Immunopathology
Dr. Zainab. W. A. AlhayaliINTRODUCTION
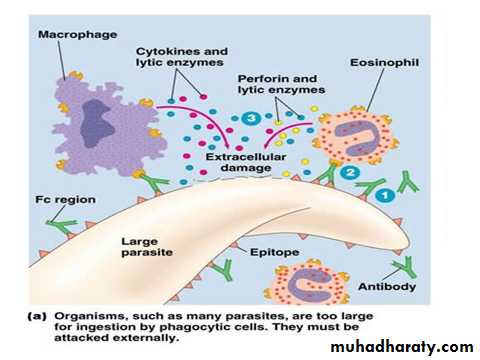
The immune system protects the host from invasion by foreign and potentially harmful agents.Immune responses can be elicited by a wide range of agents (antigens) including parasites, bacteria, viruses, chemicals, toxins, drugs, and transplanted tissues.
Immune system is like a double-edged sword. Though it is protective in most of the situations, sometimes a defect in the immune system may cause fatal diseases.
Definition
Immunity is resistance (defense mechanism) exhibited by host against invasion by any foreign antigenTypes
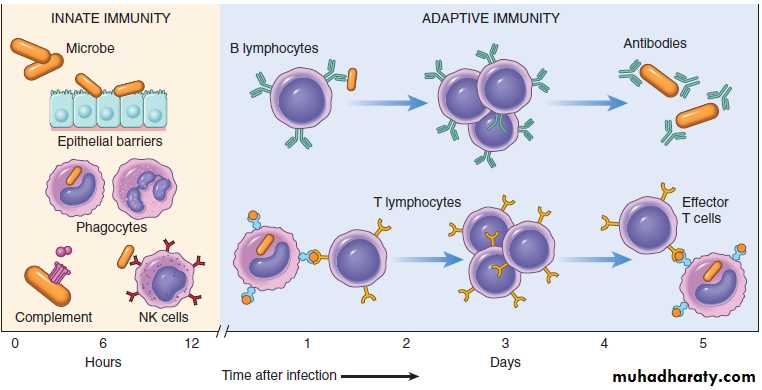
There are two types• Innate immunity.
• Adaptive immunity.
Innate (Natural/Native) Immunity
General Features
First line of defense present by birth.
Provides immediate initial protection against an invading pathogen.
Does not depend on the prior contact with foreign antigen or microbes.
Lacks specificity, but highly effective.
Triggers the adaptive immune response.
No memory is seen.
Major components of innate immunity
1. Epithelial barriers :like intact skin that blocks entry of environmental microbes2. Cellular: antigen presenting cells (macrophages, Natural killer (NK) cells, Dendritic cells).
3. Humoral:( palasma proteins of the complement system, and C-reactive protein)
Functions of Innate Immune Response
Inflammation and destruction of invading microbe.Antiviral defense is mediated by dendritic cells and NK cells.
Specific or adaptive immunity
If the innate immune system fails to provide effective protection against invading microbes, the adaptive immune system is activated.General Features
Acquired in nature
Second line of defense.
Takes more time to develop and is more powerful than innate immunity.
Prior exposure to antigen is present.
Capable of recognizing both microbial and nonmicrobial substances.
Long-lasting protection (Memory)
specific
It has 2 main components:
a) Humoral: consisting of antibodies formed by B cells.
b) Cellular: mediated by T cells.
Functions of Adaptive Immune Response
Antibodies: Protection against extracellular microbes in the blood, mucosal secretions and tissues.T lymphocytes:
– Defense against viruses, fungi and intracellular bacteria.
– Important immunoregulatory role, orchestrating and regulating the responses of other components of the immune system.
The principal components of innate immunity and adaptive immunity
ORGANS OF IMMUNE SYSTEM
a) Primary lymphoid organs:i) Thymus
ii) Bone marrow
b) Secondary lymphoid organs:
i) Lymph nodesii) Spleen
iii) MALT (Mucosa-Associated Lymphoid Tissue located in the respiratory tract and GIT).
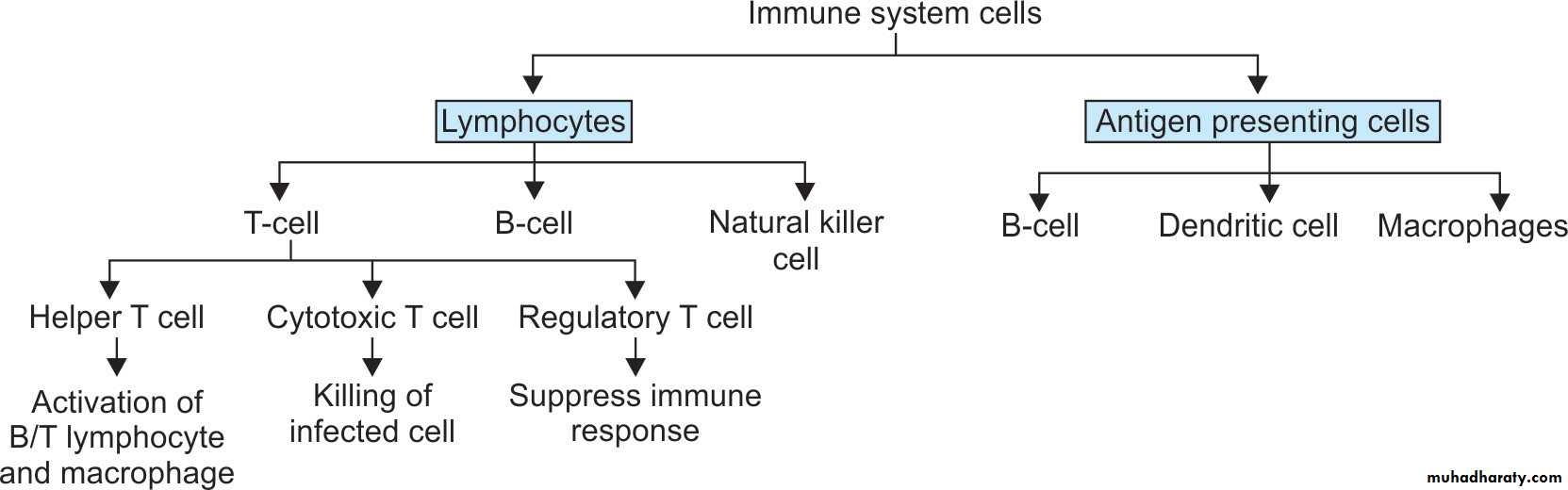
CELLS OF IMMUNE SYSTEM
i) Lymphocytes
ii) Monocytes, macrophages and dendritic cells.
iii) Mast cells and basophils
iv) Neutrophils and Eosinophils
CD4
CD8
• Lymphocytes
T lymphocytesB lymphocytes
NK lymphocytes
• T Lymphocytes
Development: T (thymus derived) lymphocytes develop from precursors in the thymus.Distribution: Mature T cells are found in
– Peripheral blood (60% to 70%) of lymphocytes
– paracortical region of lymph node and periarteriolar sheaths of spleen.
T-cell Receptor: T cell recognizes a specific cell-bound antigen by means of an antigen specific T-cell receptor (TCR).
Subsets of T lymphocytes:
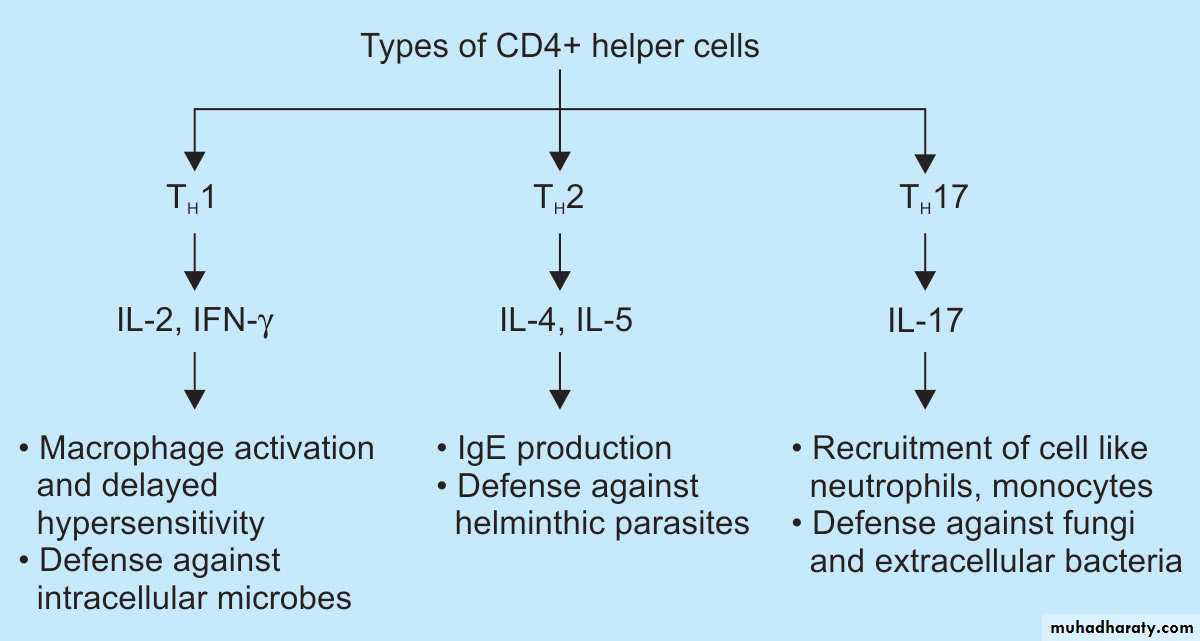
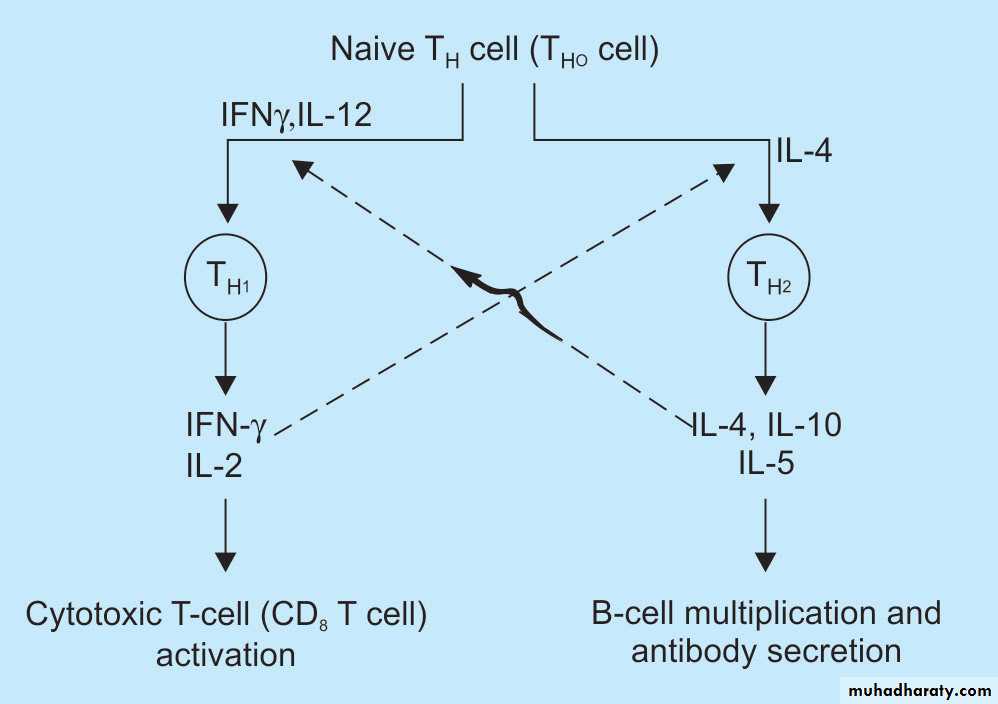
CD4+ T cell: These subset of T cells have CD4 molecule and are called as helper T cells.They constitute about 60% of mature T cells. The CD4 cells function as cytokine-secreting helper cells that help macrophages and B lymphocytes to combat infections.
They are subcategorized as TH1, TH2 and TH17 .
CD8+ T cell: These subset of T cells have CD8 molecule and are called as cytotoxic/killer T cells. They constitute about 30% of T cells.
CD8+ T cells function as cytotoxic (killer) T lymphocytes (CTLs) to destroy host cells harboring microbes and tumor cells.
Regulatory T lymphocytes: suppress the immune response
II. B LymphocytesDevelopment: B (bone marrow derived) lymphocytes .
Distribution:
– Peripheral blood: Mature B cells constitute 10% to 20% of the circulating peripheral lymphocyte population
– Peripheral lymphoid tissues: Lymph nodes (cortex), spleen (white pulp), and mucosa associated lymphoid tissues .
B-cell Receptor (BCR): B cells have receptors composed of IgM and IgD on their surface and has unique antigen specificity.
Functions of B cells:
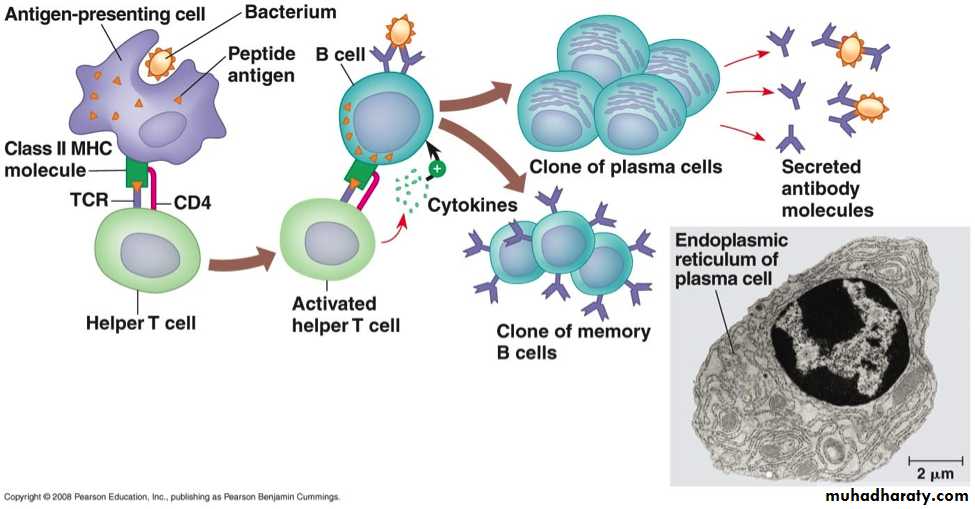
– Production of antibodies: The primary function of B cells is to produce antibodies. After stimulation by antigen and other signals, B cells develop into plasma cells. These cells secrete antibodies like IgG, IgM, IgD, IgA and IgE. Initially, the first antibody produced by the plasma cell is IgM and later, other antibodies like IgG, IgA etc
– Antigen presenting cell: B cells also serve as APCs and are very efficient at antigen processing.
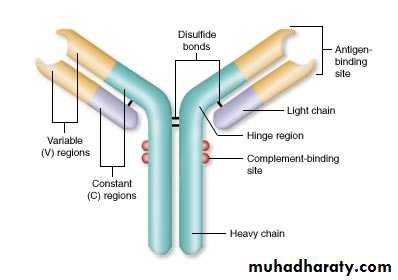
Fab region
Fc portionIII. Natural Killer Cells
Non-phagocytic lymphocytes also called ‘Large granular lymphocytes’ as they are morphologically larger than both T and B lymphocytes and contain azurophillic granules (which are absent in both T and B lymphocytes)Markers: They do not bear the markers for T or B cells. Two cell surface molecules, CD16 and CD56, are commonly used to identify them.
