
Hussien Mohammed Jumaah
CABMLecturer in internal medicine
Mosul College of Medicine
2016

learning-topics
Liver andbiliary tract diseaseThe liver weighs 1.2–1.5 kg , produces 8–14 g of albumin and1–2 L of bile daily (contains bile acids (formed from cholesterol), phospholipids, bilirubin and cholesterol.
Liver cells
Hepatocytes comprise 80% of liver cells. The remaining 20% are the endothelial cells lining the sinusoids, epithelial cells lining the intrahepatic bile ducts, cells of the immune system (macrophages (Kupffer cells) and atypical lymphocytes), and non-parenchymal cells called stellate or Ito cells. Endothelial cells line the sinusoids , a network of capillary vessels that differ from other capillary beds in the body in that there is no basement membrane.
The endothelial cells have gaps between them (fenestrae) of about 0.1 micron in diameter, allowing free flow of fluid and particulate matter to the hepatocytes.
Individual hepatocytes are separated from the leaky sinusoids by the space of Disse, which contains
stellate cells that store vitamin A and play an important part in regulating liver blood flow. They may also be immunologically active and defence against pathogens. The key role of stellate cells in terms of pathology is in the development of hepatic fibrosis, the precursor of cirrhosis.
They undergo activation in response to cytokines produced following liver injury, differentiating into myofibroblasts, which are the major producers of the collagen-rich matrix that forms fibrous tissue .
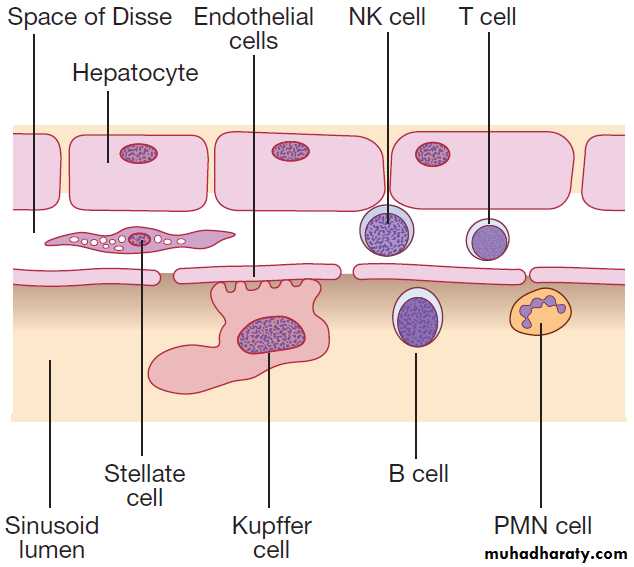
Non-parenchymal liver cells. (B = B lymphocytes;
NK = natural killer cells; PMN = polymorphonuclear leucocytes;T = T lymphocytes).
Blood supply
The blood supply to the liver constitutes 25% of theresting cardiac output , receives a dual blood supply; ~20% of the blood flow is oxygen-rich blood from the hepatic artery, and 80% is nutrient-rich blood from the portal vein arising from the stomach, intestines, pancreas, and spleen.
The dual perfusion, can have important effects on the clinical expression of liver ischaemia (which typically exhibits a less dramatic pattern than ischaemia in other organs),and can raise practical challenges in liver transplant surgery.
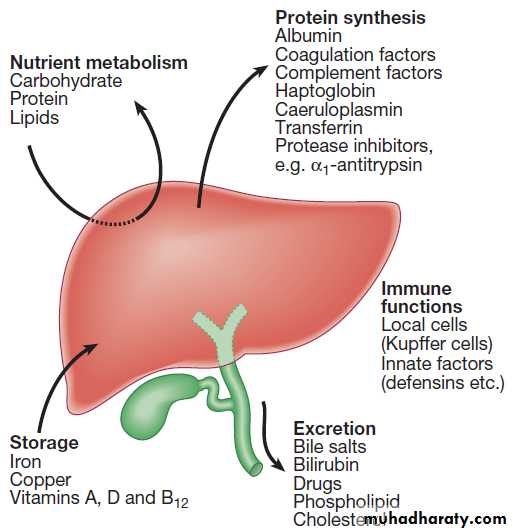
Important liver functions.
The liver produces 8–14 g of albumin per day, and this plays a critical role in maintaining oncotic pressure in the vascular space and in the transport of small molecules like bilirubin, hormones and drugs throughout the body.Amino acids that are not required for the production of new proteins are broken down, with the amino group being
converted ultimately to urea.
• Following a meal, more than half of the glucose absorbed is taken up by the liver and stored as glycogen or converted to glycerol and fatty acids, thus preventing hyperglycaemia.
• During fasting, glycogen is broken down to release glucose (gluconeogenesis), thereby preventing
Hypoglycaemia.
• The liver plays a central role in lipid metabolism,
producing VLDL and further metabolising LDL and HDL . Dysregulation of lipid metabolism is thought to have a critical role in the pathogenesis of NAFLD. Lipids are now recognised to play a key part in the pathogenesis of hepatitis C, facilitating viral entry into hepatocytes.
Clotting factors
The liver produces key proteins that are involved in the
coagulation cascade. Many of these coagulation factors (II, VII, IX and X) are post- translationally modified by vitamin K-dependent enzymes, and their synthesis is impaired in VK deficiency . PT or INR , International Normalised Ratio, is therefore one of the most important clinical tools for the assessment of hepatocyte function.
Note that the deranged PT or INR in liver disease may not directly equate to increased bleeding risk, as these tests do not capture the concurrent reduced synthesis of protein C and S anticoagulant. In general, therefore, correction of PT using blood products should be guided by clinical risk rather than the absolute value of the PT. The liver plays a central role in the metabolism of bilirubin and is responsible for the production of bile. In the blood 250–300 mg/ day of unconjugated bilirubin is produced from the catabolism of haem. Bilirubin is normally almost all unconjugated and, because it is not water-soluble, is bound to albumin and does not pass into the urine. Unconjugated bilirubin is taken by hepatocytes, conjugated in the endoplasmic reticulum by UDP-glucuronyl transferase, producing bilirubin mono- and diglucuronide.
These bilirubin conjugates are water-soluble and are exported into the bile canaliculi by specific carriers on the hepatocyte membranes. The conjugated bilirubin is excreted in the bile and passes into the duodenal lumen.
Once in the intestine, conjugated bilirubin is metabolised by colonic bacteria to form stercobilinogen, which may be further oxidised to stercobilin. Both stercobilinogen and stercobilin are then excreted in the stool, contributing to its brown colour. Biliary obstruction results in reduced stercobilinogen in the stool, and the stools become pale.
4 mg/day of stercobilinogen is absorbed from the bowel, passes through the liver, and is excreted in the urine, where it is known as urobilinogen or, following further oxidisation, urobilin.
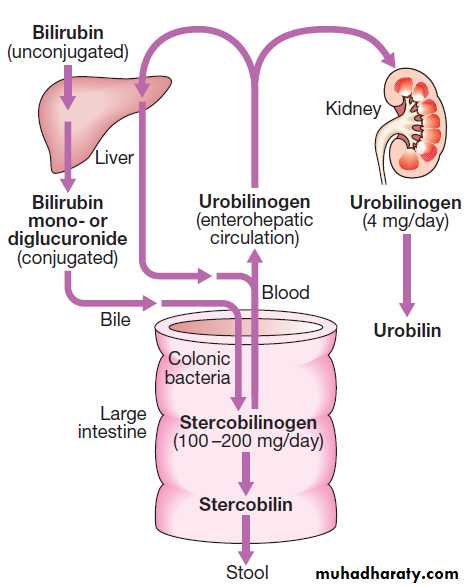
Pathway of bilirubin excretion.
Storage of vitamins and mineralsVitamins A, D and B12 are stored by the liver in large
amounts, while others, such as vitamin K and folate, are
stored in smaller amounts and disappear rapidly if
dietary intake is reduced. The liver metabolise vitamins to more active compounds, e.g. 7-dehydrocholesterol to 25(OH) vitamin D. Also stores iron(ferritin and haemosiderin) and copper, which is excreted in bile.
Immune regulation
Approximately 9% of the normal liver is composed of
immune cells . Cells of the innate immune system include Kupffer cells derived from blood monocytes, macrophages and natural killer (NK) cells, as well as ‘classical’ B and T cells of the adaptive immune response.
The enrichment of such cells in the liver reflects the unique importance of the liver in preventing microorganisms from the gut entering the systemic circulation.
Kupffer cells constitute the largest single mass of
tissue-resident macrophages in the body and account
for 80% of the phagocytic capacity of this system. They remove aged and damaged red blood cells, bacteria, viruses, antigen–antibody complexes and endotoxin.
They also produce a wide variety of inflammatory mediators that can act locally or may be released into the systemic circulation.
Biliary system and gallbladder
Hepatocytes provide the driving force for bile flow. Bile is secreted by hepatocytes and flows from cholangioles to the biliary canaliculi. The canaliculi join to form largerintrahepatic bile ducts, which in turn merge to form the
right and left hepatic ducts, join as they emerge from the liver to form the common hepatic duct, which becomes the common bile duct after joining the cystic duct . The common bile duct is approximately 5 cm long and
4–6 mm wide. The distal portion of the duct passes through the head of the pancreas and usually joins the pancreatic duct before entering the duodenum through the ampullary sphincter (sphincter of Oddi). The anatomy of the lower common bile duct can vary widely.
