~1
~
Third Stage Internal Medicine Dr. Fadhil
Clinical Immunology 2
Immune Deficiency
Mechanisms of Immunodeficiency
Loss or reduction of:
Cell type
Cell numbers
Cell function
Loss of Cell Function:
Receptors
Cell signaling
Cytokine production
Ig production
Co stimulation impairment
Intracellular killing
Extravasation impairment
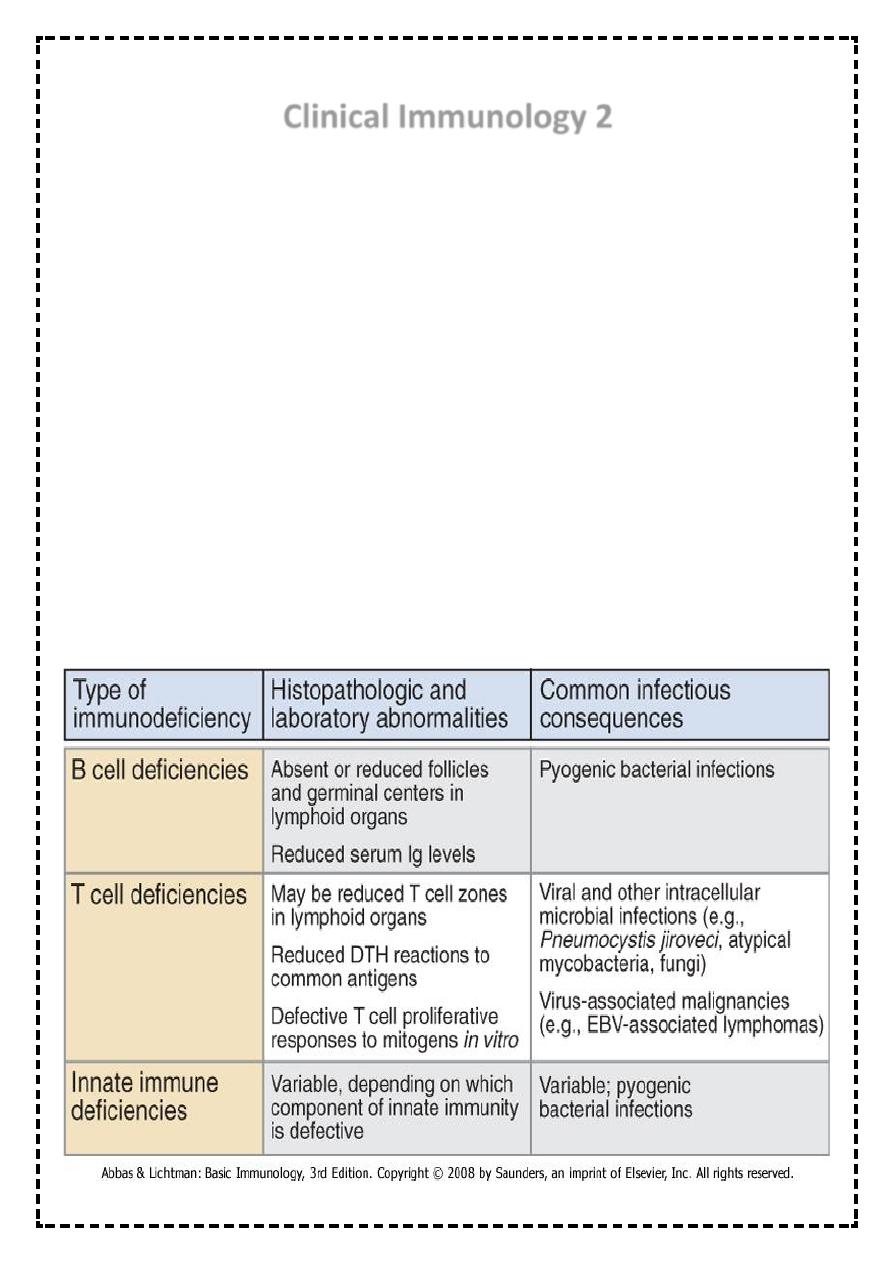
Features of immunodeficiency diseases

~2
~
Primary or congenital immunodeficiencies
Present at birth
Result from genetic abnormalities in one or more components of the immune
system
Secondary or acquired immunodeficiencies
Later in life
Much more common than primary immunodeficiency
Result from infections, malnutrition, or treatments that cause loss or inadequate
function of various components of the immune system
Most common is acquired immunodeficiency syndrome, or AIDS
Primary Immunodeficiency
Myeloid lineage
Congenital agranulocytosis
Leukocyte-adhesion deficiency
Lymphoid lineage
Severe combined immunodeficiency (SCID)
B cells
Agammaglobulinemia
Hypogammaglobulinemia
Specific Ig Deficiencies
T cells
DiGeorge Syndrome
Wiskott Aldrich Syndrome
Myeloid lineage disorders – phagocytic cell defects
1/ Quantitative – decreased numbers of granulocytes –neutrophil elastase mutation
Congenital chronic agranulocytosis
Cyclic agranulocytosis (neutropenia)
2/ Qualitative – phagocytes functional disorders, various enzyme deficits, inability of
phagocytes to degrade the ingested material