Comprise about 5% to 15% of human peripheral lymphoid cells.
Function: kills a variety of virus-infected cells and tumor cells, without prior exposure to or activation by these microbes or tumors .
They recognize abnormal cells in two ways:
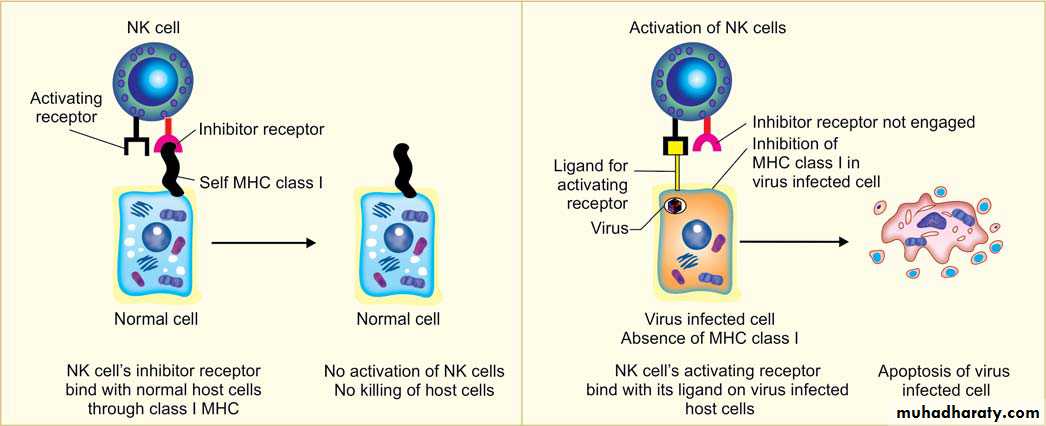
1- Antibody-dependent cellular cytotoxicity (ADCC): NK cells bear (CD16) is an Fc receptor for IgG, and it confers on NK cells the ability to lyse IgG-coated target cells.2- Perforin-granzymes system : NK cells have a variety of surface receptors for MHC (major histocompatibility complex) class I. These are activating and inhibitory receptors.
◆ Inhibitory receptors: MHC class I molecules are normally expressed on healthy/ normal host cells. NK cell inhibitory receptors recognize self–class I MHC molecules. They prevent NK cells from killing normal host cells by inhibiting the death pathway (apoptosis).
◆ Activating receptors: If the target cell with which NK cells interact, do not have MHC molecules on their surface. These activating receptors makes holes in the target cell membrane by secreting perforins and cause apoptosis of target cell .
surface receptors of natural killer (NK) cells.
2) Dendritic Cells
As the name suggests these cells have numerous fine cytoplasmic processes that resemble dendrites. These are important antigen presenting cells (APC) in the body .Types: Two functionally different types of dendritic cells
– Interdigitating dendritic cells (IDC) : They are the most important antigen-presenting cells (APCs) for initiating primary T-cell responses against protein antigens.
◆ Location:
• Common location is below the epithelial lining: Immature dendritic cells within the epidermis are known as Langerhans cells.
• 2) Interstitia of all tissues.
– Follicular dendritic cell:
◆ Location: It is present in the germinal centers of lymphoid follicles in the spleen and lymph nodes (hence named as follicular dendritic cell).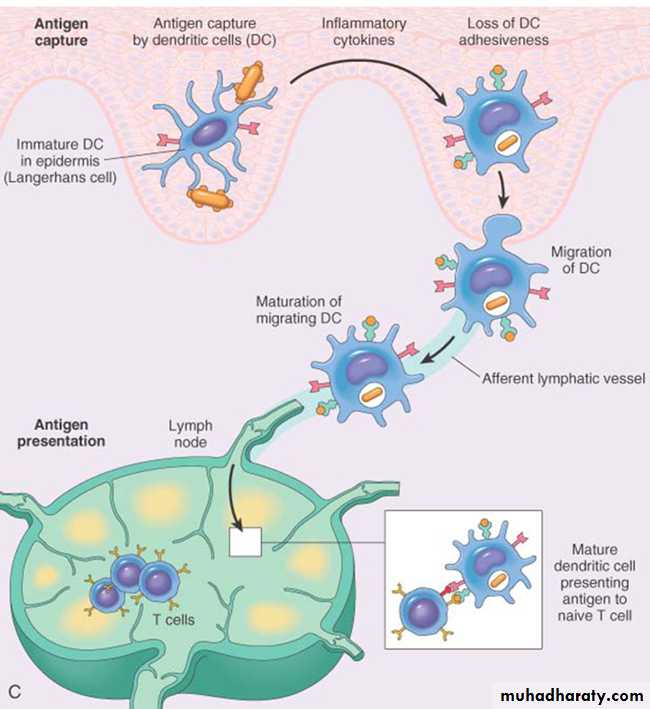
3) MONOCYTES AND MACROPHAGES
Circulating monocytes are immature macrophages and constitute about 5% of peripheral leucocytes.Functions of macrophages are as follows:
1. Antigen recognition2. Phagocytosis
3. Secretory function :
(i) Cytokines (IL-1, IL-2, IL-6, IL-8, IL-10, IL-12, tumour necrosis factor-a) and prostaglandins (PGE, thromboxane-A, leukotrienes)
(ii) Secretion of proteins involved in wound healing e.g. collagenase, elastase, fibroblast growth factor, angiogenesis factor
(iii) Acute phase reactants e.g. fibronectin, microglobulin, complement components.
4. Antigen presentation
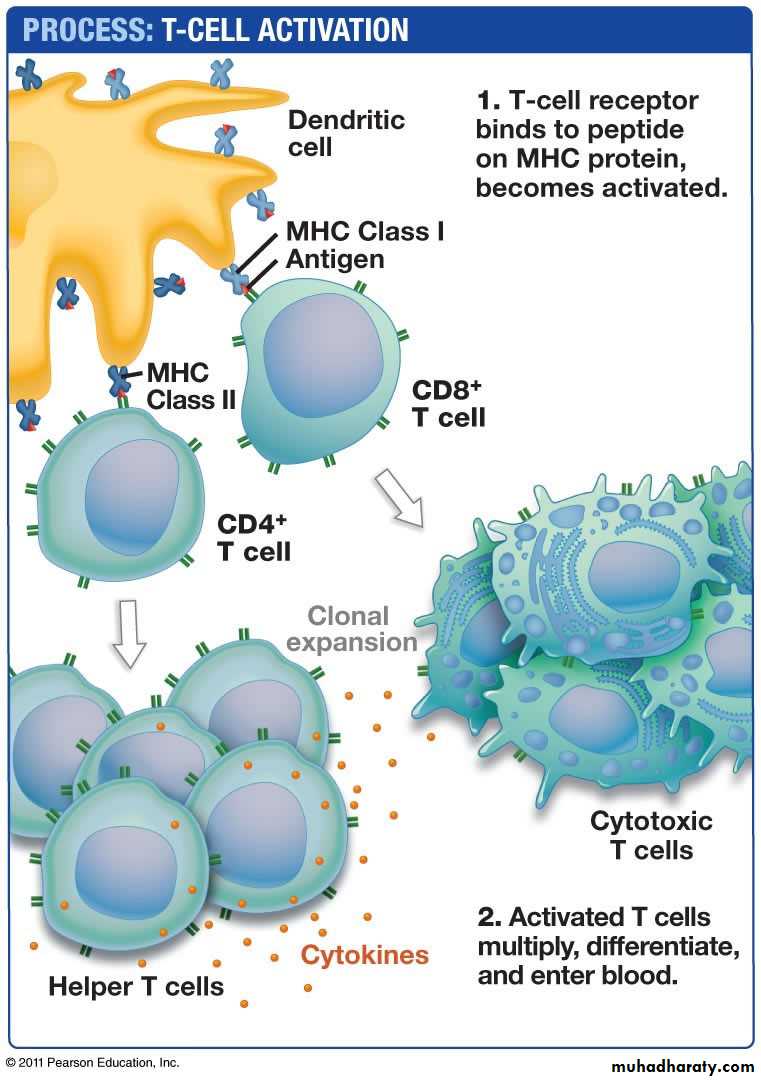
APC
T cells activation
CYTOKINES
Short-acting soluble proteins.secreted by haematopoietic and non-haematoopoietic cells in response to various stimuli.
These cytokines represent the messenger molecules of the immune system and mediate communications between various cells of the immune system.
Classification
Most of the cytokines have a many effects and can be classified depending on their functions.I.Cytokines of Innate Immunity
These cytokines are produced rapidly in response to microbes and other stimuli
Mainly secreted by macrophages, dendritic cells, and NK cells
Mediate inflammation and anti-viral defense
These cytokines include TNF, IL-1, IL-12 and chemokines.
II.Cytokines of Adaptive Immunity
These cytokines are produced mainly by CD4+ T lymphocytes in response to antigen and other signalsThey promote lymphocyte proliferation and differentiation and activate effector cells
This category include IL-2, IL-4, IL-5, IL-17, and IFN-.
III.Colony-Stimulating Factors
These cytokines stimulate hematopoiesis and are assayed by their ability to stimulate formation of blood cell colonies from bone marrow progenitors.
They increase leukocyte numbers during immune and inflammatory responses.
Function
Cytokines are involved in following actions:1. Activation of immune system
2. Inflammatory mediators3. Regulation of cell growth
Major histocompatibility complex (MHC)
All human cells have a series of molecules on their surfaces that are recognized by other individuals as foreign antigens.Major histocompatibility complex (MHC) molecules were discovered as products of genes that evoke rejection of transplanted organs and responsible for tissue compatibility between individuals.
The human major histocompatibility complex (MHC) are commonly called the human leukocyte antigen (HLA) complex is the name of cluster of genes located on short arm of chromosome 6 (6p)
They were named HLA because in humans MHC-encoded proteins were initially detected on leukocytes by the binding of antibodies
Importance of MHC:
1) in organ/tissue transplantation
2) HLA is linked to many autoimmune diseases.
HLA-DP, HLADQ, HLA-DR
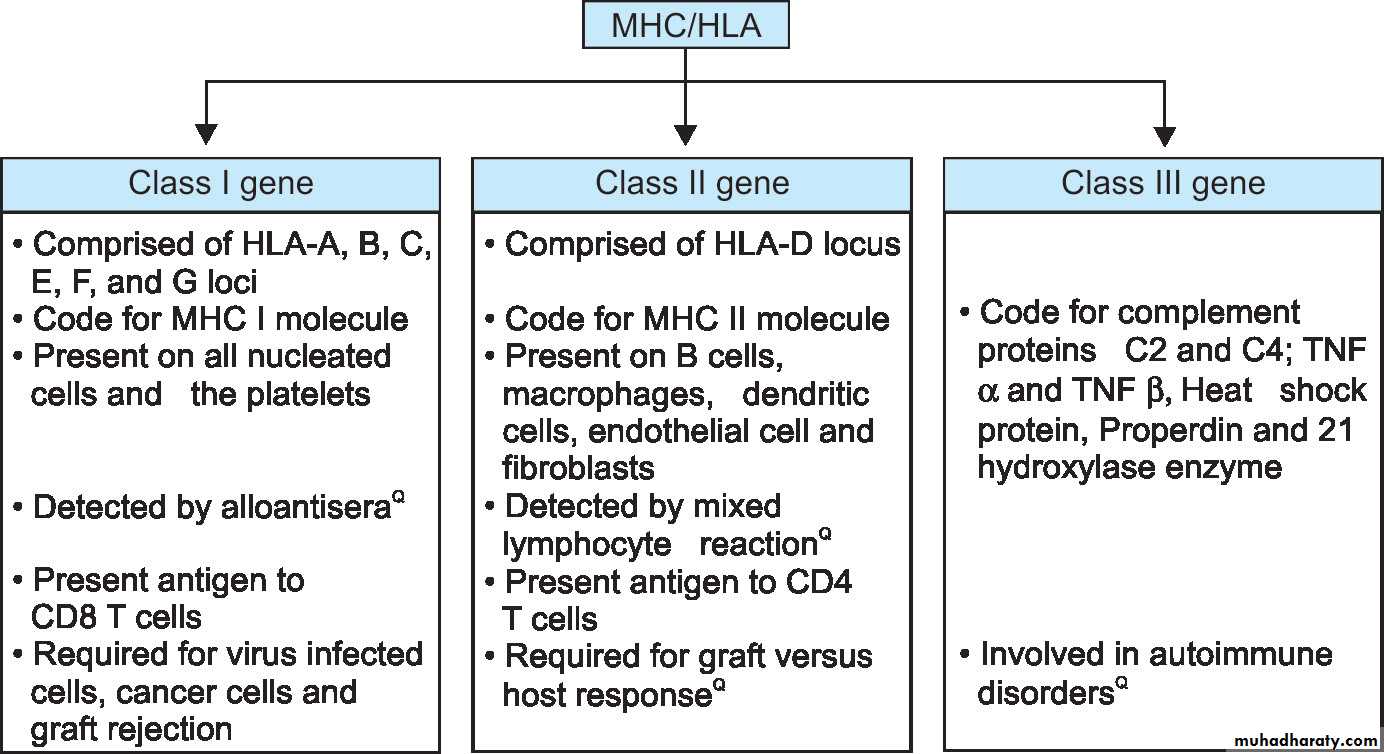
Classification of MHC MoleculesClass I MHC Molecules
They are the products of MHC class I genes and are expressed on all nucleated cells and platelets (except erythrocytes and trophoblasts).
They are encoded by three closely linked loci, designated HLA-A, HLA-B, and HLA-C.
Functions: Products of MHC class I gene are integral participants in the immune response to intracellular infections, tumors, and allografts.
Class I molecules interact with CD8+ T lymphocytes during antigen presentation and are involved in cytotoxic reactions. CD8+ T lymphocytes recognize antigens only in he context of self- class I molecules .
Class II MHC Molecules
They are encoded in a region called HLA-D, which has three sub-regions: HLA-DP, HLADQ, and HLA-DR.Class II antigens are expressed only on professional antigen presenting cells (B lymphocytes, monocytes/macrophages, Langerhans’ cells, dendritic cells) and activated T cells.
Function: This locus contains genes that encode many proteins involved in antigen processing and presentation. The class II–peptide complex is recognized by CD4+ T cells (function as helper cells) and these CD4 molecule acts as the co-receptor.
CD4+ T cells can recognize antigens only in the context of self–class II molecules .
Class III MHC Molecules
Their gene encode components of the complement system, cytokines, tumor necrosis factor (TNF), lymphotoxin, and some proteins without apparent role in the immune system.
HLA and Disease Association
Some diseases are associated with the inheritance of certain HLA alleles and these diseases can be broadly grouped into:Inflammatory diseases: e.g. ankylosing spondylitis most striking associated with HLA-B27.
Autoimmune diseases: e.g. autoimmune endocrinopathies associated with alleles at the DR locus.
Inherited errors of metabolism: e.g. hereditary hemochromatosis (HLA-A).

DISEASES OF IMMUNITY
Diseases of the immune system which are broadly classified into the following 4 groups:• Hypersensitivity reactions are characterized by hyperfunction or inappropriate response of the immune system.
2. Autoimmune diseases occur when the immune system fails to recognize ‘self’ from ‘non-self’.
3. Immunodeficiency disorders are characterized by deficient or absent cellular and/or humoral immune functions.
4. Possible immune disorders in which the immunologic mechanisms are suspected in their etiopathogenesis. Classical example of this group is amyloidosis.
1. Hypersensitivity reactions
Definition: it is a pathological, hyperfunction, and injurious immune response to antigen leading to tissue injury, disease or sometimes death in a sensitized individual.General Features of Hypersensitivity Disorders:
Priming or sensitization: It occurs in individuals who had previous contact with the antigen (allergen).Nature of antigens: It may be exogenous or endogenous origin.