Common bile duct pressure is maintained by rhythmic
contraction and relaxation of the sphincter of Oddi; thispressure exceeds gallbladder pressure in the fasting
state, so that bile normally flows into the gallbladder,
where it is concentrated tenfold by resorption of water
and electrolytes.
The gallbladder is a pear-shaped sac typically lying
under the right hemiliver, with its fundus located anteriorly
behind the tip of the 9th costal cartilage. It concentrate, and provide a reservoir for, bile. Gallbladder tone is maintained by vagal activity, and cholecystokinin released from the duodenal mucosa during feeding causes gallbladder contraction and reduces sphincter pressure, so that bile flows into the duodenum.
INVESTIGATION OF LIVER AND HEPATOBILIARY DISEASE
• identification of the presence of liver disease
• establishing the aetiology
• understanding disease severity
Aetiology is typically established through a combination
of history, specific blood tests and, where appropriate,
imaging and liver biopsy.
Staging of disease (in essence, the identification of
cirrhosis) is largely histological, although there is
increasing interest in non-invasive approaches, including
novel imaging modalities, serum markers of fibrosis
and the use of predictive scoring systems.
Prognostic scores: the Child–Pugh and MELD scores in
cirrhosis , the Glasgow score in alcoholic hepatitis .
Liver blood biochemistry (LFTs)
Serum bilirubin, aminotransferases, alkaline phosphatase, gamma-glutamyl transferase and albumin.Most analytes measured by LFTs are not truly ‘function’ tests but, given that they are released by injured hepatocytes, instead provide biochemical evidence
of liver cell damage.
Liver function per se is best assessed by the serum albumin, PT and bilirubin because of the role played by the liver in synthesis of albumin and clotting factors and in clearance of bilirubin. Serum albumin often low in liver disease, due to a change in the volume of distribution of albumin, and reduced synthesis. Since the plasma half-life of albumin is about 2 weeks, levels may be normal in acute liver failure but are reduced in chronic liver failure.
Alanine (ALT) and aspartate aminotransferase (AST) are located in the the hepatocyte. Although both transaminase enzymes are widely distributed, expression of ALT outside the liver is relatively low and this enzyme is therefore considered more specific for hepatocellular damage. Alkaline phosphatase (ALP) main sites of production are the liver, GIT , bone, placenta and kidney. ALP enzymes in the liver are located in cell membranes of the hepatic sinusoids and the biliary canaliculi. Accordingly, levels rise with intrahepatic and extrahepatic biliary obstruction.
Gamma-glutamyl transferase (GGT) is a microsomal
enzyme found in many cells and tissues of the body.The highest concentrations are located in the liver. The function of GGT is to transfer glutamyl groups from gamma-glutamyl peptides to other peptides and amino acids.
The pattern of a modest increase in aminotransferase
activity and large increases in ALP and GGT activity
favours biliary obstruction and is commonly described
as ‘cholestatic’ or ‘obstructive’ . Isolated elevation
of the serum GGT is relatively common, and may
occur during ingestion of microsomal enzyme-inducing
drugs, including alcohol , but also in NAFLD.
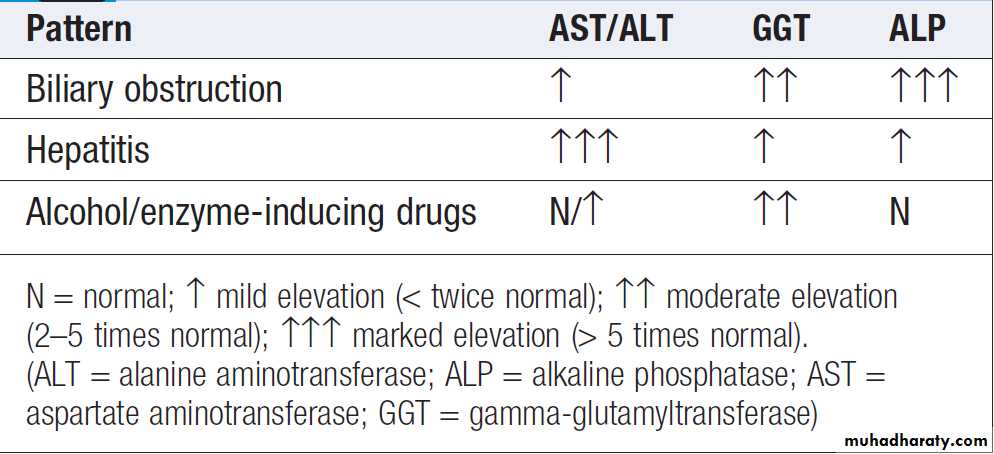
‘Hepatitic’ and ‘cholestatic’/‘obstructive’

Drugs that increase gamma- glutamyltransferase
Other biochemical tests
• Hyponatraemia occurs in severe liver disease due
to increased production of antidiuretic hormone
• Serum urea may be reduced in hepatic failure,
whereas levels of urea may be increased following
gastrointestinal haemorrhage.
• When high levels of urea are accompanied by raised
bilirubin, high serum creatinine and low urinary sodium, this suggests hepatorenal failure, carries a grave prognosis.
• Significantly elevated ferritin suggests haemochromatosis. Modest rise in inflammatory disease and alcohol excess.
Haematological tests
Blood count• A normochromic normocytic anaemia may reflect
recent gastrointestinal haemorrhage, whereas chronic blood loss is characterised by a hypochromic microcytic anaemia secondary to iron deficiency. A high erythrocyte mean cell volume (macrocytosis) is associated with alcohol misuse,
but target cells in any jaundiced patient also result
in a macrocytosis.
• Leucopenia may complicate portal hypertension and
hypersplenism, whereas leucocytosis may occur with cholangitis, alcoholic hepatitis and hepatic abscesses. Atypical lymphocytes are seen in infectious mononucleosis.
• Thrombocytopenia is common in cirrhosis and is due to reduced platelet production, and increased breakdown because of hypersplenism. Thrombopoietin, required for platelet production, is produced in the liver and levels fall with worsening liver function.
Thus platelet levels are usually more depressed than white cells and haemoglobin in the presence of hypersplenism.
Thrombocytosis is unusual but may occur in those with active GI haemorrhage and, rarely,
in hepatocellular carcinoma.
Coagulation tests
The normal half-lives of the VK-dependent factors (5–72 hs), VKdeficiency, as may occur with so changes in the PT occur relatively quickly following liver damage; provide valuable prognostic information in acute and chronic liver failure. Increased PT is evidence of severe liver damage in chronic liver disease. VK does not reverse VKdeficiency if it is due to liver disease, but will correct if the cause is biliary obstruction (non-absorption of fat-soluble Vs).
Immunological tests
The presence of liver-related autoantibodies can be suggestive of the presence of autoimmune liver disease (although false-positive results can occur in non-autoimmune inflammatory disease such as NAFLD). Elevation in overall serum immunoglobulin levels can also be suggestive of autoimmunity.

Chronic liver disease screen
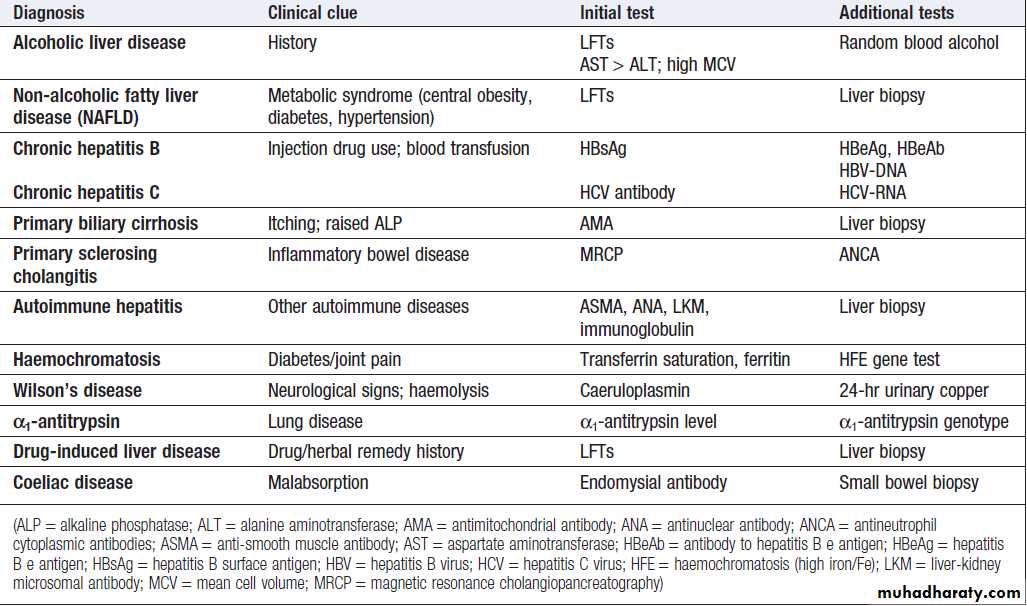
How to identify the cause of LFT abnormality
ImagingSeveral imaging techniques used to determine the site and general nature of structural lesions in the liver and biliary tree. In general, however, imaging unable to identify inflammation and have poor sensitivity for liver fibrosis unless cirrhosis with portal hypertension is present.
Ultrasound
Non-invasive used as a ‘first-line’ test to identify gallstones, biliary obstruction or thrombosis in the hepatic vasculature, splenomegaly but is less effective at identifying diffuse parenchymal disease. Focal lesions, may not be detected if they are <2 cm in diameter. Doppler allows blood flow in the hepatic artery, portal vein and hepatic veins to be investigated. Endoscopic ultrasound provides high-resolution images of the pancreas, biliary tree and liver .
