
Dr. Dlawer Abdul Hammed AL Jaff
Ph.D Pharmacology and toxicology
3/10/2016

The fourth family of receptors differs
considerably from the other three in that the
receptor is entirely intracellular and,
therefore, the ligand must diffuse into the cell
to interact with the receptor
This places constraints on the physical and
chemical properties of the ligand in that it
must have sufficient lipid solubility to be able
to move across the target cell membrane
3/10/2016

Because these receptor ligands are lipid
soluble, they are transported in the body
attached to plasma proteins, such as
albumin.
Binding of the ligand with its receptor follows
a general pattern in which the receptor
becomes activated
The activated ligandâ
€“receptor complex
migrates to the nucleus, where it binds to
specific DNA sequences, resulting in the
regulation of gene expression.
3/10/2016

The time course of activation and response of
these receptors is much longer than that of
the other mechanisms
Because gene expression and, therefore,
protein synthesis is modified, cellular
responses are not observed until considerable
time has elapsed (thirty minutes or more),
and the duration of the response (hours to
days) is much greater than that of other
receptor families
3/10/2016

Radioligand binding studies have shown that
the receptor numbers do not remain constant
but change according to circumstances.
When tissues are continuously exposed to an
agonist, the number of receptors decreases
(down-regulation)
and this may be a cause of
tachyphylaxis (loss of efficacy with frequently
repeated doses), e.g. in asthmatics who use
adrenoceptor agonist bronchodilators
excessively. Prolonged contact with an
antagonist leads to formation of new
receptors
(up-regulation).
3/10/2016

Indeed, one explanation for the worsening of
angina pectoris or cardiac ventricular
arrhythmia in some patients following abrupt
withdrawal of a (beta-adrenoceptor blocker is
that normal concentrations of circulating
catecholamines now have access to an
increased (up-regulated) population of beta-
adrenoceptors
3/10/2016

Drugs that activate receptors do so because
they resemble the natural transmitter or
hormone, and so to act for longer than the
natural substances (endogenous ligands); for
this reason
bronchodilation produced by salbutamol lasts
longer than that induced by adrenaline
3/10/2016

Antagonists (blockers) of receptors are
sufficiently similar to the natural agonist to
be 'recognised' by the receptor and to occupy
it without activating a response, thereby
preventing (blocking) the natural agonist
from exerting its effect.
Drugs that have no activating effect whatever
on the receptor are termed
pure antagonists
3/10/2016

A receptor occupied by a low efficacy agonist
is inaccessible to a subsequent dose of a high
efficacy agonist, so that, in this specific
situation, a low efficacy agonist acts as an
antagonist.
This can happen with opioids.
3/10/2016

Partial agonists. Some drugs, in addition to
blocking access of the natural agonist to the
receptor, are capable of a low degree of
activation, i.e. they have both antagonist and
agonist action. Such substances are said to
show
partial agonist activity
(PAA).
The beta- adrenoceptor antagonists pindolol
and oxprenolol have partial agonist activity
(in their case it is often called
intrinsic
sympathomimetic activity)
(ISA), while
propranolol is devoid of agonist activity, i.e. it
is a pure antagonist
3/10/2016

A patient may be as extensively '(3- blocked'
by propranolol as by pindolol, i.e. exercise
tachycardia is abolished, but the resting heart
rate is lower on propranolol; such differences
can have clinical importance.
3/10/2016

Inverse agonists.
Some substances produce effects that are
specifically opposite to those of the agonist.
The agonist action of benzodiazepines on the
benzodiazepine receptor in the CNS produces
sedation, anxiolysis, muscle relaxation and
controls convulsions; substances called
–beta
carbolines which also bind to this receptor
cause stimulation, anxiety, increased muscle
tone and convulsions; they are inverse
agonists.
3/10/2016

Receptor binding (and vice versa).
If the forces that bind drug to receptor are weak
(hydrogen bonds, van der Waals bonds,
electrostatic bonds), the binding will be easily
and rapidly reversible; if the forces involved are
strong (covalent bonds), rests on their greater
capacity to resist degradation then binding will
be effectively irreversible.
An antagonist that binds reversibly to a receptor
can by definition be displaced from the receptor
by mass action of the agonist (and vice versa). If
the concentration of agonist increases
sufficiently above that of the antagonist the
response is restored.
3/10/2016

This phenomenon is commonly seen in
clinical practice
— patients who are taking a
beta-adrenoceptor blocker, and whose low
resting heart rate can be increased by
exercise, are showing that they can raise their
sympathetic
drive
to
release
enough
noradrenaline (agonist) to diminish the
prevailing degree of receptor blockade.
3/10/2016