• – Exogenous antigens: Examples, antigens in dust, pollens, foods, drugs, microbes, chemicals, and few blood products.• – Endogenous antigens: Self antigens .
Genetic susceptibility: Hypersensitivity diseases are usually associated with the inheritance of particular susceptibility genes (e.g. HLA genes).
Causes of Hypersensitivity Reactions
• Autoimmunity: reactions against self antigens. Normally, the immune system does not react against self-generated antigens. This phenomenon is called self tolerance, implying that the body “tolerates” its own antigens. On occasion, self-tolerance fails, resulting in reactions against the body’s own cells and tissues .• Reactions against microbes
• Reactions against environmental antigens. Most healthy people do not react strongly against common environmental substances (e.g., pollens, animal danders, or dust mites), but almost 20% of the population are “allergic” to these substances.Classification of hypersensitivity reactions
• Type I (immediate) hypersensitivity.
• Type II hypersensitivity ( antibody-mediated disorder)• Type III hypersensitivity ( immune complex mediated disorders)
• Type IV hypersensitivity ( cell-mediated or delayed type).
TYPE I (IMMEDIATE) HYPERSENSITIVITY REACTIONS
Usually known as allergic or atopic disorders and the environmental antigens that elicit these reactions are known as allergens.Definition: Type I hypersensitivity reaction is a type of immunological tissue reaction, which occurs rapidly (within 5-10 minute) after the interaction of antigen (allergen) with a IgE antibody bound to the mast cells in a sensitized person.
Characteristics
Immediate reaction occurring within minutes (5–10 minutes).Antibodies: Mediated by IgE antibody.
Develops after the interaction of an antigen with IgE bodies bound to mast cells.
Genetic susceptibility: Occurs in genetically susceptible individuals previously sensitized to the antigen.
Antigens (allergens): Many allergens (e.g. house-dust mite, pollens, animal danders
or moulds) in the environment are harmless for majority of individuals. Allergens elicit
significant IgE reactions only in genetically predisposed individuals, who are said to be
atopic.
Sequence of Events
A. During Initial Exposure to Antigen (Sensitization)
In a genetically susceptible individual, the following events occur:
1. Exposure to sensitizing antigen: Individuals are exposed to environmental allergens and may be introduced by: 1) inhalation, 2) ingestion or 3) injection.
2. Presentation of the antigen: The sensitizing antigen (allergen) is presented to T cells. Antigen is captured by the antigen presenting cells and presented to the T cell which then differentiates into TH2 cell.
3. Activation of TH2 cells: In genetically susceptible individual, antigens (allergens) activates TH2 subset of CD4+ helper T cells secrets cytokines (e.g. IL-4, IL-5 and IL-13).
4. Production of IgE antibody: IL-4 secreted by TH2 cells stimulates B cells to secrete cytotropic IgE antibodies. IL-5 activates eosinophils and IL-13 stimulates epithelial cells to secrete mucus.
5. Sensitization of mast cells by IgE antibody:
Mast cells are mainly concentrated near blood vessels and nerves and in subepithelial tissues (common sites of type I hypersensitivity).
Mast cells possess Fc- receptor, which have high affinity for IgE antibodies.
IgE antibodies attach to the Fc-R on the mast cells.
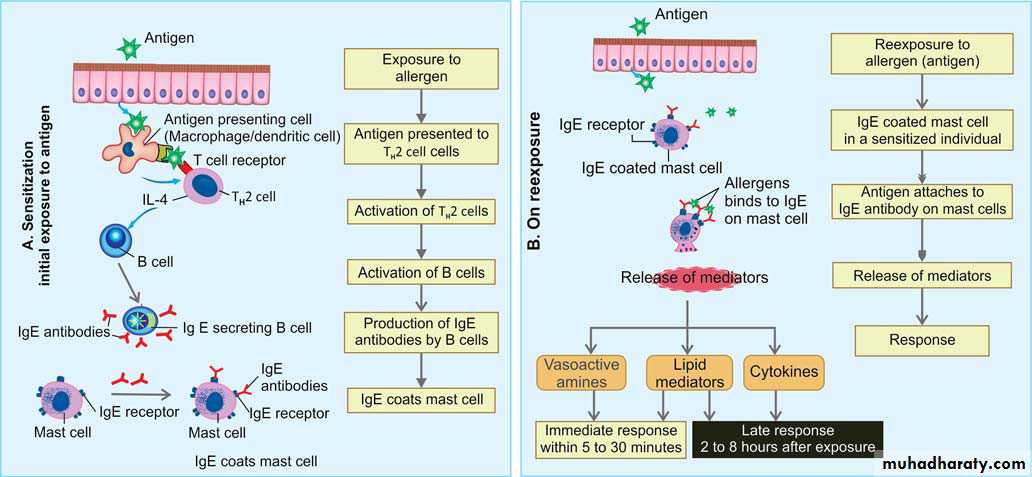
B. During re-exposure Exposure to Antigen
In sensitized individual, following events occur:Mast cell activation: The antigen (allergen) binds to the more than one IgE antibody molecules on mast cells generate signals causes mast cell degranulation ,secretion of preformed (primary) mediators that are stored in the granules.
Two phases: IgE triggered reactions can be divided into two phases:
1. Immediate phase response :
Develops within 5 to 30 minutes after exposure to an allergen and subside in 60 minutes.
there is release of preformed mediators of histamine, proteases and chemotactic factors
Characterized by vasodilation, vascular leakage, and smooth muscle spasm or glandular secretions.
2. Late-phase reaction:
Develops in 2 to 8 hours after the exposure to antigen which may last for several days.
release of secondary mediators from the mast cells that include prostaglandins, leukotrienes, cytokines and platelet activating factor (PAF).
Characterized by infiltration of tissues with eosinophils, neutrophils, basophils, monocytes, and TH2 cells. It also shows mucosal epithelial cell damage.
Mast cell mediators
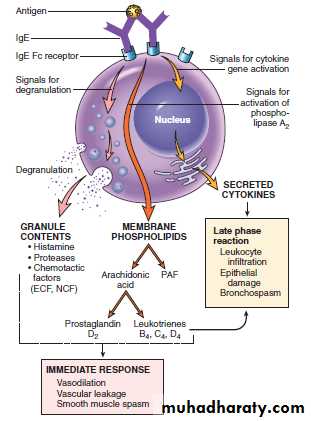
Mediators of Type I Hypersensitivity Reactions
1. Preformed mediators (primary mediators): They are stored in mast cell granules. Their biological effects start immediately following their release. These include:
Vasoactive amines: Most important being histamine, which causes
• – Vasodilatation
• – Increased vascular permeability
• – Smooth muscle contraction
• – Increased secretion of mucus by nasal, bronchial, and gastric glands.
Enzymes: It includes neutral proteases (chymase, tryptase) and several acid hydrolases. These enzymes cause tissue damage and activates components of complement (e.g. C3a) by acting on their precursor proteins.
Proteoglycans: It includes heparin (anticoagulant), and chondroitin sulfate.
chemotactic factors : for Neutrophil and eosinophil (NCF and ECF).
2. Newly synthesized (Secondary mediators) :
A. Lipid mediators: They are synthesized and secreted by mast cells, includes leukotrienes and prostaglandins.
Leukotrienes C4 and D4 : These are the most powerful (several thousand times than histamine) and cause increased vascular permeability and bronchial smooth muscle contraction
Leukotriene B4 It is chemotactic for neutrophils, eosinophils, and monocytes.
Prostaglandin D2: It causes bronchospasm and increased mucus secretion.
B. Cytokines: which include:
TNF, IL-1, and chemokines promote leukocyte recruitment (during the late-phase reaction).IL-4 and IL-5 amplifies the TH2 response and IL-13 stimulates mucus secretion by epithelial cells.
Examples of type I hypersensitivity includes:

Systemic Hypersensitivity
follows injection of an antigen to which the host has become sensitized. Often within minutes, a state of shock is produced which is fatal (anaphylactic shock) e.g., pencillin or bee venom when administered as parenteral injection result in systemic anaphylaxis.Within minutes of an exposure in sensitized host, itching, urticaria and skin erythema appears followed rapidly by profound respiratory difficulty (due to bronchoconstriction and increased mucus production) and laryngeal edema, systemic vasodilatation may follow causing anaphylactic shock and patient may progress to circulatory failure and death within minutes.
Local Hypersensitivity
depends on the portal of entry of antigen e.g. localized cutaneous swelling (skin allergy), nasal and conjunctival discharge (allergic rhinitis and conjunctivitis), bronchial asthma, allergic gastroenteritis ( food allergy).
chronic inflammation due to type I hypersensitivity reaction: Nasal mass with edematous stroma with chronic inflammatory cell infiltrate ( mainly eosinophils) covered by ciliated columnar epitheliumallergic nasal polyp
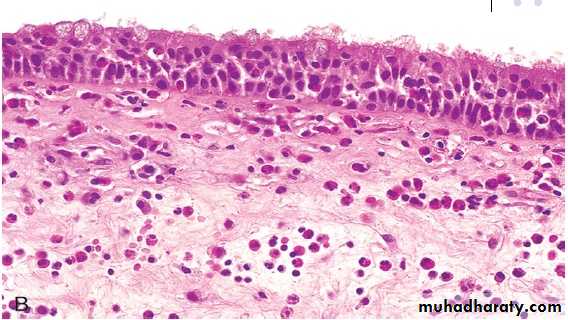
Bronchial asthma is an example of type I hypersensitivity reaction & is characterized by eosinophilic infiltrate, mucus collection & congestion.
2. ANTIBODY-MEDIATED (TYPE II) HYPERSENSITIVITY REACTIONS
Definition: it is a reaction by antibodies directed toward specific antigens fixed on cell surfaces cause lysis of target cells.Characteristics:
Antibodies: IgG (usually) and IgM (rarely) .Antigen: It may be endogenous or exogenous
Endogenous antigens: It may be normal molecules intrinsic to the cell membrane or extracellular matrix (e.g. autoimmune diseases).
Exogenous antigens: These antigens may get adsorbed on a cell surface or extracellular matrix may cause altered surface antigen (e.g. drug metabolite) .
Mechanisms of Injury
1. Complement-dependent reactions
a. Cell lysis (antibody dependent)
Antibody (IgM) directed against antigen on the cell membrane activates the complement system, leading to lysis of the cell .
Example: Transfusion reaction, incompatible blood transfusion for either ABO or Rh incompatibility when RBCs from incompatible donor enter the recipient circulation, they become coated with antibodies directed against RBC antigen of the donor and are lysed.
b. Phagocytosis
When circulating cells, such as erythrocytes or platelets, are coated (opsonized) with autoantibodies, with or without complement proteins, the cells become targets for phagocytosis (opsonization) which is phagocytosis of cells coated with antibodies) by macrophagesExample: penicillin attaches to RBCs → IgG antibodies are made against penicillin → splenic macrophages phagocytose the RBCs (hemolytic anemia).
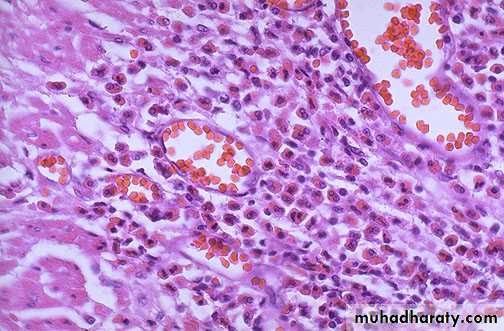
Antibody dependent cell mediated cytotoxity
b- Opsonization of cell by antibody & complement followed by phagocytosis
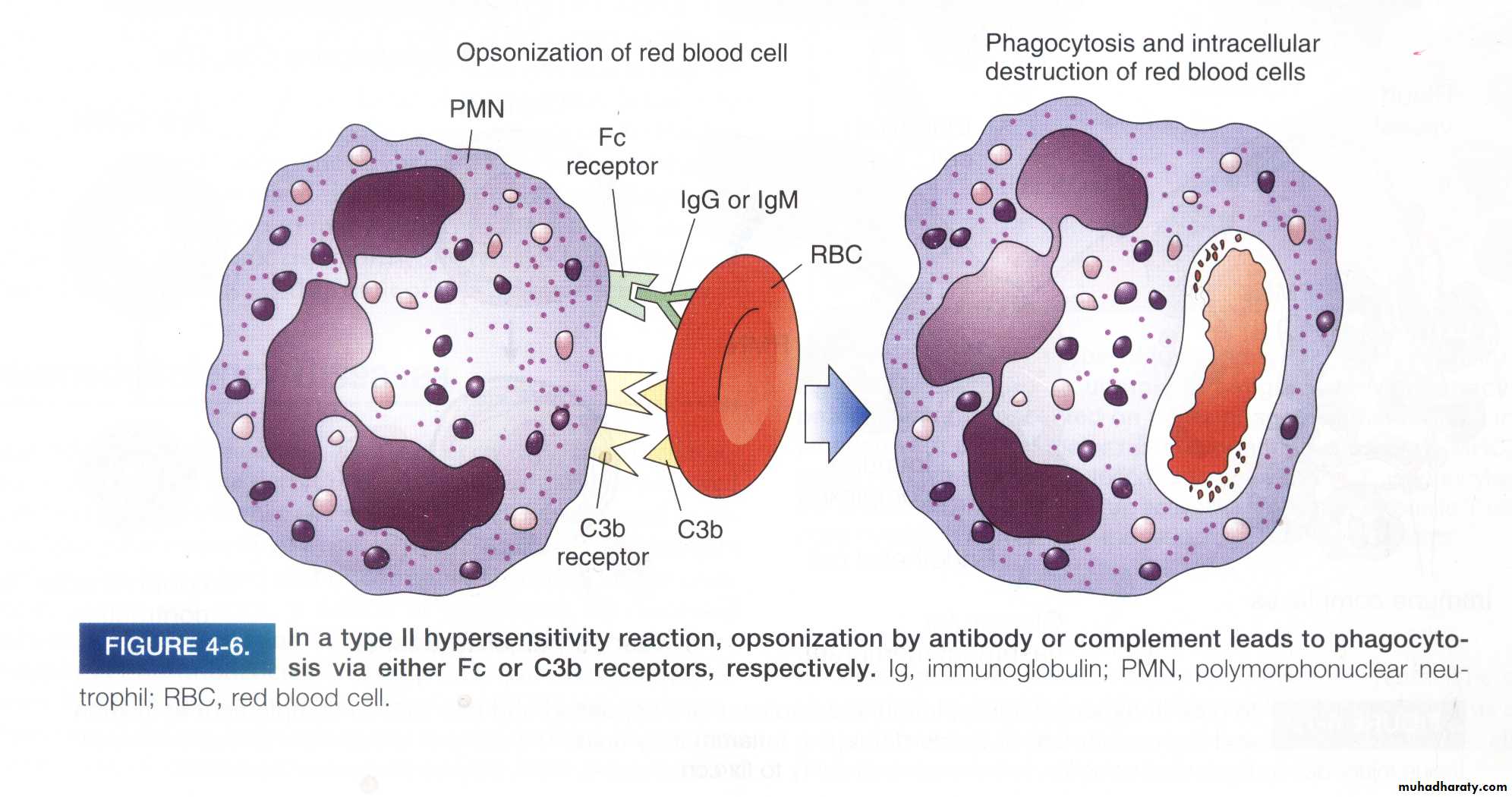
2. Complement-independent reactions
(Antibody mediated cellular dysfunction)Antibody directed against cell surface receptors impair or dysregulate function without causing cell injury or inflammation..
Example: in Graves disease IgG antibodies directed against thyroid hormone receptors stimulate the gland to synthesize excessive amounts of thyroid hormone.