Computed tomography and magnetic resonance imaging
detects smaller focal lesions, especially when combined with contrast injection . MRI can also be used. Hepatic angiography is seldom used, since CT and MRI provide images of vasculature, but it still has a therapeutic role in the embolisation of vascular tumours.Cholangiography by MR cholangiopancreatography endoscopy ( MRCP) or the percutaneous approach (percutaneous transhepatic cholangiography, PTC). MRCP is as good as ERCP at providing images of the biliary tree but has fewer complications and is the diagnostic test of choice. Both allow therapeutic interventions, such as the insertion of biliary stents across malignant bile duct strictures. The PTC is used if it is not possible to access the bile duct endoscopically.
Histological examination
US-guided liver biopsy can confirm the severity of liver damage and provide aetiological information. Performed percutaneously with Menghini needle,through an intercostal space under local anaesthesia, or radiologically using a transjugular approach,is a relatively safe ,mortality of about 0.01%. The main complications are abdominal and/or shoulder pain, bleeding and biliary peritonitis.
Liver biopsies can be carried out in patients with defective haemostasis if:
• the defect is corrected with fresh frozen plasma and platelet transfusion
• biopsy is obtained by the transjugular route, or
• the procedure is conducted under US control and the needle track is then plugged with procoagulant material.

Conditions required for safe percutaneous liver biopsy
Non-invasive markers of hepatic fibrosisCan reduce the need for liver biopsy to assess the extent of fibrosis in some settings. Serological markers of hepatic fibrosis, such as α2-macroglobulin, haptoglobin and routine clinical biochemistry tests, are used in the Fibrotest®. The ELF® (Enhanced Liver Fibrosis) serological assay uses a combination of hyaluronic acid, procollagen peptide III (PIIINP) and tissue inhibitor of metalloproteinase 1(TIMP1). An alternative to serological markers is transient elastography in which ultrasound-based shock waves are to measure liver stiffness as a surrogate for hepatic fibrosis. These tests are good at differentiating severe fibrosis from mild scarring, but is limited in its ability to detect subtle changes.
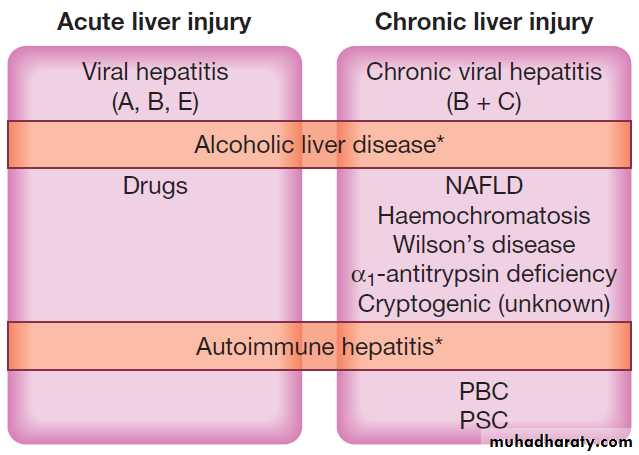
Causes of acute and chronic liver injury.
Acute liver injurymay present with non-specific symptoms of fatigue and abnormal LFTs, or with jaundice and acute liver failure.
Chronic liver injury hepatic injury,
inflammation and/or fibrosis occurring in the liver for > 6 months.
(NAFLD = non-alcoholic fatty liver disease; PBC = primary biliary cirrhosis; PSC = primary sclerosing cholangitis).
Acute liver failure
Serious condition.The presentation is with progressive deterioration in liver function and mental changes progressing from confusion to coma.
The syndrome was originally defined further as occurring within 8 weeks of onset of the precipitating illness, in the absence of evidence of preexisting liver disease. This distinguishes it from instances in which hepatic encephalopathy represents a deterioration in chronic liver disease (CLD). More recently, newer classifications have been developed divides acute liver failure into hyperacute, acute and subacute, according to the interval between onset of jaundice and encephalopathy .
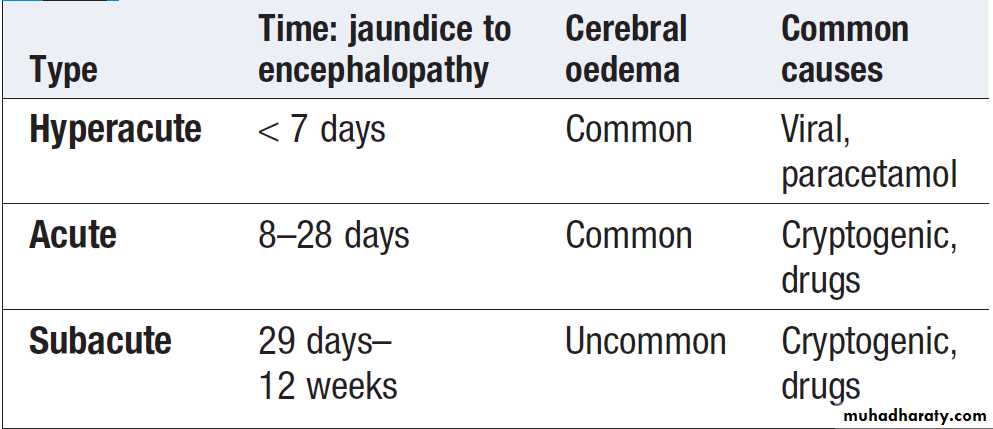
Classification of acute liver failure
PathophysiologyAcute viral hepatitis is the most common cause worldwide,
whereas paracetamol toxicity is the most frequent cause in the UK, occasionally with other drugs, or from Amanita phalloides (mushroom) poisoning, in pregnancy, in Wilson’s disease, following shock and, rarely, in extensive malignant disease of the liver. In 10% of cases the cause of acute liver failure remains unknown and these patients are often labelled as having ‘non-A–E
viral hepatitis’ or ‘cryptogenic’ acute liver failure.
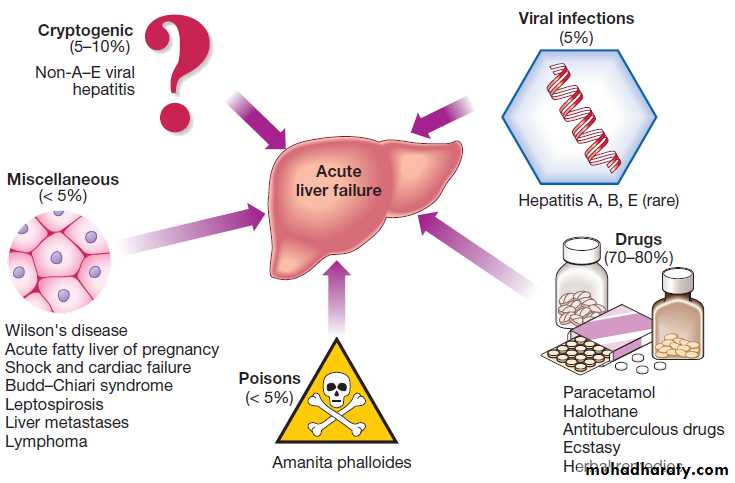
Causes of acute liver failure in the UK.
Clinical assessmenthepatic encephalopathy and/or cerebral oedema is the cardinal manifestation. The initial features are often subtle, reduced alertness and concentration, restlessness and aggression, to drowsiness and coma . Cerebral oedema may occur (increased ICP), causing abnormal pupil reaction, fixed pupils. Hypertension , bradycardia, hyperventilation, profuse sweating, focal fits or decerebrate posturing. Papilloedema rare, weakness, nausea and vomiting. Right hypochondrial discomfort. Occasionally, death occur before jaundice develops. Fetor hepaticus The liver is normal size but later becomes smaller. Hepatomegaly is unusual and, in the presence of acute ascites, suggests venous outflow obstruction (Budd– Chiari syndrome). Splenomegaly is uncommon . Ascites and oedema are late.
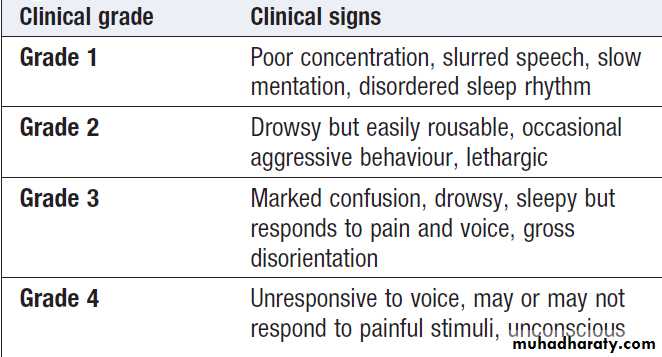
How to assess clinical grade of hepatic encephalopathy
InvestigationsTo determine the cause of the liver failure and prognosis .
Hepatitis B core IgM antibody is the best screening test for acute hepatitis B infection, as liver damage is due to the immunological response to the virus, which has often been eliminated, and the test for HBsAg may be negative.
The PT rapidly prolonged and itis of greatest prognostic value and should be carried out at least twice daily. Factor V levels can be used instead of the PT to assess liver impairment. Aminotransferase is particularly high after paracetamol overdose, reaching 100–500 times normal, but falls as liver damage progresses and is not helpful in determining prognosis. Plasma albumin remains normal unless the course is prolonged. Percutaneous liver biopsy is contraindicated (severe coagulopathy).