Increasing the dose of beta-adrenoceptor
blocker will limit or abolish exercise induced
tachycardia, showing that the degree of
blockade is enhanced as more drug becomes
available to compete with the endogenous
transmitter.
Since agonist and antagonist compete to
occupy the receptor according to the law of
mass action, this type of drug action is
termed
competitive antagonism.
3/10/2016

When
receptor-mediated
responses
are
studied either in isolated tissues or in intact
man, a graph of the logarithm of the dose
given (horizontal axis), plotted against the
response obtained (vertical axis), commonly
gives an S-shaped (sigmoid) curve, the
central part of which is a straight line.
If the measurements are repeated in the
presence of an antagonist, and the curve
obtained is parallel to the original but
displaced to the right, then antagonism is
said to be competitive and the agonist to be
surmountable.
3/10/2016

Drugs that bind
irreversibly
to receptors
include
phenoxybenzamine
(to
the
a-
adrenoceptor). Since such a drug cannot be
displaced from the receptor, increasing the
concentration of agonist does not fully
restore the response and antagonism of this
type is said to be
insurmountable
3/10/2016

The log-dose-response curves for the
agonist in the absence of and in the presence
of a noncompetitive antagonist are not
parallel. Some toxins act in this way, e.g.
bungaro toxin, a constituent of some snake
and spider venoms, binds irreversibly to the
acetylcholine receptor and is used as a tool to
study it. Restoration of the response after
irreversible binding requires elimination of
the drug from the body and synthesis of new
receptor, and for this reason the effect may
persist long after drug administration has
ceased. Irreversible agents find little place in
clinical practice
3/10/2016

Physiological (functional) antagonism
An action on the same receptor is not the only
mechanism by which one drug may oppose the effect
of another. Extreme bradycardia following overdose
of a p-adrenoceptor blocker can be relieved by
atropine which accelerates the heart by blockade of
the parasympathetic branch of the autonomic
nervous system, the cholinergic tone of which (vagal
tone) operates continuously to slow it.
Bronchoconstriction produced by histamine released
from mast cells in anaphylactic shock can be
counteracted by adrenaline (epinephrine), which
relaxes bronchial smooth muscle (P2-adrenoceptor
effect) or by theophylline. In both cases, a
pharmacological effect is overcome by a second drug
which acts by a different physiological mechanism,
i.e. there is physiological or functional antagonism.
3/10/2016

ENZYMES
Interaction between drug and enzyme is in many
respects similar to that between drug and
receptor.
Drugs may alter enzyme activity because they
resemble a natural substrate and hence compete
with it for the enzyme. For example, enalapril is
effective in hypertension because it is structurally
similar to that part of angiotensin I which is
attacked by angiotensin-converting enzyme
(ACE); by occupying the active site of the enzyme
and so inhibiting its action enalapril prevents
formation of the pressor angiotensin II.
3/10/2016

Carbidopa competes with levodopa for dopa decarboxylase and
the benefit of this combination in Parkinson's disease is reduced
metabolism of levodopa to dopamine in the blood (but not in the
brain because carbidopa does not cross the blood-brain barrier).
Ethanol prevents metabolism of methanol to its toxic metabolite,
formic acid, by competing for occupancy of the enzyme alcohol
dehydrogenase; this is the rationale for using ethanol in
methanol poisoning.
The above are examples of competitive (reversible) inhibition of
enzyme activity.
Irreversible inhibition
occurs with organophosphorus insecticides which combine
covalently with the active site of acetylcholinesterase; recovery of
cholinesterase activity depends on the formation of new enzyme.
Covalent binding of aspirin to cyclo-oxygenase (COX) inhibits the
enzyme in platelets for their entire lifespan because platelets
have no system for synthesising new protein and this is why low
doses of aspirin are sufficient for antiplatelet action.
3/10/2016

Continuous or repeated or administration of a
drug is often accompanied by a gradual
diminution of the effect it produces.
Tolerance is said to have been
acquired
when it
becomes necessary to increase the dose of a
drug to get an effect previously obtained with a
smaller dose
By contrast, the term
tachyphylaxis
describes the
phenomenon of progressive lessening of effect
(refractoriness) in response to frequently
administered doses it tends to develop more
rapidly than tolerance.
3/10/2016

Tolerance is readily observed with opioids
Tolerance is acquired rapidly with nitrates
used to prevent angina, possibly mediated by
the generation of oxygen free radicals from
nitric oxide
it can be avoided by removing transdermal
nitrate patches for 4-8 h, e.g. at night.
3/10/2016