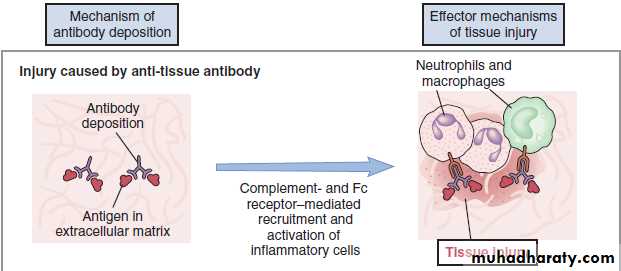
Complement-dependent reactions
Complement-independent reactions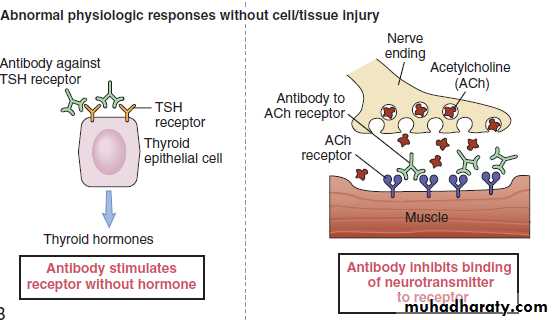
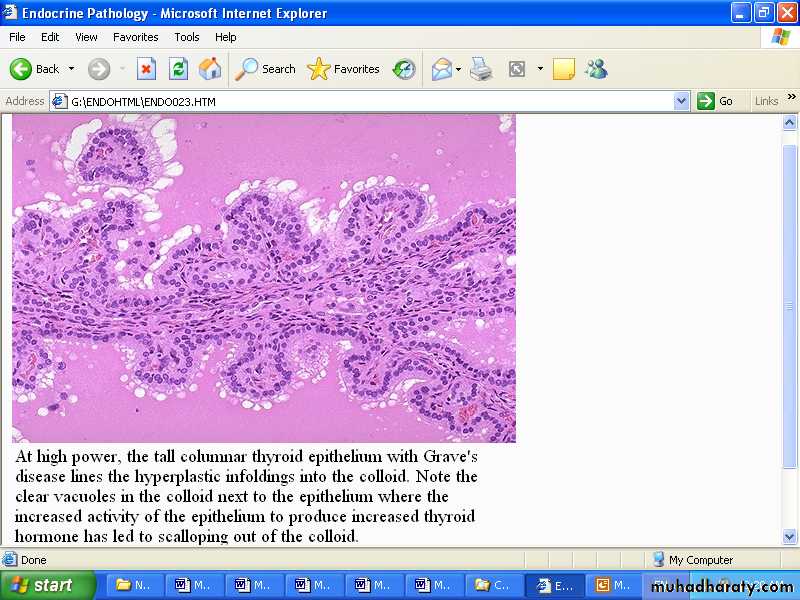
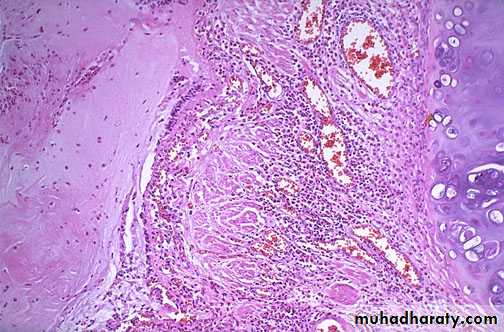
Antibodies to TSH-receptors activate thyroid cells in Grave’s disease leading to hyperplastic infolding into the colloid which show clear vacules next to the epithelium
EXAMPLES OF TYPE II REACTION
Examples of type II reaction are mainly on blood cells and some other body cells and tissues.1. Antibodies to blood cells e.g.
i) Autoimmune haemolytic anaemia
ii) Transfusion reactions
iii) Haemolytic disease of the newborn (erythroblastosis foetalis)
iv) Idiopathic thrombocytopenic purpura (ITP)
v) Drug-induced cytotoxic antibodies
2. Antibodies to tissue components e.g.
i) Graves’ disease (primary hyperthyroidism),
ii) myasthenia gravis
iii) type 1 diabetes mellitus, islet cell autoantibodies are formed
iv) In hyperacute rejection reaction, antibodies are formed against donor antigen.
3. IMMUNE COMPLEX–MEDIATED (TYPE III) HYPERSENSITIVITY REACTIONS
Definition: it is characterized by formation of immune (antigen and antibody) complexes in the circulation that may deposited in blood vessels, leading to complement activation and acute inflammation.The inflammatory cells recruited (neutrophils and monocytes) release lysosomal enzymes that generate toxic free radicals and cause tissue damage.
Characteristics
Antibodies: Complement-fixing antibodies namely IgG, IgM, and occasionally IgA.Antigen:
Exogenous: Various foreign proteins, e.g. foreign serum protein injected (e.g. diphtheria antitoxin, horse anti-thymocyte globulin) or produced by an infectious microbe.Endogenous: Antibody against self-components (autoimmunity), e.g. nucleoproteins.
Sites of Antigen-antibody Formation
Circulating immune complexes: They are formed within the circulation.In situ immune complex: They formed at extravascular sites where antigen might have been previously planted.
Sites of Immune Complex Deposition
Systemic: Circulating immune complexes may be deposited in many organs.
Localized: Immune complexes may be deposited or formed in particular organs/tissues: kidney (glomerulonephritis), joints (arthritis), small blood vessels of the skin.
Cause of Tissue Damage
Activation of complementThe inflammatory cells recruitment (neutrophils and monocytes) at the sites of deposition
Immune complex disease has the following phases:
Phase I or Immune Complex FormationPhase II or Immune Complex Deposition
Phase III or Immune complex mediated inflammation
EXAMPLES OF TYPE III REACTION

Systemic Immune Complex Disease— Serum Sickness
This was a frequent sequel to the administration of large amounts of foreign serum (e.g. serum from immunized horses used for protection against diphtheria). Nowadays it is infrequent.Pathogenesis
Divided into three phases:
1. Formation of immune complexes:
Introduction of protein antigen: It initiates an immune response.
Formation of antibody: It usually forms a week (7 to 12 days) after the injection of the foreign protein and are secreted into the blood.
Formation of immune complexes: They are formed in the circulation when antibodies react with the antigen.
2. Deposition of immune complexes:
Immune complexes of medium size and with slight antigen excess are the most pathogenic.
Sites of deposition:
– Blood vessels: It causes vasculitis.
– Renal glomeruli: It causes glomerulonephritis.
– Joints: It causes arthritis.
3. Inflammatory reaction and tissue injury: Mechanism of tissue injury include
Inflammatory reaction: Immune complexes in the tissues activates complement, the products (e.g chemotactic C5a) of which causes chemotactic recruitment of acute inflammatory cells (neutrophils and monocytes) to the site.
Tissue damage: Activated inflammatory cells (leukocyte) release lysosomal enzymes, arachidonic acid products and reactive oxygen species which produce tissue damage.
Clinical features: Fever, urticaria, joint pains (arthralgias), lymph node enlargement, and proteinuria appear during this phase.
A glomerulus with subendothelial deposites of immune complexes (PAS stain)
IgG antibodies detected in granular pattern by immunoflourescenceLocal Immune Complex Disease—Arthus Reaction
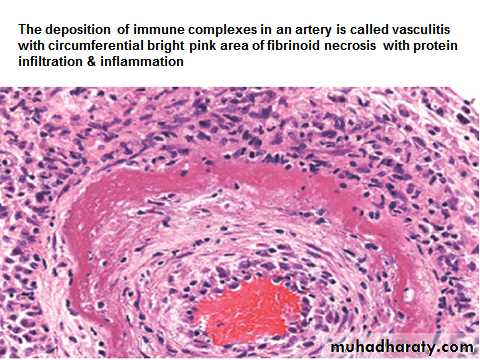
Arthus reaction is a local area of tissue necrosis usually in the skin, resulting from acute immune complex vasculitis.Arthus reaction can be experimentally produced by intracutaneous injection of an antigen to a previously immunized animal (with circulating antibodies against the antigen). As the antigen diffuses into the vascular wall, it locally binds to the antibody, and form large immune complexes at the site of injection.
Immune complexes deposited in the vessel walls, cause fibrinoid necrosis, and thrombosis leading to ischemic injury.
4. T Cell–MEDIATED (TYPE IV) HYPERSENSITIVITY REACTIONS
Type IV hypersensitivity reaction is mediated by T lymphocytes including CD4+ and CD8+ T cells.
It develops in response to antigenic exposure in a previously sensitized individual.
Reaction is delayed by 48 to 72 hours after exposure to antigen. Hence also called as delayed-type hypersensitivity (DTH).
This hypersensitivity reaction is involved in several autoimmune diseases (e.g. rheumatoid arthritis, Hashimoto thyroiditis), pathological reactions to environmental chemicals (e.g. poison ivy, nickel) and persistent microbes (e.g. tuberculosis, leprosy).
Types: Two types of Type IV hypersensitivity reaction namely:
1) Delayed type hypersensitivity reaction mediated by CD4+ T cells produce Cytokines2) Direct cell toxicity mediated by CD8+ T cells.
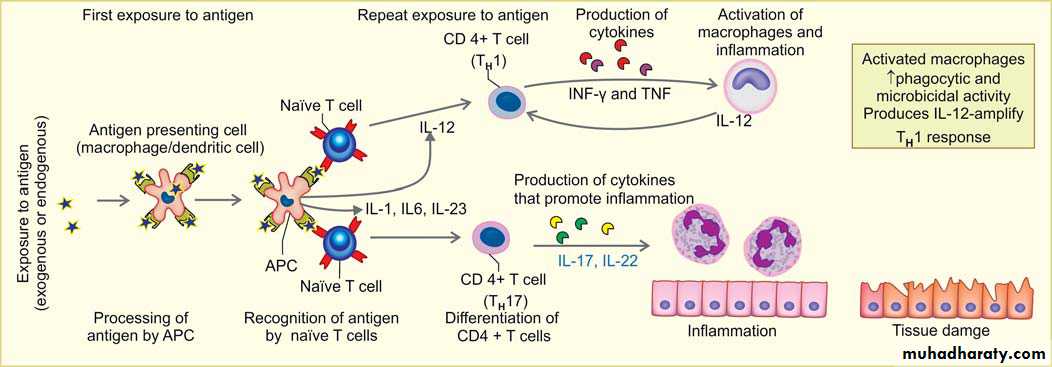
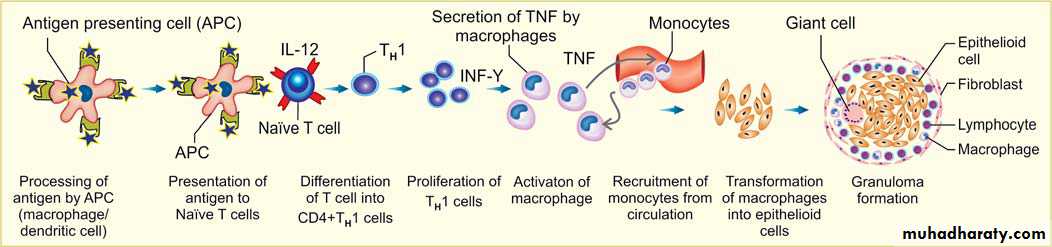
1. Delayed type hypersensitivity reaction mediated by CD4+ T cells produce Cytokines.
A. First exposure to antigenType of antigen: Antigen may be either exogenous environmental antigens or endogenous (self-antigens causing autoimmune disease).
Processing of antigen: by the antigen presenting cells (APC- dendritic cells or macrophages)
presenting of antigen to T cells, because T cells cannot directly recognize the antigen.
Recognition of antigen by naïve CD4+ T cells in association with II MHC molecules on APC.
Differentiation of CD4+ T cells:
-If the APCs secrete IL-12, the naïve CD4+ T cells differentiate into effector cells of TH1 type.
– If the APCs secrete IL-1, Il-6, or IL-23 (instead of IL-12), the naïve CD4+ T cells differentiate into effector cell of TH17 type.
B. On repeat exposure to an antigen:
TH1 cells enter the circulation and remain in the memory pool of the body. When there is re-exposure of the same individual to the antigen for the subsequent time,there is release of cytokinesTH1 cells , production of cytokines (e.g. IFN-γ, and TNF). IFN-γ, (most powerful macrophage activating cytokine) , activates macrophages.
– Activated macrophages have increased phagocytic and microbicidal power. They secrete IL-12 which amplify the TH1 response.
TH17 cells produce cytokines which promote inflammation by recruiting more neutrophils to the site of reaction.
The activated macrophages give rise to epithelioid cells and these cells surrounded by a collar of lymphocytes all around lead to formation of a granuloma. This granuloma formation is seen with tuberculin test, and other intracellular pathogens like mycobacterium, fungi and some parasites. Delayed type hypersensitivity reaction is also important in transplant rejection.
Delayed type hypersensitivity reaction mediated by CD4+ T cells produce Cytokines.
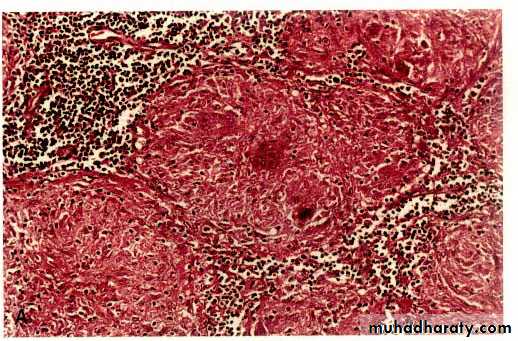
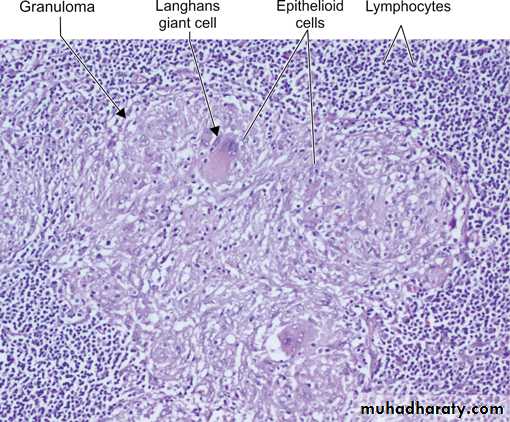
Granuloma
Prolonged DTH reaction against persistent microbes (e.g. tubercle bacilli) or other nondegradable (foreign bodies) injurious agent may produce a special microscopic reactionknown as granulomatous inflammation.
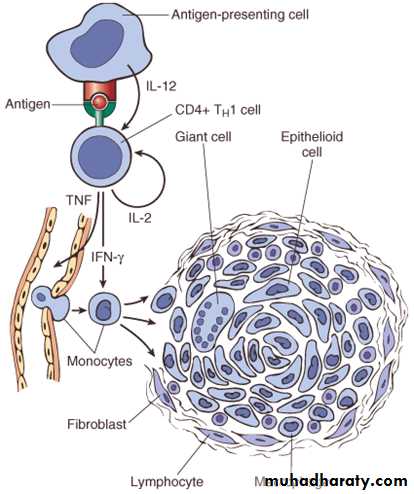
Cell mediated (type IV) hypersensitivity : the antigen presented by dendritic cell ( antigen presenting cell) leads to activation of TH1 cell which secrete cytokines that activate macrophages to form epitheliod cells & granuloma.
The granuloma is composed of localized collection of epithelioid cells surrounded by lymphocytes with langhans type giant cell.