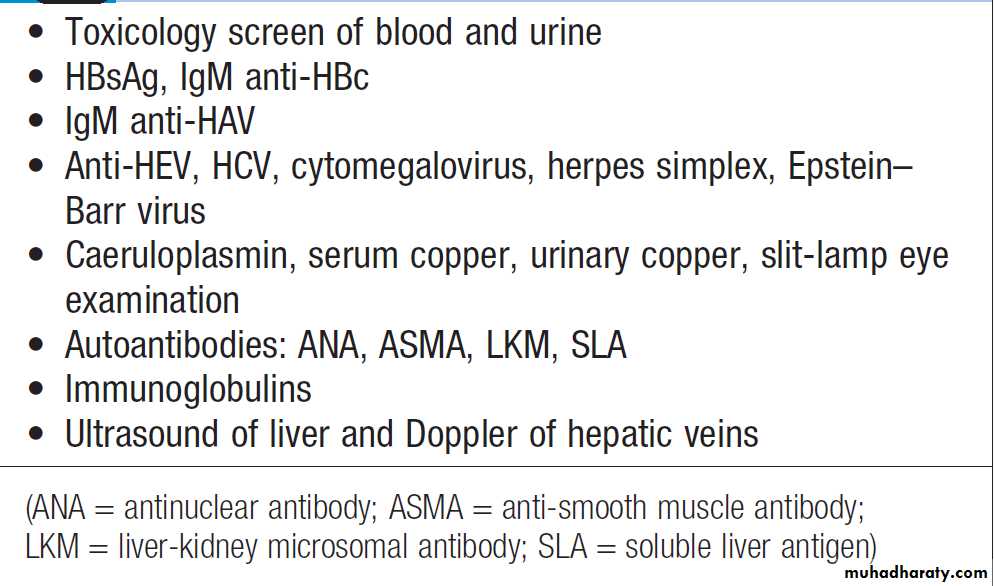
Investigations to determine the cause of
acute liver failure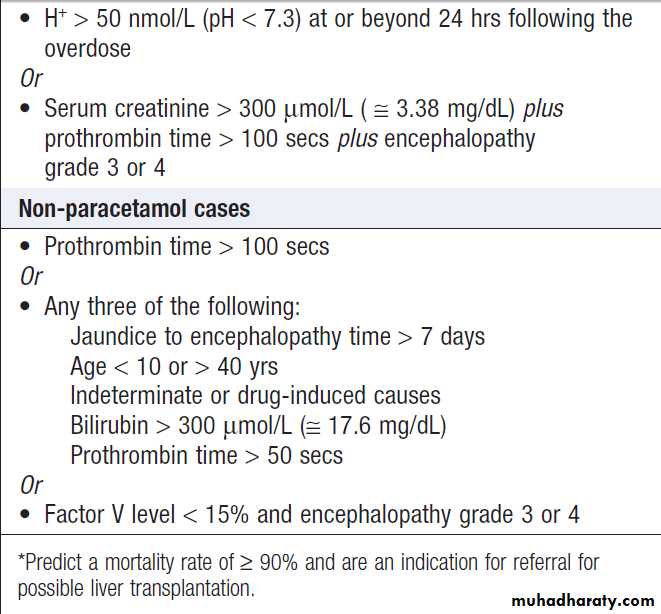
Adverse prognostic criteria in acute liver failure*
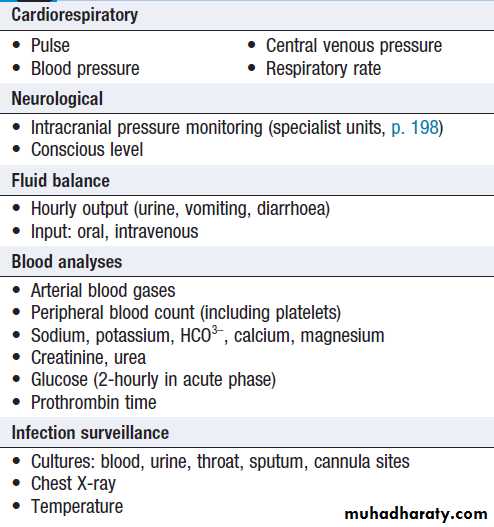
Monitoring in acute liver failure
Management
Patients with acute liver failure should be treated in ahigh-dependency or intensive care unit as soon as
progressive prolongation of the PT occurs or hepatic
encephalopathy is identified , so that prompt treatment of complications can be initiated . Conservative treatment aims to maintain life in the hope that regeneration will occur, but early transfer to a specialised transplant unit should always be considered. N- acetylcysteine may improve outcome, particularly in acute failure due to paracetamol poisoning. Liver transplantation is an increasingly important treatment option for acute liver failure. Survival following liver transplantation is improving, 1-year survival rates of about 60%.

Complications of acute liver failure
Abnormal liver function testsThe prevalence has been reported to be as high as 10%. The most common abnormalities are alcoholic or non-alcoholic fatty liver disease . When LFTs are measured routinely prior to elective surgery, 3.5% are discovered to have mildly elevated transaminases. Although transient mild abnormalities may not be clinically significant, the majority of patients with persistently abnormal LFTs do have significant liver disease. Biochemical abnormalities in CLD often fluctuate over time, and therefore even mild abnormalities can indicate significant underlying disease and so warrant follow-up and investigation.
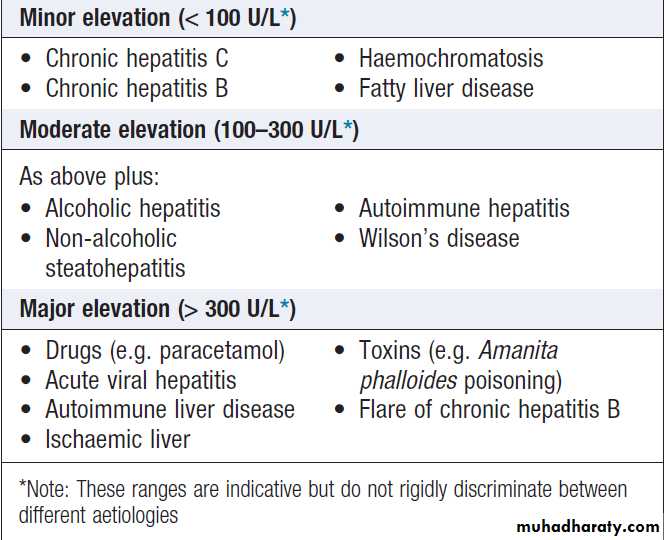
Common causes of elevated serum transaminases
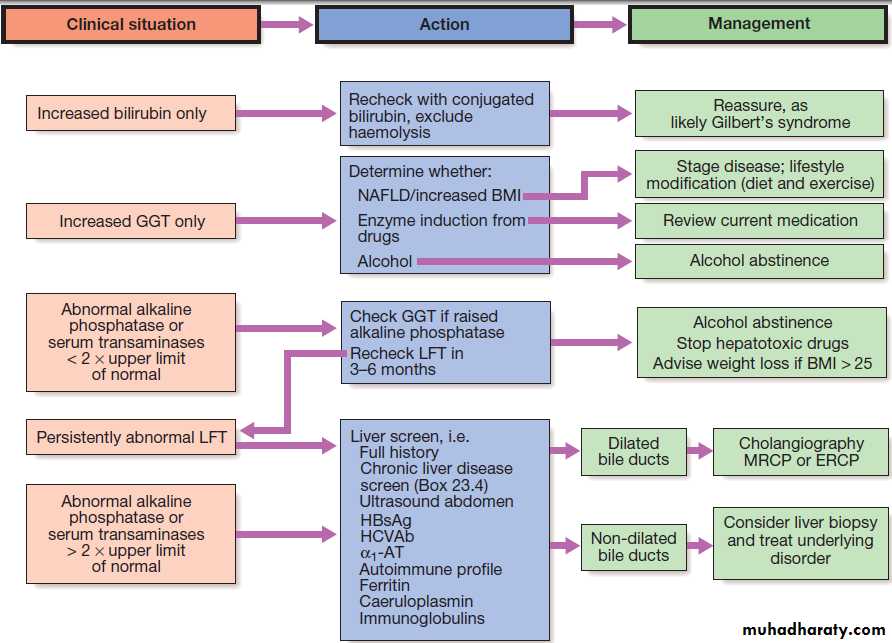
Suggested management of abnormal LFTs in asymptomatic patients. *No further investigation needed.
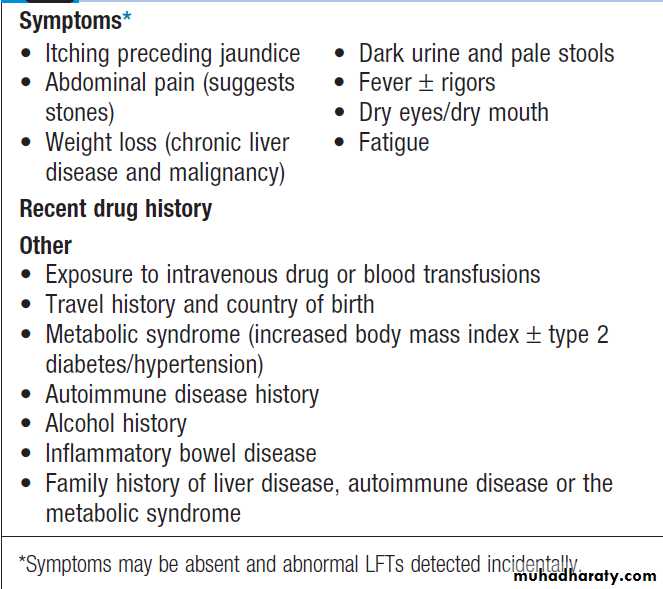
Key history points in patients with jaundice
JaundiceDetectable clinically when the plasma bilirubin exceeds ~2.5 mg/ dL . It is useful to consider whether the cause pre-hepatic, hepatic or post-hepatic
Pre-hepatic jaundice
Caused either by haemolysis or congenital , and is characterised by an isolated raised bilirubin level. Jaundice due to haemolysis is usually mild because a healthy liver can excrete a bilirubin load six times greater than normal before unconjugated bilirubin accumulates in the plasma. The most common form of non- haemolytic hyperbilirubinaemia is Gilbert’s.