~3
~
Chronic Granulomatous Disease (CGD)
• Approximately in 60% X-linked
• Enzymatic inability to generate toxic oxygen metabolites (H2O2) during oxygen
consumption) - result of defect in neutrophilic cytochrome b (part of complex containing
NADPH oxidase)
• Inability to kill bacteria such as Staph. aureus, Pseud.aeruginosa that produce catalase
• Clinical features: granulomas of skin, organs
• Treatment: long-term antibiotic administration
Another example of primary phagocyte def. are disorders of phagocyte migration because of
failure to express adhesion molecules results in the inability of phagocytes to exit(go out) of
blood stream. They cause recurrent bacterial infections &sites of infection lack pus or
neutrophils infiltration.
Peripheral blood neutrophil count may be very high during acute infection because of the
failure of mobilized neutrophils to exit blood vessels.
Defects in cytokine &cytokine receptors also result in failure of intracellular killing& such
patients are susceptible to mycobacterial infection
Defects of common &classical pathway result in the susceptibility to infection by
encapsulated bacteria like Neisseria
Mannose-binding lectin pathway def. is common& occur in 5% of general population
&associated with infection in only individuals who are immune compromised..
Genetic def. Of C1,C2&C4 classical pathway result in autoimmune disease of sever type like
SLE.
Def. Of regulatory protein C1 inhibitor cause angioedema.
SEVERE COMBINED IMMUNODEFICENCY
In about 50% of SCID patients the immunodeficiency is x-linked whereas in the other half
the deficiency is autosomal.
They are both characterized by an absence of T cell and B cell immunity and absence (or
very low numbers) of circulating T and B lymphocytes.
Patients with SCID are susceptible to a variety of bacterial, viral, mycotic and protozoan
infections.
Diagnosis
Is based on enumeration of T and B cells and immunoglobulin measurement.
Severe combined immunodeficiency can be treated with bone marrow transplant

~4
~
B cell disorders
Selective IgA deficiency
• Disorder of B cell function
• Recurrent mild/moderate infections (respiratory, GIT, urinary tract) or asymptomatic
• Risk of reaction to live attenuated vaccines or generation of anti-IgA antibodies after a
blood transfusion
Selective IgG subclasses or specific IgG deficiency
• B cell function disorder
• Onset of symptoms in childhood, mostly respiratory tract infections caused by
encapsulated bacteria (H.influenzae, Pneumococci)
Transient hypogammaglobulinemia of infancy
Common Variable Immunodeficiency (CVID)
There are defect in T cell signaling to B cells
Acquired a gammaglobulinemia in the 2
nd
or 3
rd
decade of life
May follow viral infection
Pyogenic infection
80% of patients have B cells that are not functioning
B cells are not defective. They fail to receive signaling from T lymphocytes
Unknown
X-linked a gammaglobulinaemia
In X-LA early maturation of B cells fails
Affect males
Few or no B cells in blood
Very small lymph nodes and tonsils
No Ig
Small amount of Ig G in early age
Recurrent pyogenic infection
With an exception of selective IgA def., immunization is generally not effective because of the
defect in IgG antibody production.
As with all primary immune deficiencies, live vaccines should be avoided.

~5
~
T-cell disorders
DiGeorge syndrome
• Disorder of prethymocytes maturation due to absence of thymus (disorder of
development of 3
rd
and 4
th
branchial pouch)
• Congenital heart diseases
• The onset of symptoms after the birth – hypocalcemic spasms and manifestations of
cong.heart disease
• Immunodeficiency could be only mild, the numbers of T lymphocytes later usually
become normal
• Treatment symptomatic
Wiskott-Aldrich syndrome:
• Associated with normal T cell numbers with reduced functions, which get progressively
worse.
• IgM concentrations are reduced but IgG levels are normal
• Both IgA and IgE levels are elevated.
• Boys with this syndrome develop severe eczema.
• They respond poorly to polysaccharide antigens and are prone to pyogenic infection.
Patients with suspected T-cell immune deficiencies should receive anti-Pneumocystis and
antifungal prophylaxis, and require aggressive management of specific infection. Thymic
transplantation may be used for treatment of DiGeorge syndrome.
Secondary Immunodeficiency
Causes of secondary immune deficiency
1-physiological immune deficiency.
Ageing pregnancy prematurity
2-infection
HIV measles TB

~6
~
3-Iatrogenic
Immunosuppressive therapy , antineoplastic agents , corticosteroids , stem cell transplantation&
radiation therapy
4-malignancy
B-cell malignancies including leukemia, lymphoma & multiple myeloma, solid tumors &
thymoma
5-Biochemical and nutritional disorders
Malnutrition, renal insufficiency/dialysis ,DM, specific mineral deficiencies, eg. iron , zinc.
6- other conditions
Burns, asplenia / hyposplenism .
Warning signs of immune deficiency
1-8 respiratory tract infections/year in a child, or more than 4 respiratory tract infections/year in
an adult
2->1 infection requiring hospital admission or IV antibiotics
3-infections with unusual organisms.
4- infections at unusual sites
5-chronic infection unresponsive to usual treatment.
6-early end organ damage(eg. Bronchiectasis).
7-family history of immune deficiency.