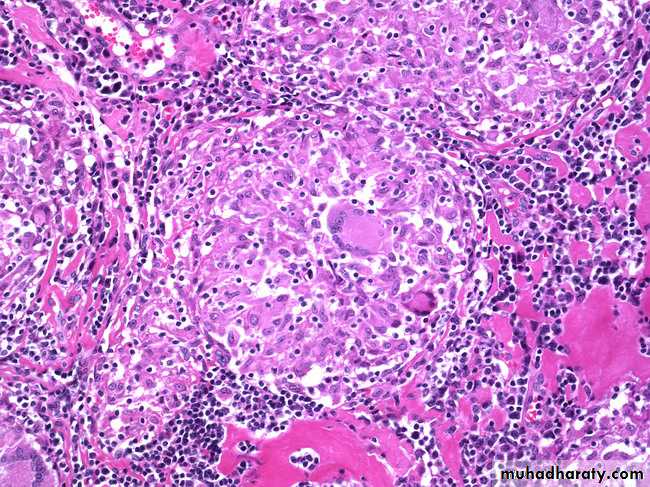
The granuloma is composed of localized collection of epitheliod cells surrounded by lymphocytes
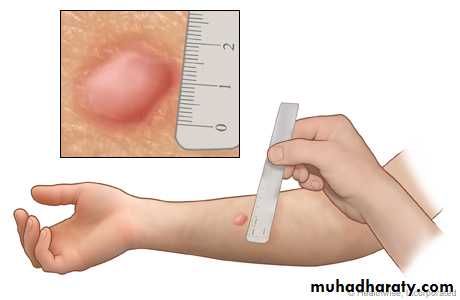
Tuberculin Reaction (Montoux Test)
Tuberculin reaction is a classical example for delayed-type hypersensitivity.It is produced by the intracutaneous injection of purified protein derivative (PPD, also called tuberculin), a protein-containing antigen of the tubercle bacillus.
In a previously sensitized individual, the injection site becomes red and indurated in 8 to 12 hours, reaches a peak (usually 1 to 2 cm in diameter) in 24 to 72 hours, and thereafter slowly subsides.
Microscopically, the injected site shows perivascular accumulation “cuffing” of CD4+ T cells and macrophages.
Mechanisms of granuloma formation
2. Direct Cell Toxicity Mediated By CD8+ T Cells
It is a type of T-cell mediated tissue injury due to CD8+ T lymphocytes (also called as cytotoxic T lymphocytes- CTLs), which kill antigen-bearing target cells. Example—killing of virus infected cells (e.g. in viral hepatitis) and some tumor cells.Mechanism of Cytotoxic T Cell–Mediated Killing
In this type of hypersensitivity, CD8+ cytotoxic T cells (CTLs) kill antigen- bearing target cellsby two mechanisms:
1. Perforin-granzymes system : Main mechanism of T cell-mediated killing of
target cells
CTLs have lysosome-like granules containing preformed mediators perforins and
granzymes
CTLs that recognize the target cells secrete perforin and granzymes
Perforin is a transmembrane pore-forming molecule, which allows the entry of granzymes into the cytoplasm of target cells
Granzymes are proteases, which cleave and activate cellular caspases (effector pathway of apoptosis)
Activated caspases induce apoptosis of the target cells.
2. Through Fas ligand : Activated CTLs also express Fas ligand (a molecule with
homology to TNF), which can bind to Fas expressed on target cells and cause apoptosis byextrinsic pathway.
Mechanism of Cytotoxic T Cell–Mediated Killing
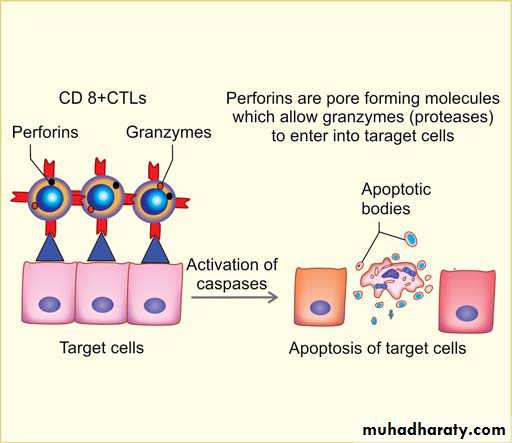
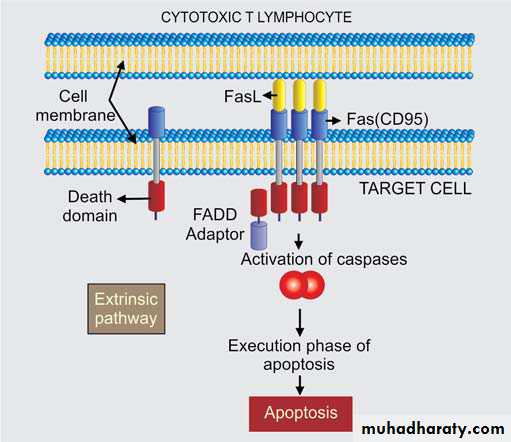
Perforin-granzymes system
Through Fas ligandEXAMPLES OF TYPE IV REACTION These are as follows:
1. Reaction against mycobacterial infection e.g. tuberculin reaction, granulomatous reaction in tuberculosis, leprosy.2. Reaction against organ transplantation e.g. transplant rejection, graft versus host reaction
3. Reaction against virally infected cells.
4. Reaction against malignant cells in the body.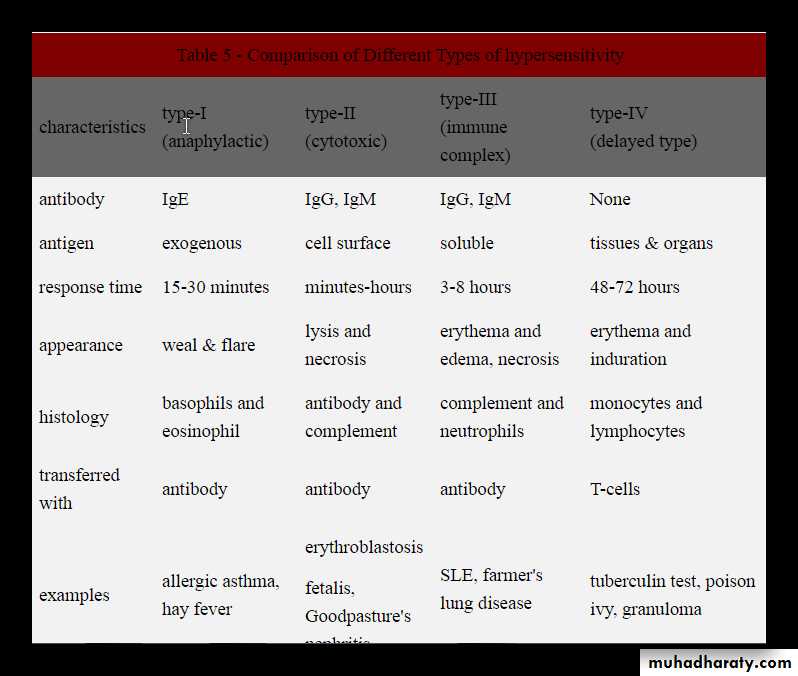
Tissue Transplantation
Tissue TransplantationGrafts are divided into 4 types
Auto graft: from one part of the body into another (e.g. transplantation of the skin from one part of the body into another).
Isograft: From one individual to another who is genetically identical like the identical twin and here survival of the graft is the role.
Allograft: Between two genetically non identical individuals from the same species and here rejection of the graft is the role if immunosuppression is not done.
Xenograft: transplantation from another species and here rejection is very severe and it is unlikely for the graft to survive.
Problems of Transplantation
1- Graft rejection (Host versus graft)2- Graft versus host reaction (GVH)
Graft Rejection
It is a complex immunologic phenomenon as responses of the host directed against MHC on the donor graft.The rejection happens because the MHC (HLA) system of the donor is different from that of the recipient so that both cell mediated immunity and antibody response will be mounted against the transplanted tissue.
The major types of hypersensitivity reactions involved are types II, III and IV.
Kidney is the most common transplanted organ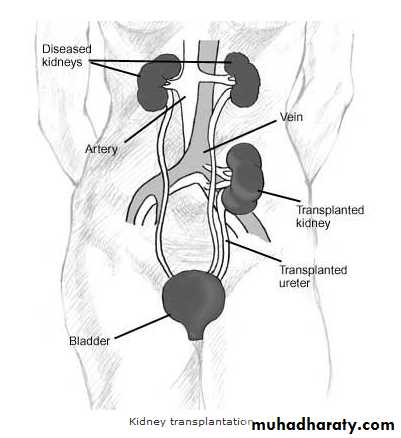
Pathology, types of graft rejection
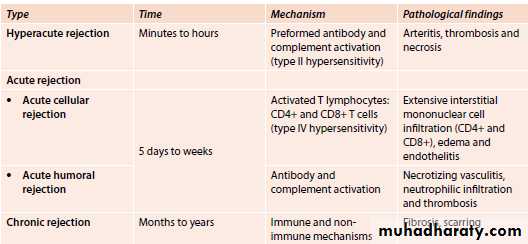
1-Hyperacute rejection:Pre-formed antidonor antibodies bind to graft endothelium immediately after transplantation.
This occurs due to previous sensitization by previous transplantation, blood transfusion, multiple pregnancies and infections.
In the transplanted kidney the surgeon will notice, just after vascular anastomosis, that the kidney will become cyanosed and secretes only few drops of bloody urine.
Microscopically, vasculitis with fibrinoid necrosis and thrombosis.
Hyper acute rejection of a kidney allograft showing endothelial vascular damage (by preformed antibodies) with formation of fibrin-platelet thrombi in the glomerular capillaries.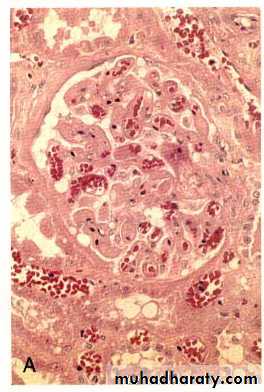
2-Acute rejection:
This occurs few days after transplantation or after stoppage of the immunosuppressive therapy.Acute cellular rejection: T cells destroy graft parenchyma (and vessels) by cytotoxicity and inflammatory reactions.
Acute humoral rejection: Antibodies damage graft vasculature.
Microscopically:
Cellular rejection is manifested by extensive interstitial mononuclear cell infiltrate.Humoral rejection is manifested by acute necrotizing vasculitis leading to narrowing of renal arterioles and infarction of renal cortex.
This is a form of acute renal transplant rejection known as acute cellular tubulointerstitial rejection because most of the inflammation is in the interstitium. The glomerulus seen here is normal, but the tubules are infiltrated by many lymphocytes at the upper right
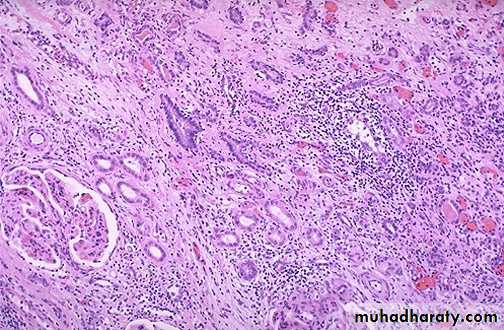
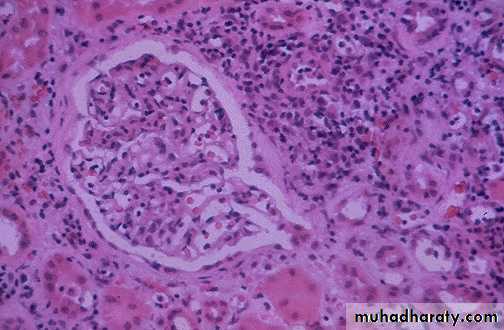
Acute vascular rejection of the kidney with inflammatory cells in the interstitium& in between the tubules
3-Chronic rejection:
Occurs after months-years after transplantation .This type is probably caused by T cell reaction and secretion of cytokines that induce proliferation of vascular smooth muscle cells, associated with parenchymal fibrosis.
Microscopically:
vasculitis with marked thickening of intima (intimal fibrosis) leading to obliteration of vessel lumen and renal ischemia with fibrosis of glomeruli, interstitial fibrosis and atrophy of tubules.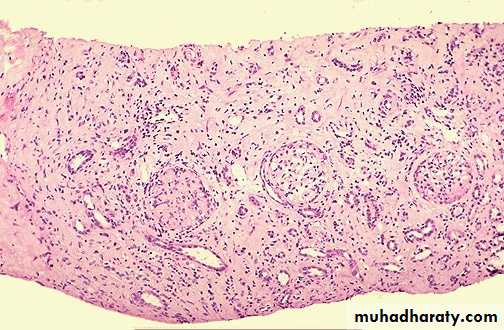
Chronic rejection of the kidney is dominated by interstitial fibrosis, tubular atrophy & loss of renal parenchyma due to arteriolosclerosis of arteries
MECHANISMS OF GRAFT REJECTION
• CELLULAR REACTIONS :These are mainly responsible for graft rejection and are mediated by T cells.
2. HUMORAL REACTIONS :
These include: preformed circulating antibodies due to pre-sensitisation of the recipient before transplantation e.g. by blood transfusions and previous pregnancies.
• Methods of increasing graft survival
• 1- The best results will be obtained from finding of HLA compatible donor but unfortunately this is not available except from an identical twin and 1:4 of brothers or sisters.• 2- When the donor is not compatible we should use immunosuppression like high doses of prednisolone or cytotoxic therapy. Both cause non-specific immunosuppression and the patient may suffer from opportunistic infections.
• 3- Now a days, antithymocytes antibodies are used.
• 4- Repeated blood transfusion in small doses from the same donor before transplantation increases the success of kidney transplantation.
• Before transplantation, we should destroy all the recipient marrow by total body irradiation and cytotoxic drugs, and this will make the patient (recipient) immunodeficient.
• GVHD occurs most commonly during bone marrow transplantation and transplantation of solid organs rich in lymphoid cells.
When such recipients receive normal bone marrow cells from allogenic donors, the immunocompetent T cells present in the donor marrow recognize the recipient’s HLA antigen as foreign antigen and reacts against them. Both CD4+ and CD8+T cells recognize and attack host tissues by generating DTH and cell mediated cytotoxicity.
Graft Versus Host Disease (GVHD)
• The donor cells will attack the actively proliferating cells of the recipient resulting in gastroenteritis (nausea & vomiting), skin rash, jaundice, etc…
The main cells involved in GVHD are CD8+ T-lymphocytes
GVHD is a lethal and can be minimized by:Proper HLA typing
Donor T cells can be depleted before marrow transplantation.
Immunological tolerance
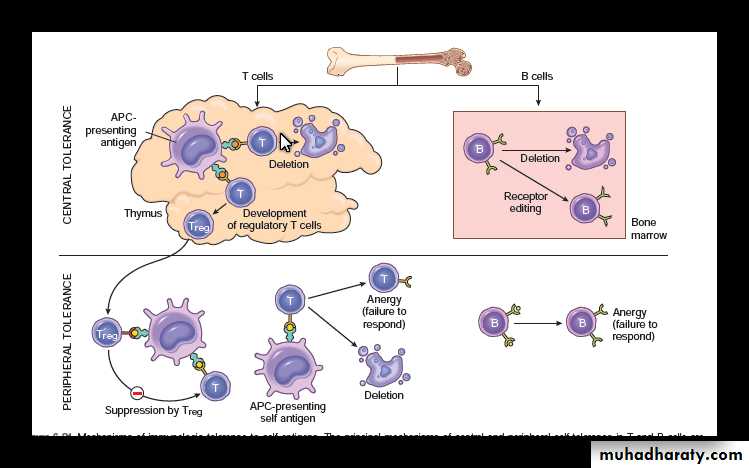
Immunological tolerance is the phenomenon in which there is no immune response to self antigens.Self-tolerance: It is absence of immune response to an individual’s own antigens.