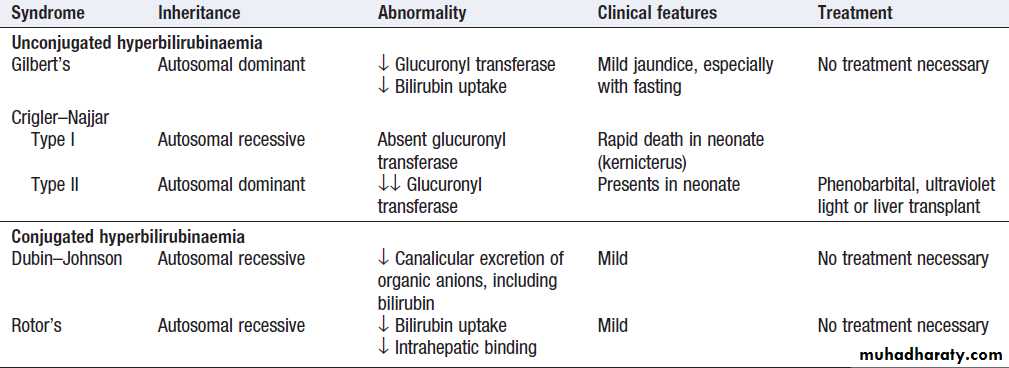
Congenital non-haemolytic hyperbilirubinaemia
Hepatocellular jaundiceResults from an inability of the liver to transport
(at any point between uptake of unconjugated bilirubin into the cells and transport of conjugated bilirubin into the canaliculi) bilirubin into the bile.
In hepatocellular , both unconjugated and conjugated bilirubin in the blood increase. Characteristically associated with increases in transaminases . Acute jaundice in the presence of an ALT of > 1000 U/L is highly suggestive of an infectious cause (e.g. hepatitis A, B), drugs (e.g. paracetamol) or hepatic ischaemia.
Obstructive (cholestatic) jaundice
May be caused by:
• failure of hepatocytes to initiate bile flow
• obstruction of the bile ducts or portal tracts
• obstruction of bile flow in the extrahepatic bile ducts between the porta hepatis and the papilla of Vater.
Clinical features , due to cholestasis, those due to secondary infection(cholangitis) and underlying condition. Extrahepatic biliary blockage is characteristically associated with pale stools and dark urine. Pruritus , skin excoriations may be a dominant feature.
Peripheral stigmata of chronic liver disease are absent.
If the gallbladder is palpable, the jaundice is unlikely to be caused by biliary obstruction due to gallstones, probably because a chronically inflamed stone-containing gallbladder cannot readily dilate. This is Courvoisier’s Law, and suggests that jaundice is due to a malignant biliary obstruction (e.g. pancreatic cancer).
Cholangitis is characterised by ‘Charcot’s triad’ of jaundice, right upper quadrant pain and fever.
Cholestatic jaundice is characterised by a relatively greater elevation of ALP and GGT than the aminotransferases.
Ultrasound is indicated to determine the cause .
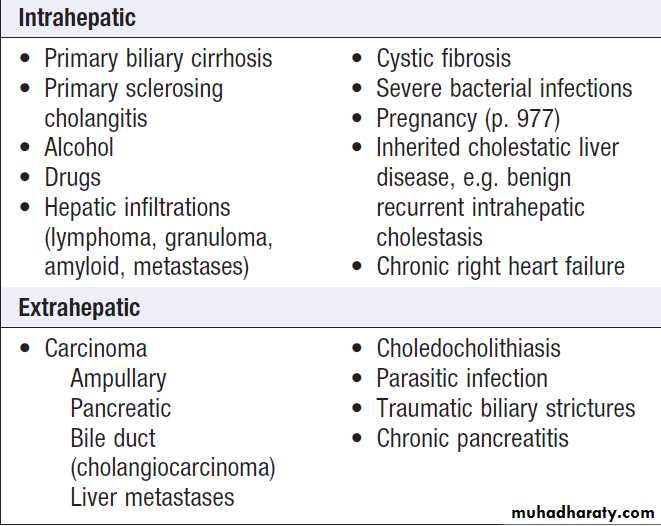
Causes of cholestatic jaundice

Clinical features and complications of
cholestatic jaundice
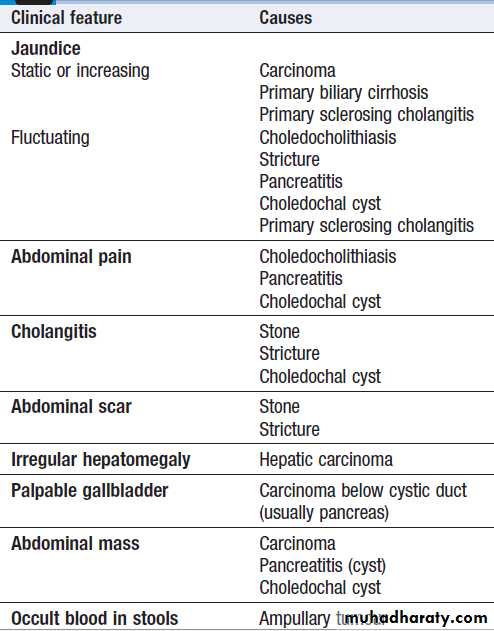
*Each of the diseases listed here can give rise to almost any of the clinical
features shown, but the box indicates the most likely cause of the clinical features listed.Clinical features suggesting an underlying
cause of cholestatic jaundice*
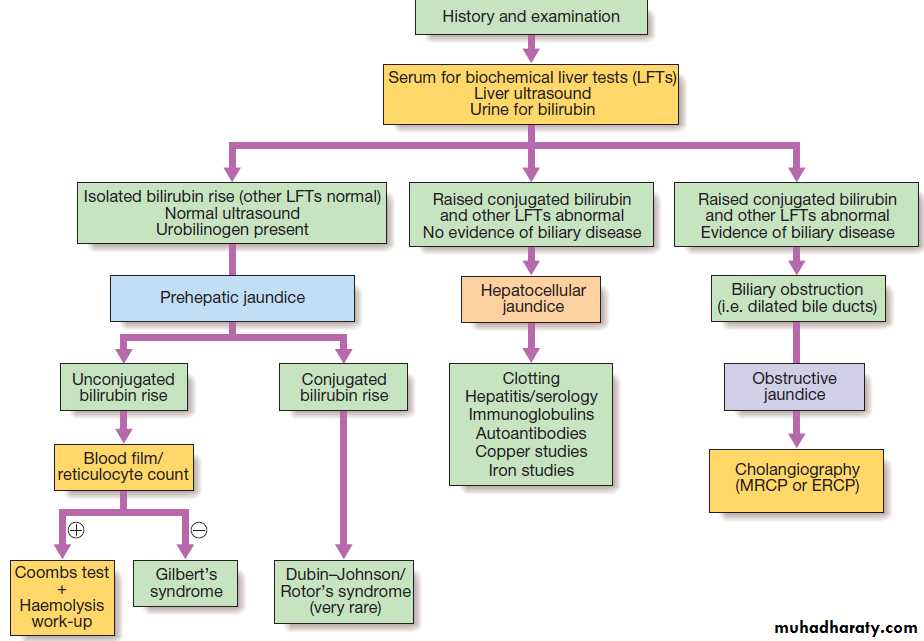
Investigation of jaundice.

Causes of change in liver size
Ascites
Free fluid in the peritoneal cavity, accumulations of (> 1 L) causes abdominal distension, fullness in the flanks, shifting dullness and when marked, a fluid thrill, eversion of the umbilicus, herniae, abdominal striae, and scrotal oedema. Dilated superficial abdominal veins may be seen due to portal hypertension. Splanchnic vasodilatation is thought to be the main factor leading to ascites in cirrhosis, mediated by vasodilators (mainly nitric oxide) that are released when portal hypertension causes shunting of blood into the systemic circulation. Systemic arterial pressure falls leads to activation of the RAS with secondary aldosteronism, increased sympathetic activity, increased atrial natriuretic hormone secretion and altered activity of the kallikrein–kinin system .
These systems tend to normalise arterial pressure
but produce salt and water retention. In this setting the combination of splanchnic arterial vasodilatation and portal hypertension alters intestinal capillary permeability, promoting accumulation of fluid within the peritoneum.Investigations
US is the best means of detecting ascites. Paracentesis (if necessary under ultrasonic guidance) can be used to obtain ascitic fluid for analysis.Pleural effusions are found in
about 10%, usually on the right side , most are small . Pleural effusions, particularly those on the left side, should not be assumed to be due to the ascites.
Measurement of the protein concentration and the
serum–ascites albumin gradient (SAAG) are used to distinguish a transudate from an exudate.