The order of reaction or process
In the body, drug molecules cross cell
membranes, are transported across cells, and
many are altered by being metabolised.
These movements and changes involve
interaction with membranes, carrier proteins
and enzymes.
The
rate
at which these movements or
changes can take place is subject to
important influences that are referred to as
the
order
of reaction or process.
3/10/2016

In biology generally, two orders of such
reactions
are
recognised
First-order
processes by which a constant
fraction
of
drug is transported/metabolised in unit time.
Zero-order processes by which a constant
amount
of drug is transported/metabolised in
unit time.
3/10/2016

In the majority of instances the rates at which
absorption, distribution, metabolism and
excretion of a drug occur are directly
proportional to its concentration in the body.
In other words, transfer of drug across a cell
membrane or formation of a metabolite is
high at high concentrations and falls in direct
proportion to be low at low concentrations
(an exponential relationship).
3/10/2016

This is because the processes follow the Law of
Mass Action, which states that the rate of
reaction is directly proportional to the active
masses of reacting substances.
In other words, at high concentrations, there are
more opportunities for crowded molecules to
interact with each other or to cross cell
membranes
than
at
low,
uncrowded
concentrations.
Processes for which rate of reaction is
proportional to concentration are
called first-
order.
In doses used clinically, most drugs are
subject to first-order processes of absorption,
distribution, metabolism and elimination. The
knowledge that a drug exhibits first-order
kinetics is useful.
3/10/2016

As the amount of drug in the body rises, any
metabolic reactions or processes that have
limited capacity become saturated. In other
words, the rate of the process reaches a
maximum amount at which it stays constant,
e.g. due to limited activity of an enzyme, and
further increase in rate is impossible despite
an increase in the dose of drug.
3/10/2016

Clearly, these are circumstances in which the
rate of reaction is no longer proportional to
dose, and processes that exhibit this type of
kinetics are described as rate-limited or
dose-dependent or zero-order or as showing
saturation
kinetics.
In
practice
enzymemediated metabolic reactions are the
most likely to show rate-limitation because
the amount of enzyme present is finite and
can become saturated.
3/10/2016

Alcohol (ethanol) is a drug whose kinetics has
considerable implications for society as well
as for the individual, as follows. Alcohol is
subject to first-order kinetics with a t1/2 of
about one hour at plasma concentrations
below 10 mg/dl [attained after drinking about
two thirds of a unit (glass) of wine or beer]
Above this concentration the main enzyme
(alcohol dehydrogenase) that converts the
alcohol into acetaldehyde approaches and
then reaches saturation, at which point
alcohol metabolism cannot proceed any
faster.
3/10/2016

Thus if the subject continues to drink, the
blood
alcohol
concentration
rises
disproportionately, for the rate of metabolism
remains the same (at about 10 ml or 8 g/h
for a 70 kg man), i.e. a constant amount is
metabolised in unit time, and alcohol shows
zero-order kinetics.
3/10/2016

Absorption
•
Enteral:
by mouth (swallowed) or by sublingual
or buccal absorption; by rectum
•
Parenteral:
by intravenous injection or infusion,
intramuscular injection, subcutaneous injection
or infusion, inhalation, topical application for
local (skin, eye, lung) or for systemic
(transdermal) effect
•
Other routes,
e.g. intrathecal, intradermal,
intranasal, intratracheal, intrapleural, are used
when appropriate.
3/10/2016

The
small intestine
is the principal site for
absorption of nutrients and it is also where
most orally administered drugs enter the
body. This part of the gut has two important
attributes, an enormous surface area due to
the intestinal villi, and an epithelium through
which fluid readily filters in response to
osmotic differences caused by the presence
of food.
3/10/2016

It follows that drug access to the small intestinal
mucosa is important and disturbed alimentary
motility can reduce absorption, i.e. if gastric
emptying is slowed by food, or intestinal transit
is accelerated by gut infection.
The colon is capable of absorbing drugs and
many sustained release formulations probably
depend on absorption there. The stomach does
not play a major role in absorbing drugs, even
those that are acidic and thus unionized and
lipid-soluble at gastric pH, because its surface
area is much smaller than that of the small
intestine and gastric emptying is speedy (t/£ 30
min).
3/10/2016