It is the result of exposure of lymphocytes to that specific antigen with elimination of self reactive T cell and B cells.
Mechanisms of Self-Tolerance
All immune and blood cells develop from multipotent hematopoietic stem cells that originate in the bone marrow.Immature T cells undergo final maturation process in the Thymus that "educates" them to distinguish between self and non-self antigens.
Immature B cells undergo final maturation process in the Bone marrow.
In normal persons, the autoreactive T- cells and B- cells are deleted or inactivated.
The mechanism by which this Self-Tolerance is achieved can be broadly classified into two groups:
• Central tolerance: Immature lymphocytes that recognize self antigens in the central (generative) lymphoid organs are killed by apoptosis.
These organs are thymus for T cells and the bone marrow for B cells.
• Peripheral tolerance: Mature lymphocytes that recognize self antigens in peripheral tissues become functionally inactive (anergic), or are suppressed by regulatory T lymphocytes, or die by apoptosis.
Anergy : irreversible inactivation ( rather than apoptosis) of lymphocytes .
AUTOIMMUNE DISEASES
Definition:Immune reactions to self antigens due to the loss of self-tolerance.
Mechanisms(causes)of Autoimmune Diseases
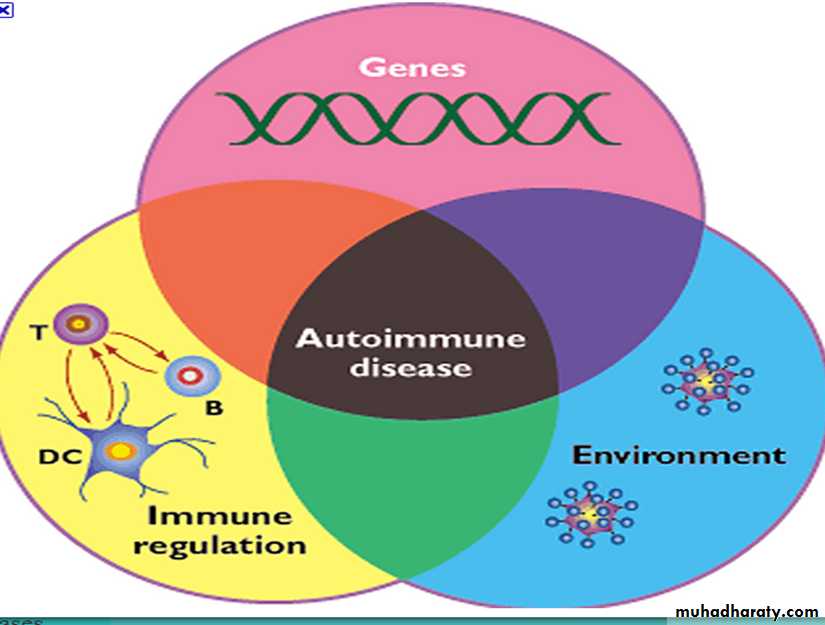
1- Failure of self tolerance
2- Environmental factors3- Genetic susceptibility
Failure of self toleranceFailure of tolerance is due to several mechanisms:
• Failure of activation induced cell death: is due to a defect in Fas-Fas ligand system which leads to failure of apoptosis of self reactive T cells induced by this system with persistence of these cells in the circulation.
2. Break down of T cell anergy:
Anergy is broken when normal cell becomes expressing costimulatory molecules required for T cell activation . (Normally these molecules are not expressed by antigen presenting cells), such induction of expression of costimulatory molecules is due to infection .3. Modification in the structure of self antigen that allow its recognition as a foreign antigen by T cells.
e.g. autoimmune hemolytic anemia occurs after use of certain drug because the drug induces change in the surface of RBCs that create an antigen which is recognized as a foreign by T cells which subsequently stimulate B cells to produce autoantibodies against RBCs leading to hemolysis.
4. Molecular Mimicry
Some infectious agents share the same epitope (shape) with self antigens, so immune response against such microbes produces similar response to the self antigen.e.g. polysaccharide coat of streptococci share the same epitope with cardiac glycoprotein so after streptococcal throat infection, antibodies formed against bacteria cross react with antigens in the heart wall leading to carditis (rheumatic heart disease) and damage of endocardium, myocardium and pericardium.
5. Polyclonal lymphocyte activation:
Some clones of anergic (inactivated) lymphocytes become activated by endotoxins of bacteria resulting in autoimmunity.
6. Release of sequestered antigens:
Any self antigen that is completely sequestered (hidden) from the immune system during development is likely to be recognized as foreign antigen of it is subsequently exposed to the immune system.e.g. spermatozoa and ocular antigens are sequestered self antigens and may be exposed to the immune system after trauma to the testis or eye and recognized as a foreign antigen resulting in autoimmunity.
Environmental factors and Autoimmunity
Role of infections in autoimmunity.1. Antigenic cross reactivity: this is due to similarity between the microbial antigenic structure and the self antigens. This is also known as ‘molecular mimicry’.
2. Upregulation of co-stimulatory molecules by infectious organisms.
3. Polyclonal B cell activation: caused by EBV and HIV resulting in production of autoantibodies.
4. Alteration of tissue antigens: infections may alter tissue self antigens so that they activate T cells and loose the property of self tolerance.
Autoimmune Diseases result in cell and tissue destruction by:
• Type(VI) HSR: antigen-specific CD8 cytotoxic T cells• Type II and III HSR: autoantibodies (antibodies to self-proteins) and the accompanying inflammatory process.
Examples of Autoimmune Diseases
Organ specific autoimmune disorders
Type I diabetes mellitus
Pernicious anemia
Gravis disease
Hypothyroidism
Autoimmune hepatitis and primary biliary cirrhosis
Multisystem autoimmune diseases
Rheumatoid arthritisSystemic lupus erythematosus (SLE)
Polyarteritis nodosa
Auto immune hemolytic anemia
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
SLE is a systemic autoimmune disease caused by autoantibodies produced against numerous self-antigens and the formation of immune complexes having following characteristics:• Protean manifestation and variable behavior.
• More commonly seen in the females around the age of 20-30’s3. Remission and relapses.
4. Multisystemic involvement: Mainly affects skin, kidneys, joints, serous membranes and heart.
5. Broad spectrum of autoantibodies: The auto-antibodies are formed against DNA, histones, non histone proteins bound to RNA and nucleolar antigens. These are collectively called as antinuclear antibodies (ANA).
Etiology
SLE is an autoimmune disease in which fundamental defect is failure of self-tolerance. It leads to production of many autoantibodies that damages the tissue either directly or indirectly by immune complex deposits.
Mechanisms of Tissue Injury
Type III hypersensitivity: It occurs with deposition of immune complexes. It is the most common cause of tissue injury and visceral lesions.Type II hypersensitivity: Autoantibodies against cell surface antigens specifi c for RBCs, white cells, and platelets, opsonize these cells, promote their phagocytosis and lysis, cytopenias.
Pathogenesis of SLE
Genetic FactorsEvidence to support genetic predisposition are:
1. Familial association:
Family members of SLE patients have an increased risk of SLE. About 20% of unaffected first-degree relatives may show autoantibodies.
High rate of concordance (>25%) in monozygotic twins when compared with dizygotic twins (1%–3%).
2. HLA association: Risk is more with HLA-DR2 or HLA-DR3.
Environmental Factors1. Ultraviolet (UV) radiation: Exposure to sunlight exacerbates the lesions of the disease.
Mechanism: UV irradiation causes apoptosis of host cells, increases burden of nuclear antigens and promote inflammation.
2. Cigarette smoking: It is associated with development of SLE.
3. Sex hormones: SLE is 10 times greater during the reproductive period (17 through 55 years) in women than in men. SLE shows exacerbation during normal menses and pregnancy.
4. Drugs: Examples include hydralazine
Immunological Abnormalities
failure of self-tolerance with several immunological abnormalities of both innate and adaptive immune system have been observed in SLE.
The clinical criteria for the diagnosis of SLE
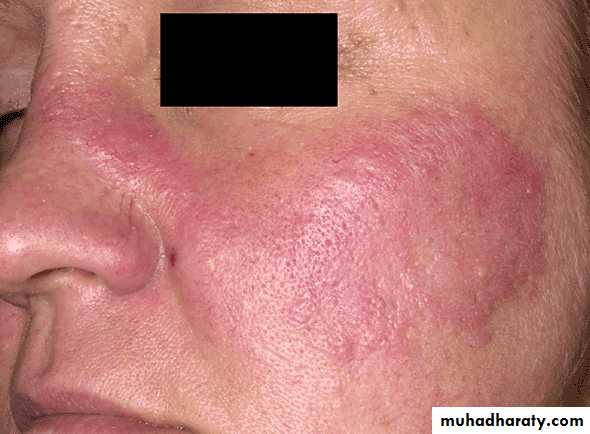
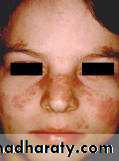
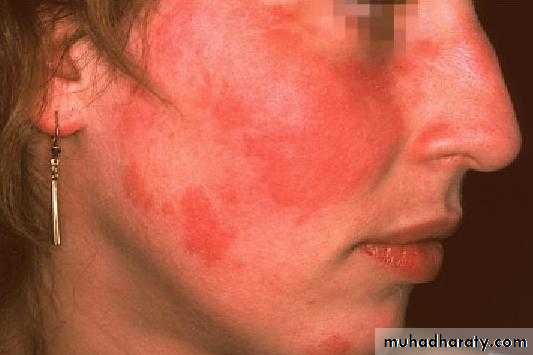
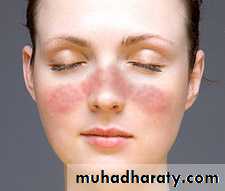
A person is considered to suffer from SLE if at least 4 of 11 clinical and laboratory criteria are identified simultaneously or cumulatively at any interval of observation.1. Malar (‘‘butterfly’’) rash over the face
2. Discoid rash3. Photosensitivity (rash as a result of sun exposure)
4. Oral ulcers
5. Arthritis (nondeforming polyarthritis)
6. Serositis (pleuritis and/or pericarditis)7. Renal disorder (proteinuria of >0.5 g/day; cellular casts as signs of glomerulonephritis)
8. Neurologic disorder (seizures and/or psychosis)
9. Hematologic disorder (leukopenia, lymphopenia, thrombocytopenia, and hemolytic anemia)
10. Immunologic disorder (antibodies to native DNA or Smith antigen [anti-Sm]; antiphospholipid antibodies)
11. Antinuclear antibody (in the absence of drug-induced lupus)
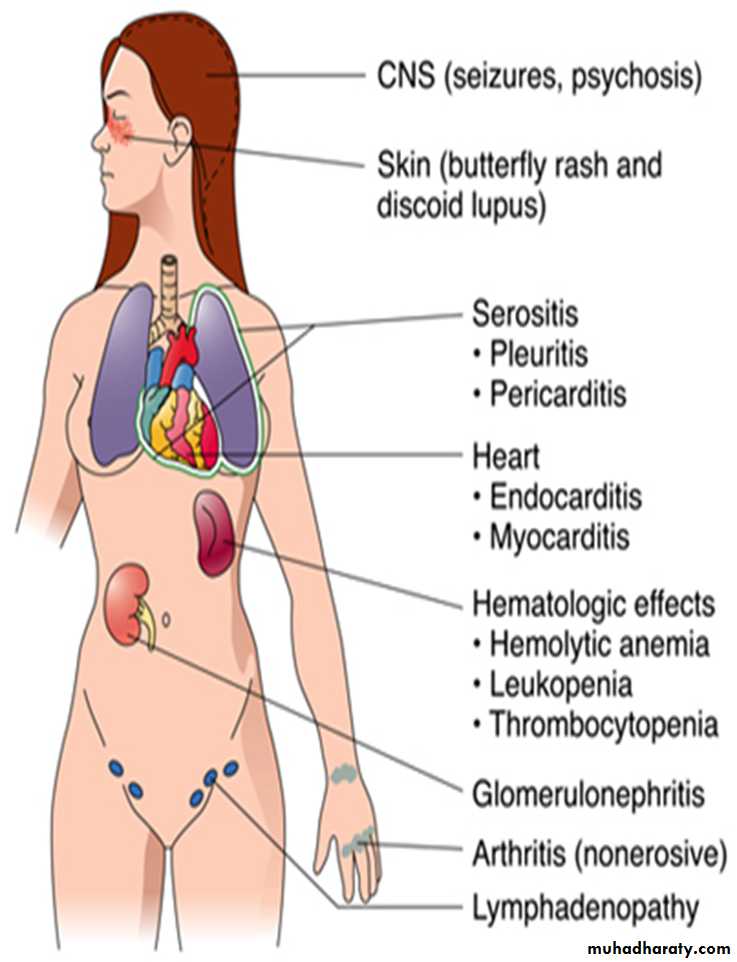
Any organ in the body can be affected & the result is inflammation of the skin, joints, kidneys, lung, heart, serosal surfaces….



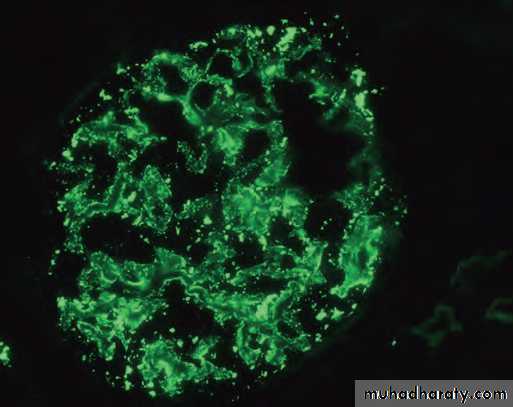
Deposition of IgG antibody in a granular
pattern, detected by immunofluorescence.
Lupus glomerulonephritis
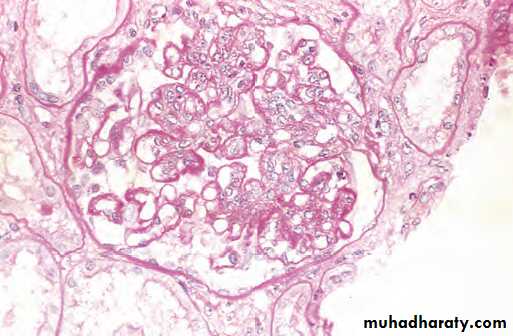
A glomerulus with several “wire loop” lesions representing extensive subendothelial deposits of immune complexes
(periodic acid-Schiff stain).
IMMUNODEFICIENCY SYNDROMES
IMMUNODEFICIENCY SYNDROMESImmunodeficiency is defect in immunity characterized by deficient or absent cellular and/or humoral immune functions.
Classification
Primary immunodeficiency due to an inherited defects affecting immune system development.Secondary immunodeficiency which may arise as complications of an underlying condition like cancers, infections, malnutrition, or immunosuppression, irradiation, or chemotherapy for cancer and other diseases.