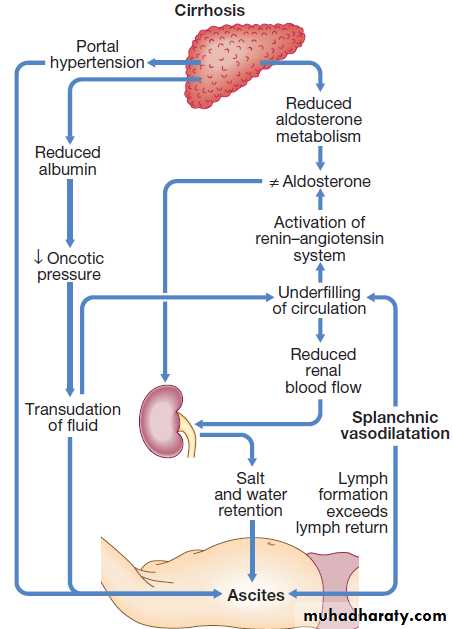
Pathogenesis of ascites.
Causes of ascites

Ascitic fluid: appearance and analysis
Cirrhotic develop a transudate with a total protein concentration< 25 g/L and relatively few cells. However, in up to 30% of patients, the total protein concentration is more than 30 g/L. In these cases, it is useful to calculate the SAAG by subtracting the concentration of the ascites fluid albumin from the serum albumin. A gradient of >11 g/L is 96% predictive that ascites is due to portal hypertension. Venous outflow obstruction due to cardiac failure or hepatic venous obstruction also cause a transudate, gradient > 11 g/L but, unlike in cirrhosis, the total protein is usually > 25 g/L. Exudative ascites (ascites protein concentration > 25 g/L or a SAAG < 11 g/L) raises the possibility of infection (tuberculosis), malignancy, hepatic venous obstruction, pancreatic or hypothyroidism.Ascites amylase activity above 1000 U/L identifies pancreatic ascites, and low ascites glucose concentrations suggest malignant disease or tuberculosis.
Cytological examination may reveal malignant cells
(one-third of cirrhotic patients with a bloody tap have a
hepatocellular carcinoma). Polymorphonuclear leucocyte
counts above 250 × 106/L strongly suggest infection
(spontaneous bacterial peritonitis). Laparoscopy can be valuable in detecting peritoneal disease.
Management
Successful treatment relieves discomfort but does not
prolong life; if over-vigorous, it can produce serious disorders of fluid and electrolyte balance and precipitate
hepatic encephalopathy .
Treatment of transudative ascites is based on restricting sodium and water intake, diuretics and, if necessary, removing ascites directly by paracentesis.
Exudative ascites due to malignancy is treated with
paracentesis. During management of ascites, the patient
should be weighed regularly. Diuretics should be titrated to remove no >1 L of fluid daily, so body weight should not fall by >1 kg daily to avoid excessive fluid depletion. Sodium and water restriction
Restriction of sodium intake to 100 mmol/day (‘no added salt diet’) is usually adequate. Drugs containing relatively large amounts of sodium, and those promoting sodium retention such as NSAIDs, must be avoided .
Restriction of water intake to 1.0–1.5 L/day is necessary only if the plasma sodium falls < 125 mmol/L.
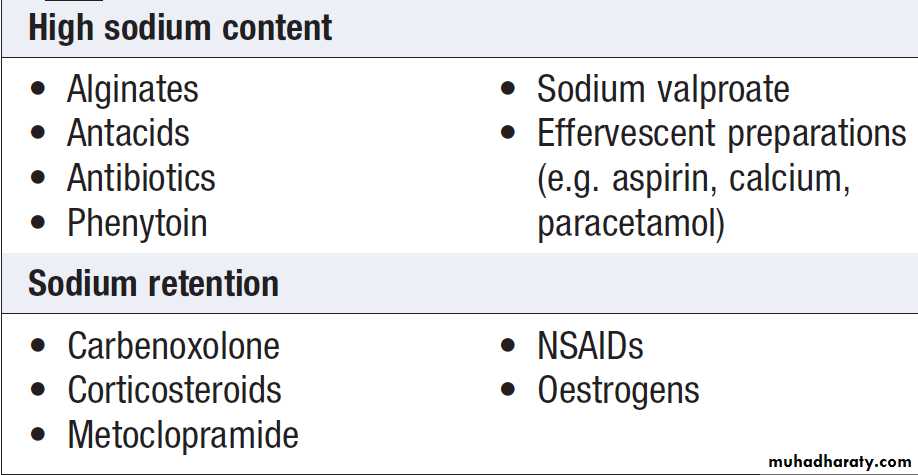
Some drugs containing relatively large amounts of sodium or causing sodium retention
Diuretics :Spironolactone (100–400 mg/day) is the first-line , it is a powerful aldosterone antagonist; it can cause painful gynaecomastia and hyperkalaemia, in which case amiloride (5–10 mg/day) can be substituted. Some also require loop diuretics. Patients who do not respond to 400 mg spironolactone and 160 mg furosemide, or unable to tolerate these doses due to hyponatraemia or renal impairment, are considered to have refractory or diuretic-resistant ascites and should be treated by other measures.Paracentesis : First-line treatment of refractory ascites is large-volume paracentesis. Paracentesis to dryness is safe, provided the circulation is supported with an IV colloid such as human albumin , 6–8 g per litre of ascites removed, Paracentesis can be used as an initial therapy or when other treatments fail.
Transjugular intrahepatic portosystemic stent shunt (TIPSS) can relieve resistant ascites but does not prolong life; it may be an option where the only alternative
1.is frequent, large-volume paracentesis.
2. in patients awaiting liver transplantation, but can aggravate encephalopathy in those with poor function.
Peritoneo-venous shunt
A long tube with a non-return valve running subcutaneously from the peritoneum to the internal jugular vein in the neck; it allows fluid to pass directly into the systemic circulation.
Complications, including infection, SVC thrombosis, pulmonary oedema, bleeding from oesophageal varices and DIC. limit its use.
Complications of ascites
1. Renal failure
It can be pre-renal due to vasodilatation from sepsis and/or diuretic therapy, or hepatorenal syndrome.
2. Hepatorenal syndrome
Occurs in 10% with advanced cirrhosis complicated by ascites. There are two clinical types; mediated by renal vasoconstriction.
Type 1 oliguria, a rapid rise of creatinine and a very poor prognosis .Treatment , albumin infusions with terlipressin. Haemodialysis should not be used routinely because it does not improve the outcome. Patients who survive should be considered for liver transplantation.
Type 2 occurs in patients with refractory ascites, moderate and stable increase in s creatinine, has a better prognosis.
3. Spontaneous bacterial peritonitis (SBP)
Present with abdominal pain, rebound tenderness, absent bowel sounds , fever, encephalopathy and fever. Diagnostic paracentesis may show cloudy fluid, and ascites neutrophil count above 250 × 106/L. The source of infection cannot usually be determined, but most organisms isolated are of enteric origin and Ecoli is most frequently found. Ascitic culture in blood culture gives the highest yield of organisms. SBP needs to be differentiated from other intra-abdominal emergencies. Treatment should be started immediately with broad spectrum antibiotics, such as cefotaxime or piperacillin/ tazobactam. Recurrence is common but reduced with prophylactic quinolones such as norfloxacin or ciprofloxacin .Prognosis
10–20% of patients survive 5 years from the first appearance of ascites due to cirrhosis. The prognosis is better when a treatable cause for the underlying cirrhosis is present or when a precipitating cause for ascites, such as excess salt intake, is found.
Antibiotics and spontaneous bacterial peritonitis (SBP)
Hepatic encephalopathyA neuropsychiatric syndrome caused by liver disease.
As it progresses, confusion is followed by coma. Confusion needs to be differentiated from delirium tremens and Wernicke’s encephalopathy, and coma from subdural haematoma, which can occur in alcoholics after a fall .
Features include changes of intellect,personality, drowsiness, disorientation, slurring of speech and eventually coma. Convulsions sometimes occur. Examination usually shows a flapping tremor (asterixis), inability to perform simple mental arithmetic tasks or to draw objects such as a star (constructional apraxia), and, as the condition progresses, hyper-reflexia and bilateral extensor plantar responses.
The degree can be graded from 1 to 4.
Hepatic encephalopathy rarely causes focal neurological signs; if these are present, other causes must be sought. Fetor hepaticus, a sweet musty odour to the breath, is usually present but is more a sign of portosystemic shunting than of encephalopathy. Rarely, chronic encephalopathy gives rise to variable combinations of cerebellar dysfunction, Parkinson, spastic paraplegia and dementia.
Differential diagnosis of hepatic encephalopathy
• Intracranial bleed (subdural, extradural haematoma,
• Drug or alcohol intoxication
• Delirium tremens/alcohol withdrawal
• Wernicke’s encephalopathy
• Primary psychiatric disorders
• Hypoglycaemia
• Neurological Wilson’s disease
• Post- ictal state
Pathophysiology of Hepatic encephalopathy
Hepatic encephalopathy is thought to be provoked by circulating neurotoxins that are normally metabolised by the liver. Accordingly, most affected patients have evidence of liver failure and portosystemic shunting of blood. The ‘neurotoxins’ causing encephalopathy are unknown, but they are thought to be mainly nitrogenous substances produced in the gut, at least in part by bacterial action. These substances are normally metabolised by the healthy liver and excluded from the systemic circulation. Ammonia considered an important factor. Recent interest has focused on γ- aminobutyric acid (GABA), amino acids, mercaptans and fatty acids that can act as neurotransmitters. Disruption of the blood–brain barrier may lead to cerebral oedema.Investigations
The diagnosis can usually be made clinically, electroencephalogram (EEG) shows diffuse slowing of the normal alpha waves with eventual development of delta waves. The arterial ammonia is usually increased, however, increased concentrations can occur in the absence of clinical encephalopathy, rendering this investigation of little diagnostic value.