This system is illustrated by the bile salts, which
are conserved by circulating through liver,
intestine and portal blood about eight times a
day. A number of drugs form conjugates with
glucuronic acid in the liver and are excreted in
the bile. These glucuronides are too polar
(ionised) to be reabsorbed; they therefore remain
in the gut, are hydrolysed by intestinal enzymes
and bacteria, releasing the parent drug, which is
then reabsorbed and reconjugated in the liver.
Enterohepatic recycling appears to help sustain
the plasma concentration.
3/10/2016

When a drug is injected intravenously it
enters the systemic circulation and thence
gains access to the tissues and to receptors,
i.e. 100% is available to exert its therapeutic
effect. If the same quantity of the drug is
swallowed, it does not follow that the entire
amount will reach first the portal blood and
then the systemic blood, i.e. its availability for
therapeutic effect via the systemic circulation
may be less than 100%.
3/10/2016

The terms potency and efficacy are often used
imprecisely and therefore, confusingly
Potency is the amount (weight) of drug in relation to
its effect, e.g. if weight-for-weight drug A has a
greater effect than drug B, then drug A is more potent
than drug B, although the maximum therapeutic
effect obtainable may be similar with both drugs.
The diuretic effect of bumetanide 1 mg is equivalent
to frusemide 50 mg, thus bumetanide is more
potent
than frusemide but both drugs achieve about the
same maximum effect.
The difference in weight of drug that has to be
administered is of no clinical significance unless it is
great.
3/10/2016

Pharmacological efficacy
refers to the strength of response induced by
occupancy of a receptor by antagonists
(intrinsic activity).
3/10/2016

Half-Life
The half-life (
t
½
) is the time it takes for the plasma
concentration or the amount of drug in the body to
be reduced by 50%.
T ½ =0.693*Vd/CL
Clearance is the measure of the body's ability to
eliminate a drug; thus, as clearance decreases, owing
to a disease process, for example, half-life would be
expected to increase. However, this reciprocal
relationship is valid only when the disease does not
change the volume of distribution.
For example, the half-life of diazepam increases with
increasing age; however, it is not clearance that
changes as a function of age but rather the volume of
distribution. Similarly, changes in protein binding of a
drug may affect its clearance as well as its volume of
distribution, leading to unpredictable changes in
half-life as a function of disease.
3/10/2016

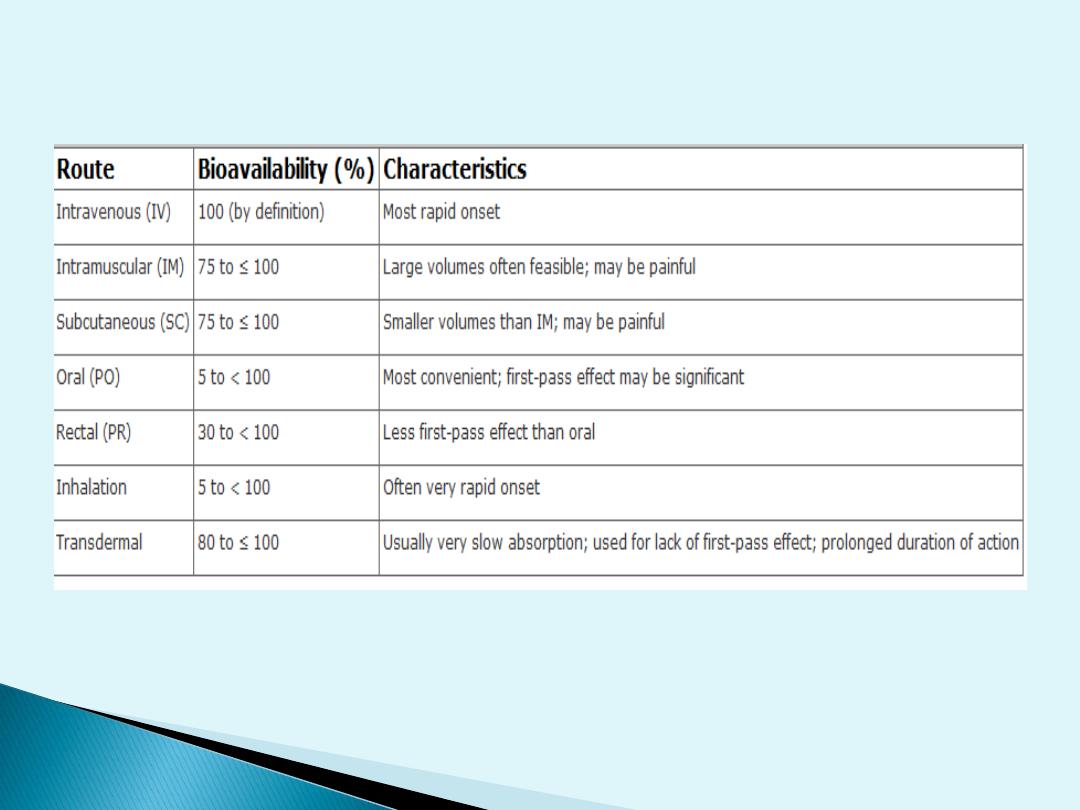
Bioavailability
Bioavailability is defined as the fraction of
unchanged drug reaching the systemic
circulation following administration by any
route.
The
area
under
the
blood
concentration-time curve (AUC) is a common
measure of the extent of bioavailability for a
drug given by a particular route.
For an intravenous dose of the drug,
bioavailability is assumed to be equal to
unity. For a drug administered orally,
bioavailability may be less than 100% for two
main
reasons
—incomplete
extent
of
absorption and first-pass elimination
3/10/2016
3/10/2016

