated infections such as sepsis,
meningitis or cellulitis
A family history of primary immune deficiency
If an immune deficiency is suspected but has not yet been formally
characterised, patients should not receive live vaccines because of the
risk of vaccine-induced disease.
1-PRIMARY IMMUMEDEFICIENCY
A-Primary deficiency in innate immune system

1-Primary phagocyte deficiencies
2-Leucocyte adhesion deficiencies
These are disorders of phagocyte migration, when failure to express
adhesion molecules on vascular endothelium results in the inability of
phagocytes to exit the blood stream.
3-Defects in cytokines and cytokine receptors
Defects of cytokines such as IFN-
γ, IL-12 or their receptors also result in
failure of intracellular killing, and individuals are particularly susceptible
to mycobacterial infections..Q WHY INTRA
4-Complement pathway deficiencies
Genetic deficiencies of almost all the complement pathway proteins
have been described.
.C.F
1-recurrent infection with encapsulated bacteria, particularly Neisseria
species.
,2 a -high prevalence of autoimmune disease, particularly systemic
lupus erythematosus
.
3-Deficiency of the regulatory protein C1 esterase inhibitor is not
associated with recurrent infections but causes recurrent angioedema.
-
B-Primary deficiencies of the adaptive immune system
1-Primary T-lymphocyte deficiencies
These are characterised by
recurrent viral, protozoal and fungal
infections . In addition, many T-cell deficiencies are associated with

defective antibody production
because of the importance of T cells in
providing help for B cells.
2-Combined B- and T-lymphocyte immune deficiencies
causes
recurrent bacterial, fungal and viral infections
soon after birth.
Bone marrow transplantation is the only current treatment option,
although specific gene therapy is under investigation.
3-Primary antibody deficienciesMORE IN ADULT
characterisedby recurrent bacterial infections, particularly of the
respiratory and gastrointestinal tract
,:
A-SELECTIVE IMMUNOGLOBULIN A DEFICIENCY
Selective IgA deficiency is the most common primary
immunodeficiency disorder and is characterized by serum
its prevalence is about 1:500 individuals.
Clinical .Features
1-Most persons are
asymptomatic
because of compensatory increases
in secreted IgG and IgM.
2-
frequent and recurrent infections
, such as sinusitis, otitis, and
bronchitis.
3-Individuals with selective IgA deficiency may have high
titers of anti-IgA antibodies and are at risk for
anaphylactic
reactions
following exposure to IgA through infusions of
plasma (or blood transfusions). These anti-IgA antibodies

develop in the absence of prior exposure to human plasma or
blood, possibly due to cross
ea ti ity to o i e IgA i o ’s
milk or prior sensitization to maternal IgA in breast milk.
TREATMENT
Some cases of IgA deficiency may spontaneously remit. Treatment with
commercial immune globulin is ineffective, since IgA and IgM are
present only in trace quantitiesin these preparations
B-Common variable immune deficiency (CVID)
#.intrinsic B cell defects that prevent terminal maturation into
antibody-secreting plasma cells.
#The absolute B cell count in the peripheral blood is normal
#It is characterized by low serum IgG levels but over time all antibody
classes (IgG, IgA, and IgM)may be affected .
and failure to make antibody responses to exogenous pathogens.
CLINICAL FEATURES
by1- an increased incidence of recurrent infections,
2-autoimmune phenomena, Paradoxically, antibody-mediated
autoimmune diseases such as idiopathic thrombocytopenic purpura
and autoimmune haemolyticanaemia are common autoimmune
endocrinopathies, seronegative rheumatic disease, and gastrointestinal
disorders are also commonly seen
3-and neoplastic diseases There is an increased propensity for the
development of B cell neoplasms

(increaserisk of lymphoma), gastric carcinomas, and skin cancers
INVESTIGATION
1-, specific antibody responses to known pathogens should be assessed
by measuring IgG antibodies against tetanus, H. influenzae and Strep.
pneumoniae.
(most patients will have been exposed to some of these antigens
through either infection or immunisation). If specific antibody levels are
low, immunisation with the appropriate killed vaccine should be
followed by repeat antibody measurement 6-8 weeks later; failure to
mount a response indicates a significant defect in antibody production.
Management
1-All patients with antibody deficiencies require aggressive treatment
of infections and prophylactic antibiotics may be indicated.
2- The mainstay of treatment is immunoglobulin replacement
(intravenous immunoglobulin, IVIgGcontains IgG antibodies to a wide
variety of common organisms. IVIgG is usually administered every 3-4
weeks with the aim of maintaining trough IgG levels within the normal
adult range. Treatment may be self-administered and is life-long.
With the exception of selective IgA deficiency, immunisation is
generally not effective because of the defect in IgG antibody
production. As with all primary immune deficiencies, live vaccines
should be avoided.