Factors precipitating hepatic encephalopathy
Management of Hepatic encephalopathyThe principles are to treat or remove precipitating causes and to suppress the production of neurotoxins by bacteria in the bowel. Dietary protein restriction is rarely needed , no longer recommended because it is unpalatable and can lead to a worsening nutritional state in already malnourished. Lactulose (15–30 mL 3 times daily) is increased until the bowels are moving twice daily. It produces an osmotic laxative effect, reduces the pH, thereby limiting colonic ammonia absorption. Rifaximin (400 mg 3 times daily) non-absorbed antibiotic that acts by reducing the bacterial content of the bowel , shown to be effective. It can be used in addition, or as an alternative to lactulose if diarrhoea becomes troublesome. Chronic or refractory encephalopathy is an indications for liver transplantation.
CIRRHOSIS
Cirrhosis is characterised by diffuse hepatic fibrosis and
nodule formation. Worldwide, the most common causes are chronic viral hepatitis, prolonged excessive alcohol consumption and NAFLD. Cirrhosis is the most common cause of portal hypertension and its complications.
It may also occur in prolonged biliary damage or obstruction, as is found in primary biliary cirrhosis (PBC), primary sclerosing cholangitis and post-surgical biliary strictures. Persistent blockage of venous return from the liver, such as occurs in venoocclusive disease and Budd– Chiari syndrome, can also result in cirrhosis.
Variceal bleeding
Acute upper gastrointestinal haemorrhage from gastrooesophageal varices is common in chronic liver disease.Investigation and management are discussed below.
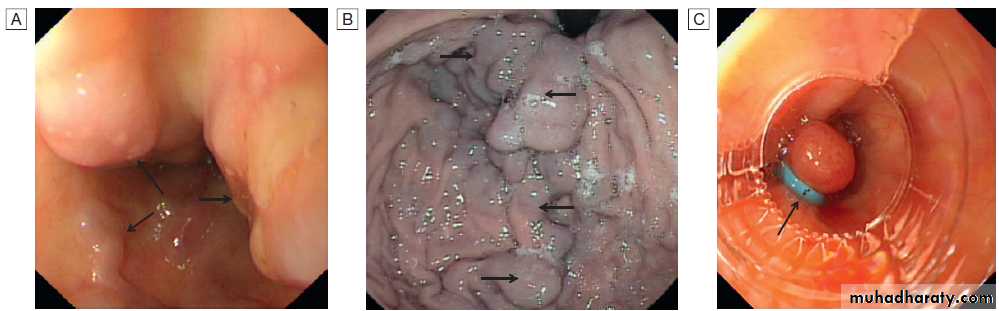
Varices: endoscopic views. A Oesophageal varices (arrows) at the lower end of the oesophagus. B Gastric varices (arrows). C Appearance of oesophageal varices following application of strangulating bands (band ligation, arrow).
Pathophysiology
Following liver injury, stellate cells in the space of Disseare activated by cytokines produced by Kupffer cells and hepatocytes. This transforms the stellate cell into a myofibroblast-like cell, capable of producing collagen, pro-inflammatory cytokines and other mediators that promote hepatocyte damage and fibrosis .
Cirrhosis is a histological diagnosis
classified histologically into:
• Micronodular cirrhosis, nodules about 1 mm in diameter and typically seen in alcoholic cirrhosis.
• Macronodular cirrhosis, larger nodules of various sizes.
Clinical features
Some patients are asymptomatic and the diagnosis is made incidentally at ultrasound or at surgery. Others present with isolated hepatomegaly, splenomegaly, signs of portal hypertension or hepatic insufficiency. When symptoms are present, they are often non-specific and include weakness, fatigue, muscle cramps, weight loss, anorexia, nausea, vomiting and upper abdominal discomfort . Cirrhosis will occasionally present because of shortness of breath due to a large right pleural effusion, or with hepatopulmonary syndrome .Hepatomegaly is common when the cirrhosis is due
to ALD or haemochromatosis.
A reduction in liver size is especially common if the cause
is viral hepatitis or autoimmune liver disease. The liver
is often hard, irregular and non-tender. Jaundice is mild
due primarily to a failure to excrete bilirubin. Palmar erythema. Spider telangiectasias. 1 to 2 mm in diameter, and are usually found only above the nipples. Florid spider telangiectasia, gynaecomastia and parotid enlargement are most common in alcoholic cirrhosis. Pigmentation in haemochromatosis and in cirrhosis with prolonged cholestasis. Pulmonary arteriovenous shunts also develop, leading to hypoxaemia and central cyanosis. Endocrine changes are noticed more readily in men, loss of male hair distribution and testicular atrophy. Gynaecomastia is common.
Easy bruising. Splenomegaly and collateral vessel formation are features of portal hypertension, in advanced disease . Ascites also signifies advanced disease.
Nonspecific features include clubbing. Dupuytren’s contracture is traditionally regarded as a complication of cirrhosis, but the evidence for this is weak. Chronic liver failure develops when the metabolic capacity of the liver is exceeded.It is characterised by the presence of encephalopathy and/or ascites. The term ‘hepatic decompensation’ or ‘decompensated liver disease’ is often used when chronic liver failure occurs.
Other features may be present these include peripheral oedema, renal failure, jaundice, and hypoalbuminaemia and coagulation abnormalities.

Causes of cirrhosis

Clinical features of hepatic cirrhosis
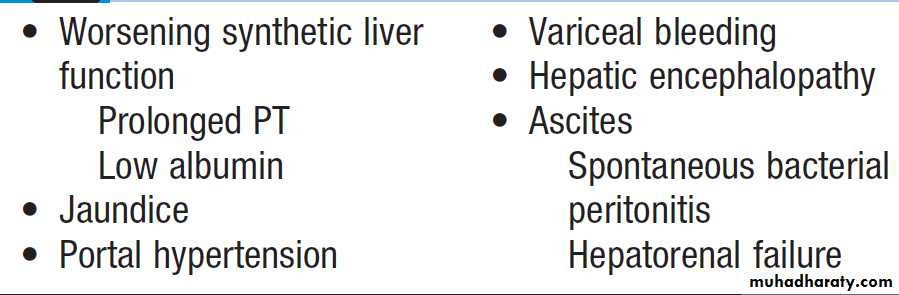
Features of chronic liver failure
Management of hepatic cirrhosis
This includes treatment of the cause, maintenance of nutrition and treatment of complications, endoscopy should be performed to screen for oesophageal varices and repeated every 2 years. As cirrhosis is associated with an increased risk of hepatocellular carcinoma (HCC), patients should be placed under regular surveillance.Chronic liver failure due to cirrhosis can also be treated by liver transplantation.
Prognosis
The overall prognosis is poor. 25% of patients survive
5 years from diagnosis but, where liver function is good, 50% survive for 5 years and 25% for up to 10 years.
The prognosis is more favourable when the underlying cause of the cirrhosis can be corrected, as in alcohol misuse, haemochromatosis or Wilson’s disease.
Deteriorating liver function, indicates a poor prognosis unless a treatable cause such as infection is found.
Increasing bilirubin, falling albumin, hyponatraemia
(< 120 mmol/L) not due to diuretic therapy, and a prolonged PT are all bad prognostic features .
The Child–Pugh score and, more recently, the MELD (Model for End-stage Liver Disease) score can be used to assess prognosis.
Although these scores give a guide to prognosis, the course of cirrhosis can be unpredictable, as complications such as variceal bleeding may occur.