EXTENT OF ABSORPTION
After oral administration, a drug may be incompletely
absorbed, eg, only 70% of a dose of digoxin reaches
the systemic circulation. This is mainly due to lack of
absorption from the gut. Other drugs are either too
hydrophilic (eg, atenolol) or too lipophilic (eg,
acyclovir) to be absorbed easily, and their low
bioavailability is also due to incomplete absorption.
If too hydrophilic, the drug cannot cross the lipid cell
membrane; if too lipophilic, the drug is not soluble
enough to cross the water layer adjacent to the cell.
Drugs may not be absorbed because of a reverse
transporter associated with P-glycoprotein.
This process actively pumps drug out of gut wall
cells back into the gut lumen. Inhibition of P-
glycoprotein and gut wall metabolism, eg, by
grapefruit juice, may be associated with substantially
increased drug absorption
3/10/2016

FIRST-PASS ELIMINATION
Following absorption across the gut wall, the
portal blood delivers the drug to the liver prior to
entry into the systemic circulation. A drug can be
metabolized in the gut wall (eg, by the CYP3A4
enzyme system) or even in the portal blood, but
most commonly it is the liver that is responsible
for metabolism before the drug reaches the
systemic circulation.
In addition, the liver can excrete the drug into
the bile. Any of these sites can contribute to this
reduction in bioavailability, and the overall
process is known as first-pass elimination. The
effect of first-pass hepatic elimination on
bioavailability is expressed as the extraction ratio
(ER):
3/10/2016

Volume of Distribution
Volume is a second fundamental parameter
that is useful in considering processes of
drug disposition. The volume of distribution
(
V
) relates the amount of drug in the body to
the concentration of drug (
C
) in the blood or
plasma depending on the fluid measured.
This volume does not necessarily refer to an
identifiable physiological volume but rather
to the fluid volume that would be required to
contain all the drug in the body at the same
concentration measured in the blood or
plasma:
3/10/2016

A drug's volume of distribution therefore
reflects the extent to which it is present in
extravascular tissues and not in the plasma.
The plasma volume of a typical 70-kg man is
3 L, blood volume is about 5.5 L, extracellular
fluid volume outside the plasma is 12 L, and
the
volume
of
total-body
water
is
approximately 42 L.
V=amount of the drug in the body /C
3/10/2016

Many drugs exhibit volumes of distribution
far in excess of these values. For example, if
500 g of the cardiac glycoside
digoxin
were
in the body of a 70-kg subject, a plasma
concentration of approximately 0.75 ng/ml
would be observed. Dividing the amount of
drug in the body by the plasma concentration
yields a volume of distribution for digoxin of
about 667 L, or a value approximately 10
times greater than the total-body volume of a
70-kg man.
3/10/2016

In fact, digoxin distributes preferentially to
muscle and adipose tissue and to its specific
receptors (Na
+
,K
+
-ATPase), leaving a very
small amount of drug in the plasma to be
measured.
For drugs that are bound extensively to
plasma proteins but that are not bound to
tissue
components,
the
volume
of
distribution will approach that of the plasma
volume because drug bound to plasma
protein is measurable in the assay of most
drugs.
3/10/2016

The volume of distribution may vary widely
depending on the relative degrees of binding
to high-affinity receptor sites, plasma and
tissue proteins, the partition coefficient of the
drug in fat, and accumulation in poorly
perfused tissues.
As might be expected, the volume of
distribution for a given drug can differ
according to patient's age, gender, body
composition, and presence of disease. Total-
body water of infants younger than 1 year of
age, for example, is 75% to 80% of body
weight, whereas that of adult males is 60%
and that of females is 55%.
3/10/2016