Clinically
patients with immune deficiency present with increased susceptibility to infections as well as to certain forms of cancer.B-cell deficiencies:
○ X-linked agammaglobulinemia (Bruton type)
○ Common variable immunodeficiency
○ Isolated IgA deficiency
T-cell deficiencies:
○ Hyper-IgM syndrome
○ DiGeorge syndrome
Severe combined immunodeficiency
Classification of Primary immunodeficiency
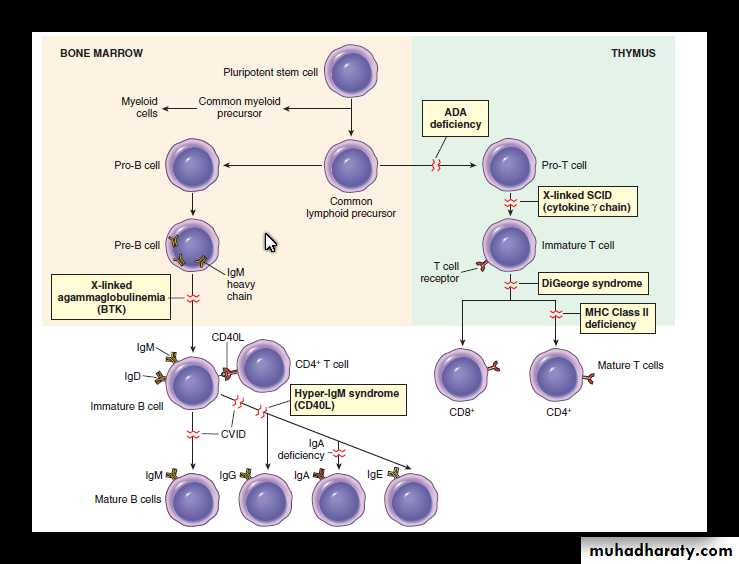
X-linked agammaglobulinemia of Bruton
Failure of B-cell precursors (pro-B cells and pre-B cells) to mature into B cells due to mutation of B-cell tyrosine kinase (Btk).B cells are absent or markedly decreased in the circulation.
Absent or markedly decreased concentration of all classes of immunoglobulins (agammaglobulinemia)
Affects boys (X-linked disease)
Symptoms appear after 6 months of age. Typically, there is an increased incidence of otitis
media, and skin and respiratory infections caused by Haemophilus influenzae, Streptococcus
pneumoniae, or Staphylococcus aureus. During the first 6 months, the infant is protected
by maternally acquired antibodies and the infections appear only after these antibodies have
been cleared from the infant’s body.
Common variable immunodeficiency
Most patients have normal or near-normal numbers of B cells in the blood and lymphoid tissues which are not able to differentiate into plasma cells.
Late-childhood onset of hypogammaglobulinemia and recurrent infections
Boys and girls affected equally
High frequency of malignancies (gastric cancer and lymphoma) later in life
Isolated IgA immunodeficiency
The most common congenital immunodeficiency, found in 1:850 people.Many of these men and women are asymptomatic.
Because IgA is the major immunoglobulin found in mucosal secretions (respiratory, gastrointestinal, and urogenital tract), IgA deficiency is characterized by recurrent sinusitis, pulmonary and urinary infections, as well as recurrent diarrhea.
DiGeorge syndrome
T-cell deficiency due to the abnormal development of thymus (hypoplasia or lack of the thymus),Part of CATCH 22 syndrome: cardiac abnormality, Abnormal facies , Thymic aplasia, cleft palate, hypocalcemia; syndrome is caused by a chromosome 22q11 deletion
X-linked hyper-IgM syndrome
Affected people have high titers of IgM and IgD antibodies but low titers of IgG, IgA, and IgE antibodies, indicating that there is a defect in isotype switching.The primary defect is in CD4 T cells, which are responsible for production of cofactors necessary for stimulation of B-cell and isotype switching.
The cause is a mutation of X-chromosome gene encoding CD40L. This protein expressed on CD4 lymphocytes acts as a ligand binding to a B-cell surface receptor (CD40). Binding of this ligand is important for the activation of B cells.
Patients suffer from pyogenic infections and are susceptible to infection with Pneumocystis jiroveci.
Severe combined immunodeficiency (SCID)
Prominent susceptibility to severe, recurrent fungal (Candida albicans), viral, and bacterial infections
Both humoral (antibodies) and cellular (T cells) immune responses severely reduced
X-chromosome linked (boys are typically affected); mutation involves the gene encoding cytokine receptors
Acquired Immunodeficiency Syndrome (AIDS)
Definition:infectious disease caused by human immunodeficiency viruses (HIV)
characterized by the triad of immunosuppression associated with opportunistic infections, secondary neoplasms and neurologic manifestations .
Properties of HIV
Non-transforming human retrovirus belonging to the lentivirus family.RNA viruses having an enzyme called reverse transcriptase, which prepares a DNA copy of the RNA genome of the virus in host cell.
Genetic forms:
HIV-1 is most common in the United States, Europe, and Central Africa,
HIV-2 is common in West Africa and India.
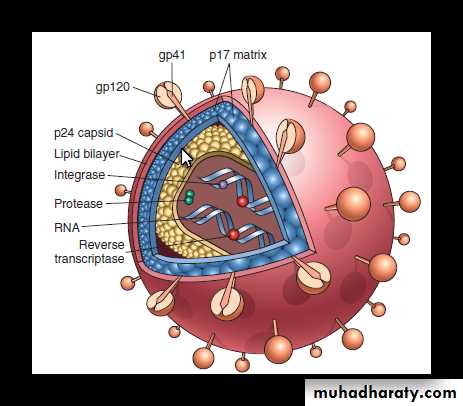
Structure of HIV
HIV-1 is spherical enveloped virus which is about 90–120 nm in diameter.It consists of electron-dense, cone-shaped core surrounded by nucleocapsid cell which is covered by lipoprotein envelope.
A. Viral core: It contains
1. Major capsid protein p24.
2. Nucleocapsid protein p7/p9.
3. Two identical copies of single stranded RNA genome.
4. Three viral enzymes:
protease
reverse transcriptase (RNA-dependent DNA polymerase)
integrase. When the virus infects a cell, viral RNA is not translated, instead transcribed by reverse transcriptase into DNA. The DNA form of the retroviral genome is called a provirus which can be integrated into the chromosome of host cell.
B. Nucleocapsid:
The viral core is surrounded by a matrix protein p17, which lies underneath the lipid envelope of the virion.
C. Lipid envelope:
The virus contains a lipoprotein envelope, which consist of lipid derived from the host cell and two viral glycoproteins. These glycoproteins are:1. gp120 which project as a knob-like spikes on the surface
2. gp41 anchoring transmembrane pedicle.
These glycoproteins are essential for HIV infection of cells.
Structure of HIV
The major routes of transmission of HIV:
Sexual contact: homosexual (male) and heterosexual (male-to-female and female-to-male).Parenteral inoculation: intravenous drug abusers and recipients of blood and blood products or through needle stick injury.
Passage from infected mother to child (vertical transmission): transplacental spread, transmission during the delivery, and breast-feeding
Life Cycle of HIV infection
Life Cycle of HIV consists of four main steps namely:1) infection of cells by HIV
2) integration of the provirus into the host cell genome
3) activation of viral replication
4) production and release of infectious virus
• Infection of Cells by HIV:
Major targets: HIV can infect many tissues, but two major targets of HIV infection are the: Immune system and Central nervous system (CNS).
Cell tropism: Primary targets for HIV are CD4 T cells, but the viruses also invade macrophages and dendritic cells.
HIV binds to the CD4 molecule, which acts as a high-affinity receptor, but the infection requires coreceptors: CXCR4 (mostly on T cells) or CCR5 (mostly on macrophages).
The HIV envelope contains two glycoproteins, surface gp120 noncovalently attached to a transmembrane protein, gp41.
Conformational change: Binding of gp120 to CD4 leads to a conformational change in the HIV, that results in the formation of a new recognition site on gp120 for the coreceptors CCR5 or CXCR4.
Penetration of host cell membrane by gp41: Binding of gp120 to the chemokine coreceptors leads to conformational changes in gp41.
Membrane fusion: The conformational change in gp41 allows HIV to penetrate the cell membrane of the target cells (e.g. CD4+ T cells or macrophages), leading to fusion of the virus with the host cell.
Entry of viral genome into cytoplasm of host cell: Once internalized, the virus core containing the HIV genome enters the cytoplasm of the host cell.
2. Integration of the proviral DNA into the genome of the host cell
After the internalization of the virus core, the RNA genome of the virus undergoes reverse transcription leading to the synthesis of double-stranded complementary DNA (cDNA/proviral DNA).Episomal form: In quiescent T cells, HIV cDNA may remain as a linear episomal form in the cytoplasm of infected cell.
Integration of cDNA: In dividing T cells, HIV cDNA enters the nucleus, and becomes integrated into the genome of the host cell using a viral integrase protein.
3. Viral replication: After the integration of proviral DNA it can either be latent or productive infection.
Latent infection: During this, the provirus remains silent for months or years.
Productive infection: In this the proviral DNA is transcribed leading to viral replication,formation of complete viral particles
4. Production and release of infectious virus: The complete virus particle formed, buds from the cell membrane and release new infectious virus. This productive infection when extensive, leads to death of infected host cells.
The virus infection remains latent for long periods in lymphoid tissues. Active viral replication is associated with more infection of cells and progression to AIDS.
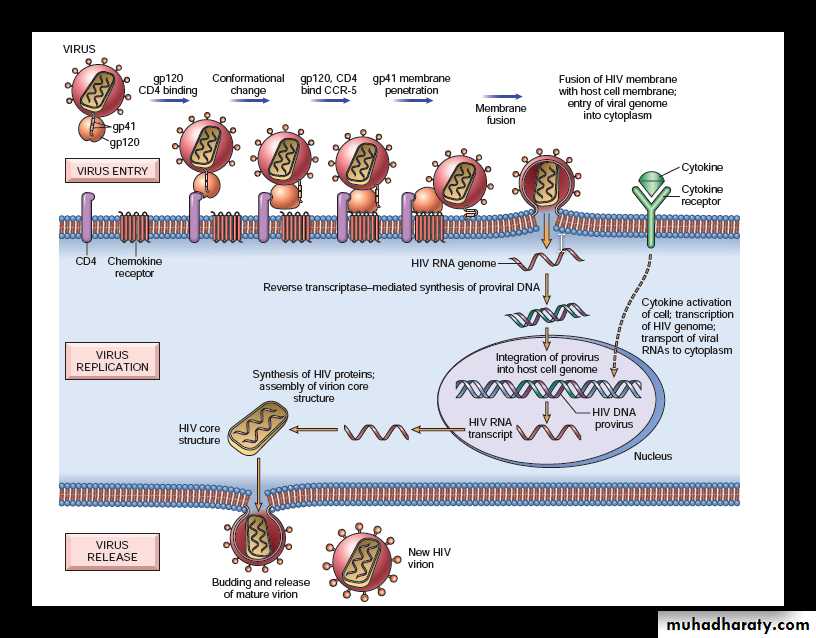
The initial infection starts in mucosal tissues, involving mainly memory CD4+ T cells and dendritic cells, and spreads to lymph nodes.
Viral replication leads to viremia and widespread seeding of lymphoid tissue. The viremia is controlled by the host immune response, and the patient then enters a phase of clinical latency. During this phase, viral replication in both T cells and macrophages continues , but there is some immune containment of virus .
There is continuous gradual erosion of CD4+ cells and ultimately, CD4+ T-cell
numbers decline, and the patient develops clinical symptoms of full-blown AIDS.
Mechanisms of immune deficiency in AIDS
• ■ Loss of CD4+ T cells: T-cell death during viral replication and budding (similar to other cytopathic infections); apoptosis as a result of chronic stimulation; decreased thymic output; functional defects.• ■ Defective macrophage and DC functions
• ■ Destruction of architecture of lymphoid tissues (late)
Pathogenesis of HIV infection
The early acute phase
It is seen for 3-12 weeks and is characterized by high levels of plasma viremia, and widespread seeding of the lymphoid tissues. The initial infection is controlled by the development of an antiviral immune response.Clinically, this stage is associated with a self-limited acute illness with nonspecific symptoms, including sore throat, myalgias, fever, rash, weight loss, and fatigue, and clinical features, such as rash, cervical adenopathy, diarrhea, and vomiting.
The middle chronic phase
This is characterized by a period of clinical latency. It is usually lasting for an average duration of 10 years. In this phase, there is a continuous battle between the virus and the host immune cells. The immune system is intact, but there is continuous HIV replication, predominantly in the lymphoid tissues, which may last for several years.Clinically, Patients are either asymptomatic or develop persistent generalized lymphadenopathy. The patients may have minor opportunistic infections as thrush and herpes zoster. Persistent lymphadenopathy with significant constitutional symptoms (fever, rash, fatigue) reflects the onset of the crisis phase.
Phases of HIV infection
The final phase or the stage of crisis
It is associated with the loss of host immune cells in the battle and the progression to AIDS.
Clinically, patient presented with a long-lasting fever (>1 month), fatigue, weight loss, and diarrhea.
AIDS-Defining lesions in Patient with HIV-Infection:
1- Opportunistic infections in AIDSOpportunistic infections account for the vast majority of deaths in patients. These infections
include:
• Pneumocystis jiroveci pneumonia
• Candida albicans infections of the mouth, esophagus, vagina, and lungs
• Cytomegalovirus enteritis and pneumonitis
• Atypical mycobacterial infection (M. avium-intracellulare) of the gastrointestinal tract
• Cryptococcus neoformans meningitis
• Cryptosporidium enteritis
2- The most common neoplasms associated with HIV infections
• Kaposi sarcoma• Non-Hodgkin lymphoma
• Carcinoma of the uterine cervix
• Squamous cell carcinoma of the skin
3- Neurologic consequences of HIV infection
Involvement of the central nervous system is common (clinically 40%–60%) and may present in several forms:
• HIV-related diseases
○ Aseptic meningitis
○ AIDS dementia complex
2. Opportunistic infections: viral (cytomegalovirus and herpes simplex virus), fungal (Coccidioides and Cryptococcus), and protozoal (Toxoplasma gondii)
3. Neoplasms (lymphoma)
Pneumocystis jiroveci pneumonia: alveoli are filled with numerous cysts
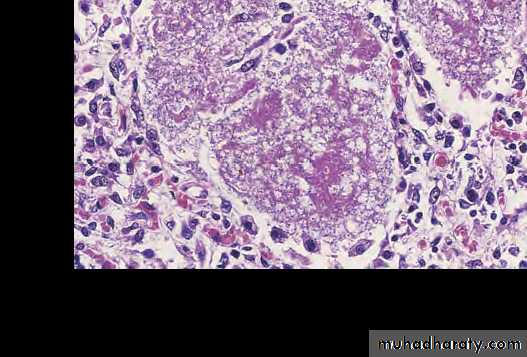
This is cytomegalovirus (CMV) infection in the lung. Note the very large cells that have large violet intranuclear inclusions with a small clear halo. Basophilic stippling can be seen in the cytoplasm.
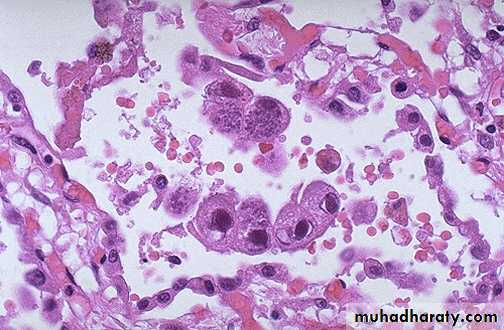
Kaposi sarcoma : atypical spindle cells enclosing slit like vascular spaces this rare tumor is a frequent finding in patient with AIDS.
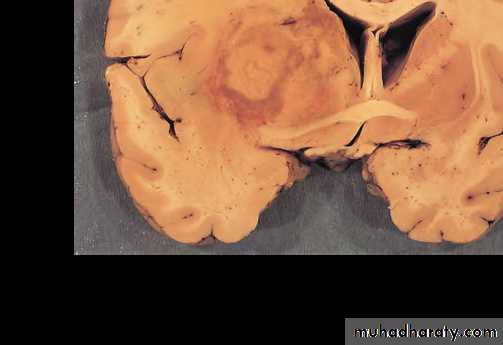
Cerebral toxoplasmosis with hemorrhagic necrotic lesion compressing brain tissue
Diagnosis of AIDS
• ELISA is used for the detection of antibodies against viral proteins. This is the most sensitive and the best screening test for the diagnosis of AIDS.• Western blot is the most specific or the confirmatory test for HIV.
• Direct detection of the viral infection is with p24 antigen capture assay, reverse transcriptase polymerase chain reaction (RT-PCR), DNA-PCR and culture of the virus from the monocytes and CD4+ T cells.
AMYLOIDOSIS
Amyloidosis: group of diseases having in common the deposition of amyloid.
Amyloid : pathologic fibrillar protein deposited in the extracellular space in various tissues and organs of the body in variety of clinical condition.
Physical and Chemical Nature of Amyloid
All types of amyloid are composed of non-branching fibrils of indefinite length and a diameter of approximately 7.5 to 10 nm by the electron microscope .About 95% of the amyloid material consists of fibril proteins.
Remaining 5% consists of proteoglycans, glycosaminoglycans, serum P components , etc.I. FIBRIL PROTEINS
It consists of two major distinct proteins and more than 20 minor forms.A. Major forms: These are AL, AA .
1. AL (amyloid light chain) protein:
Consists of complete immunoglobulin (Ig) light chains or the amino-terminal fragments of light chains, or both.
Produced by plasma cells.
Associated with monoclonal B cell proliferation (e.g. plasma cell tumors).
It is included in primary systemic amyloidosis.
2. AA (amyloid-associated) protein:
Non-immunoglobulin.Derived from a larger precursor in the serum called SAA (serum amyloid-associated) protein synthesized by the liver. Increased synthesis of SAA protein occurs under the influence of cytokines (e.g. IL-6 and IL-1) during inflammation.
Associated with chronic inflammation (called as secondary amyloidosis).
B. Minor forms:
1. Transthyretin (TTR):It is a normal serum protein that transports thyroxine and retinol.
Mutations in gene encoding TTR alter its structure , misfolds.
Found in a familial amyloid polyneuropathies, heart of aged individuals (senile systemic amyloidosis).
2. Amyloid B 2-microglobulin:
B 2-microglobulin is a normal serum protein.Amyloid fibril subunit namely AB2m is derived from B2-microglobulin and is found in amyloidosis of patients on long-term hemodialysis.
3. Amyloid B-peptide (AB)
AB is distinct from AB2M and is deposited in cerebral amyloid angiopathy and neurofibrillary tangles in Alzheimer’s disease.
AB is derived from amyloid beta precursor protein (AbPP).
4. Endocrine amyloid from hormone precursor proteins e.g. amyloid derived from pro-calcitonin (ACal), islet amyloid polypeptide (AIAPP, amylin), pro-insulin (AIns), prolactin (APro) etc.
5. Amyloid of prion protein (APrP)
It is derived from precursor prion protein which is a plasma membrane glycoprotein.Prion proteins are proteinaceous infectious particles lacking in RNA or DNA.
II. NON-FIBRILLAR COMPONENTS
Non-fibrillar components comprise about 5% of the amyloid material. These components include the following:• Amyloid P (AP)-component
synthesised in the liver and is present in all types of amyloid.derived from circulating serum amyloid P-component, a glycoprotein resembling the normal serum α1-glycoprotein
2. Apolipoprotein-E (apoE) It is a regulator of lipoprotein metabolism and is found in all types of amyloid.
3. Sulfated glycosaminoglycans (GAGs) These are constituents of matrix proteins .
Classification of Amyloidosis
A- According To the Distribution of Amyloid deposition into:
Generalized (Systemic); distant sites from the cells producing these abnormal proteins.localized; close to cells producing it.
B- According to the etiology:
Inherited (primary)Acquired (secondary)
C- According to the type of amyloid fibril protein.
Systemic amyloidosisIt is sub classified into:
• Primary Amyloidosis :
Due to blood dyscrasia e.g. multiple myeloma ( a malignant tumor of plasma cells).
The amyloid deposition is systemic and of AL type. Malignant plasma cells secrete excessive amount of immunoglobulin light chains called Bence Jones protein which is also present in serum and urine.
B) Secondary amyloidosis:
The amyloid deposition is systemic and of AA type. Secondary amyloidosis is due to:1) Infections e.g. T.B.
2) Non infectious chronic inflammatory processes :
Bronchiactasis
Chronic osteomylitis
Rheumatoid arthritis
Inflammatory bowel diseases
It is possible that chronic inflammation causes activation of macrophages resulting in secretion of IL-1 and 6 which stimulate liver cells to serum amyloid associated protein which form AA protein of amyloidosis.
C) Senile systemic amyloidosis in elderly.
characterized by the systemic deposition of amyloid in elderly patients usually between 70 to 80 years. Also called senile cardiac amyloidosis because of the symptoms related to restrictive cardiomyopathy and arrhythmias. The amyloid is composed of the normal TTR molecule.Localized amyloidosis
Amyloid deposits are limited to a single organ or tissue without involvement of any other site producing localized nodular mass e.g. lung, larynx, skin, tongue .etc. amyloid deposit is of AL type, some of these localized lesions are due to endocrine tumors ( endocrine amyloid) like medullary carcinoma of thyroid and carcinoma of stomach.Hemodialysis-associated Amyloidosis
Patients with chronic renal failure on long-term hemodialysis have high levels of B2 microglobulin in the serum because it cannot be filtered through dialysis membranes deposited as amyloid.Hereditary or Familial Amyloidosis
It constitutes a heterogeneous group, are rare and occur in certain geographic areas.1. Familial Mediterranean Fever
Autosomal recessive disorder.
Characterized by recurrent attacks of fever accompanied with inflammation of serosal surfaces (peritoneum, pleura, and synovial membrane).
The gene encodes a protein called pyrin (for its relation to fever), regulate inflammatory reactions by producing high levels of pro-inflammatory cytokines IL-1.
The amyloid fibril proteins are of AA type probably produced due to recurrent bouts of infl ammation.
2. Familial Amyloidotic Neuropathies
It is characterized by deposition of amyloid in peripheral and autonomic nerves and the fibrils are made up of mutant TTRs.
Clinical Features
Clinical manifestations initially may be nonspecific (e.g. weakness, weight loss). Specific symptoms appear later and are related to renal, cardiac, and gastrointestinal involvement.Renal involvement: It gives rise to proteinuria sometime massive enough to cause nephrotic syndrome. In advanced cases, the obliteration of glomeruli causes renal failure and uremia.
Cardiac amyloidosis: It may present as congestive heart failure, conduction disturbances and arrhythmias.
Gastrointestinal amyloidosis: It may be asymptomatic, or present as malabsorption, diarrhea, and digestive disturbances.
Morphology
Main Organs Involved:Primary amyloidosis: Heart, GI tract, respiratory tract, peripheral nerves, skin and tongue.
Secondary amyloidosis: Kidneys, liver, spleen, lymph nodes, adrenals, and thyroid.
Gross:
May or may not be apparent grossly.
If large amount accumulates affected organs are enlarged, firm and have a waxy appearance
Cut surface: If the amyloid deposits are large, painting the cut surface with iodine gives a yellow color, which is transformed to blue violet after application of sulfuric acid (which acidifies iodine).
Microscopy:
Amyloid deposits are always extracellular .
Appears as an amorphous, eosinophilic, hyaline, glassy substance.
Many other substances (e.g. collagen, fibrin) also stain eosinophilic with hematoxylin and eosin. Hence, it is necessary to differentiate amyloid from these other hyaline deposits by using special stains.
Special stains for amyloid
• Congo red: It is the most widely used specific stain for amyloid. Under light microscope, with Congo red stain, amyloid appears red but under polarized light, the Congo red stained amyloid shows apple-green birefringence.
• Thioflavin ‘T’ and ‘S’ give secondary immunofluorescence with ultraviolet light. Thioflavin T is more useful for demonstrating juxtaglomerular apparatus of the kidney.
• Metachromatic stains like crystal violet and methyl violet give rose pink appearance.
• Amyloid is PAS positive.
In kidney: Amyloid is deposited an glomeruli (in basement membrane of capillary loops) and wall of blood vessels .
In spleen: Amyloid causes marked enlargement of spleen and is deposited in splenic sinuses and splenic follicles.
In the liver: Amyloid causes marked enlargement of liver and is deposited in the wall of blood vessels and sinusoids
In the heart: Amyloid leads to cardiac enlargement in elderly and is deposited in between myocardial fibers

Macroscopically the affected organ enlarged, firm & has waxy consistency
In the heart, amyloid deposits begin to accumulate within the myocardium between muscle fibers & in the wall of blood vessels as seen by H&E as amorphous eosinophilic hyaline (right) & congo red stains as red deposits (left)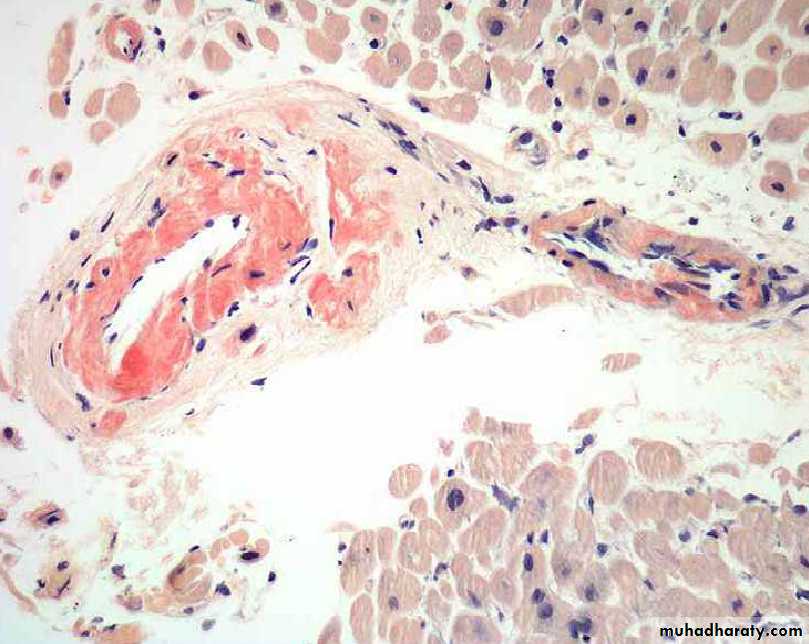
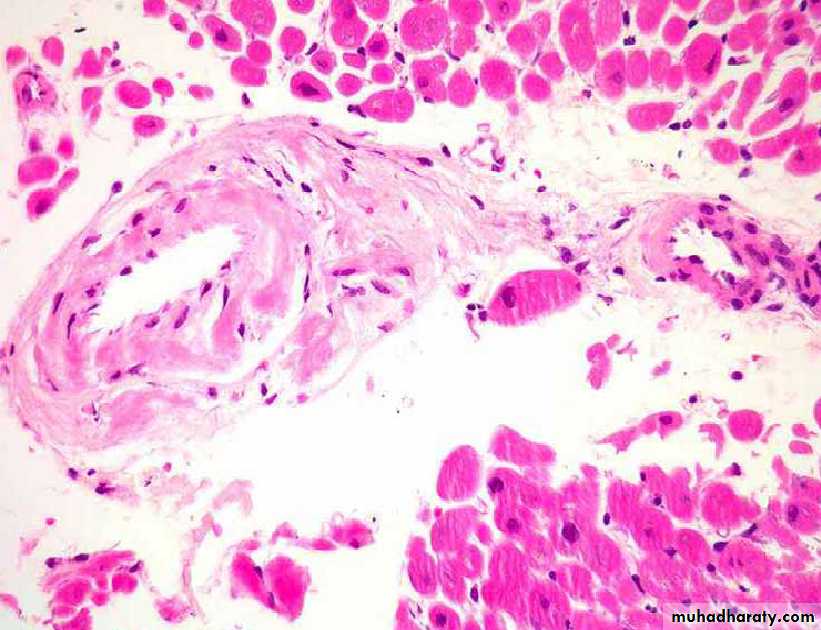
H&E stain
Congo red stainH&E the amyloid appears as an amprphous, eosinophilic hyaline extracellular substance that with progressive accumulation lead to pressure atrophy of adjacent cells

Note the yellow-green birefringence of amyloid in the heart with polarized microscope
A section of liver stained with congo red reveals pink-red deposits of amyloid in the wall of blood vessels & along the sinusoidsNote the yellow green birefringence of the deposits in the wall of blood vessels & sinusoids when observed by polarizing microscope
In adrenal gland the amyloid is deposited in the interglandular tissues & the wall of arterioles as an amorphous eosinophilic material as seen by H&E
Normal glomeruli
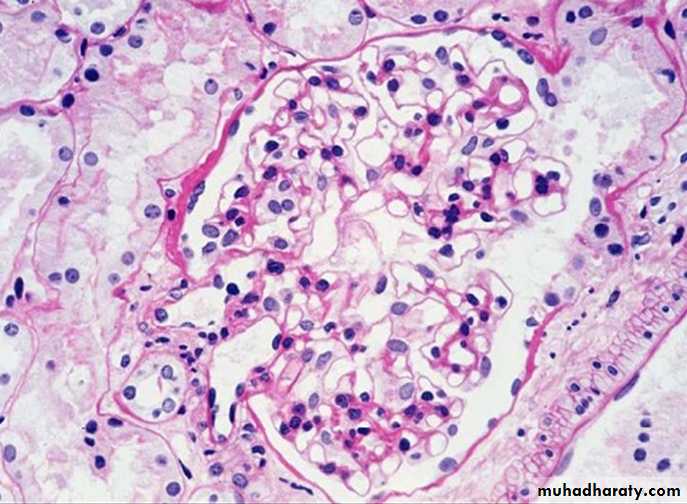
In kidney: Amyloid is deposited an glomeruli (in basement membrane of capillary loops) and wall of blood vessels
Amyloidosis of kidney-congo red stain
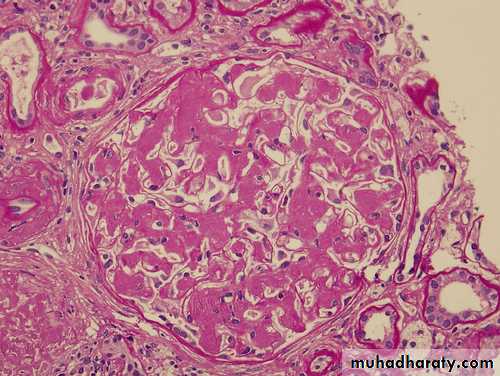
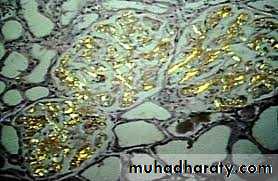
Amyloidosis of kidney- polarized light, the Congo red stained amyloid shows apple-green birefringence.
Diagnosis
Biopsy: histologic demonstration of amyloid deposits in tissues. The most common sites are the kidney, rectum or gingival tissues in systemic amyloidosis.
The rectum is the best site for taking the biopsy.
Examination of abdominal fat aspirates stained with Congo red is quite specific, but has low sensitivity .
Bone marrow aspirates may show monoclonal plasmacytosis, even in the absence of multiple myeloma in immunocyte-associated amyloidosis.
Electron microscopy show nonbranching fibrils of indefinite length and a diameter of approximately 7.5 to 10 nm.
In immunocyte-associated amyloidosis, serum and urine protein electrophoresis and immunoelectrophoresis should be done.
Scintigraphy with radiolabeled serum amyloid P (SAP) component is a rapid and specific test.
X-ray: crystallography and infrared spectroscopy Characteristic cross B pleated sheet conformation
Thank you