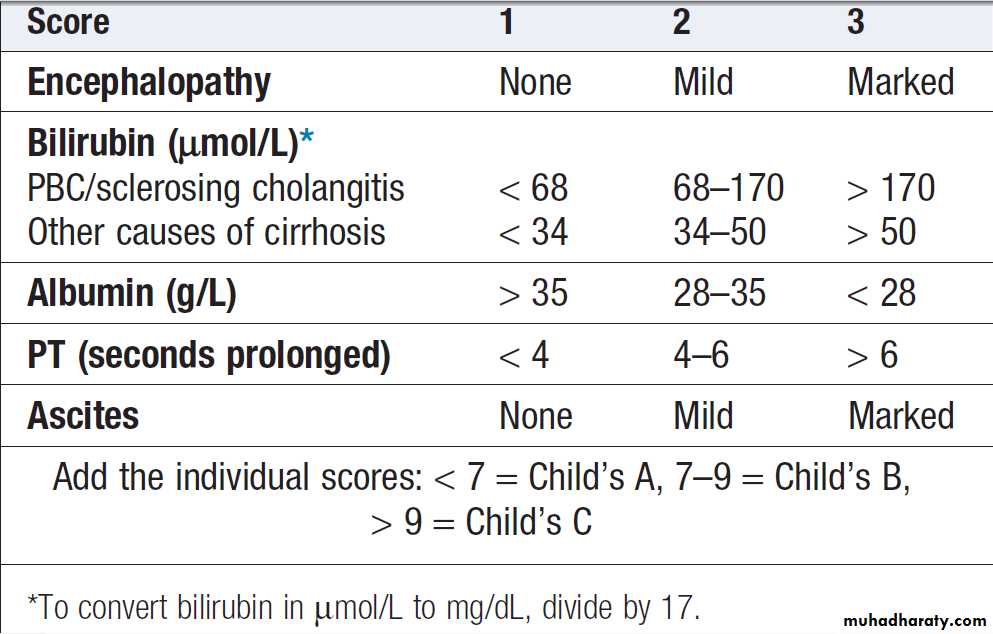
Child–Pugh classification of prognosis in cirrhosis
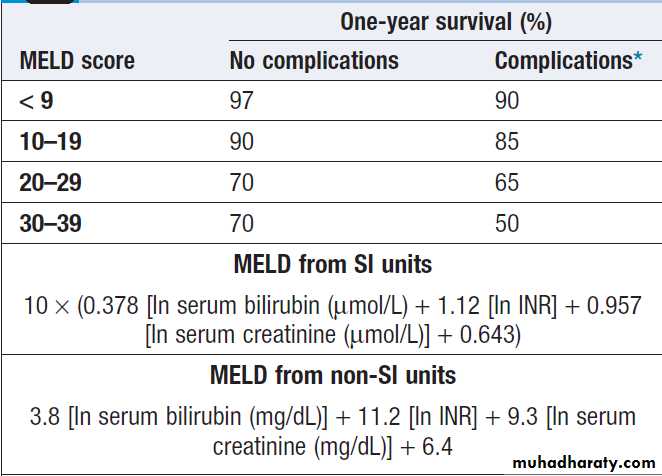
One-year survival rate depending on MELD score
PORTAL HYPERTENSIONThis frequently complicates cirrhosis. The normal hepatic venous pressure gradient (difference between the wedged hepatic venous pressure and free hepatic venous pressure) is 5–6 mm Hg. Clinically significant portal hypertension is present when the gradient exceeds 10 mm Hg and risk of variceal bleeding increases beyond a gradient of 12 mm Hg. Causes are classified in accordance with the main sites
of obstruction. Extrahepatic portal vein obstruction is the usual source of portal hypertension in childhood and adolescence, while cirrhosis causes at least 90% of cases in adults in developed countries. Schistosomiasis is the most common cause of portal hypertension worldwide but is infrequent outside endemic areas .
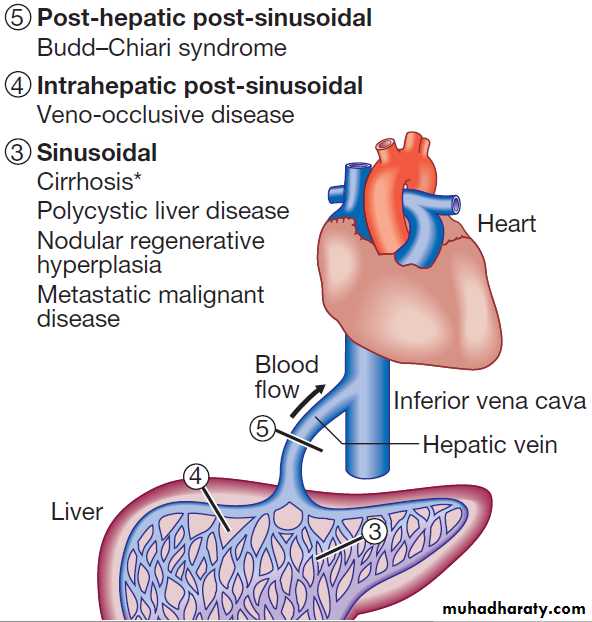
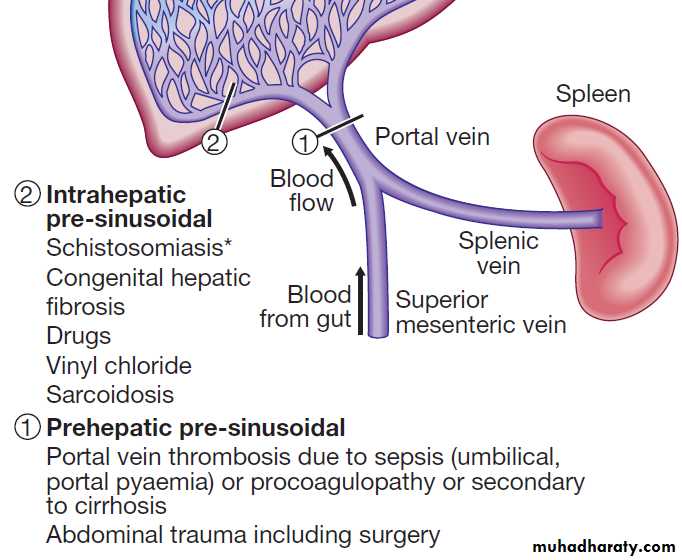
Classification of portal hypertension according to site of vascular obstruction.
*Most common cause. Note that splenic vein occlusion can also follow pancreatitis, leading to gastric varices.Clinical features
The clinical features result principally from portalvenous congestion and collateral. Splenomegaly is a cardinal finding, and a diagnosis of portal hypertension is unusual when splenomegaly cannot be detected clinically or by US. The spleen is rarely enlarged >5 cm below the left costal margin. Collateral vessels may be visible on the anterior abdominal wall and occasionally several radiate from the umbilicus to form a caput medusae. Rarely, a large umbilical collateral vessel has a blood flow sufficient to give a venous hum on auscultation (Cruveilhier– Baumgarten syndrome). The most important collateral occurs in the oesophagus and stomach, and this can be a source of severe bleeding.
Rectal varices also cause bleeding and are often mistaken for haemorrhoids . Fetor hepaticus results from portosystemic shunting of mercaptans to the lungs.
Ascites occurs as a result of renal sodium retention
and portal hypertension .The most important consequence of portal hypertension is variceal bleeding, which commonly arises from oesophageal varices located within 3–5 cm of the gastrooesophageal junction, or from gastric varices. The size of the varices, endoscopic variceal features such as red spots and stripes, high portal pressure and liver failure are all general factors that predispose to bleeding. Drugs capable of causing mucosal erosion, such as salicylates and NSAIDs, can also precipitate bleeding.

Complications of portal hypertension
PathophysiologyIncreased portal vascular resistance leads to a gradual
reduction in the flow of portal blood to the liver and
simultaneously to the development of collateral vessels,
allowing portal blood to bypass the liver and enter the
systemic circulation directly. Portosystemic shunting
occurs, particularly in the GIT especially the distal oesophagus, stomach and rectum, in the anterior abdominal wall, and in the renal, lumbar, ovarian and testicular vasculature. Stomal varices can occur at the site of an ileostomy. As collateral vessel formation progresses, > half of the portal blood flow may be shunted directly to the systemic circulation. Increased portal flow contributes to portal hypertension.
Investigations
The diagnosis is often made clinically. Portal venous pressure measurements are rarely needed for clinical assessment or routine management, but can be used to confirm portal hypertension and to differentiate Sinusoidal and pre-sinusoidal forms.Pressure measurements are made by using a balloon catheter inserted using the transjugular route (via the inferior vena cava into a hepatic vein and then hepatic venule) to measure the wedged hepatic venous pressure (WHVP).
This is an indirect measurement of portal vein pressure.
Thrombocytopenia is common (hypersplenism), usually in the region of 100 × 109/L. Leucopenia occurs occasionally but anaemia is seldom attributed to hypersplenism; if anaemia is found, a source of bleeding should be sought. The most useful investigation is endoscopy to determine whether varices are present. Ultrasonography often shows features of portal hypertension, such as splenomegaly and collateral, and can sometimes indicate the cause, such as liver disease or portal vein thrombosis.
CT and MRI angiography can identify the extent of portal vein clot and are used to identify hepatic vein patency.
Management
Acute upper gastrointestinal haemorrhage from gastrooesophageal varices is a common manifestation ofchronic liver disease. In the presence of portal hypertension,
the risk of a variceal bleed occurring within 2 years
varies from 7% for small varices up to 30% for large
varices. The mortality following a variceal bleed has
improved to around 15% overall but is still about 45%
in those with poor liver function (i.e. Child–Pugh C). The management of portal hypertension is largely focused on the prevention and/or control of variceal haemorrhage. However, it should be remembered that bleeding can also result from peptic ulceration, which is more common in patients with liver disease than in the general population.
Primary prevention of variceal bleeding
If non-bleeding varices are identified at endoscopy,β-blocker , propranolol at doses that reduce the heart rate by 25% has been shown to be effective in the primary prevention of variceal bleeding (80–160 mg/day) or nadolol is effective in reducing portal venous pressure. The efficacy of β-blockers is similar to that of prophylactic banding, which may also be considered, particularly in patients that are unable to tolerate β-blocker therapy. Carvedilol, a non- cardioselective vasodilating β-blocker, is also effective.
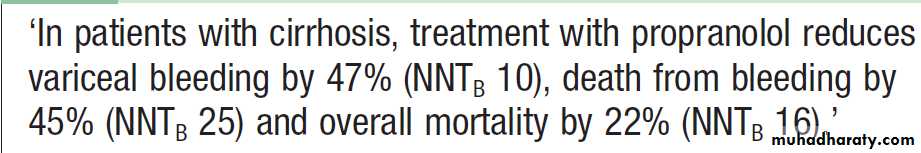
Management of acute variceal bleeding
The priority in acute bleeding is to restore the circulationwith blood and plasma, because shock reduces liver blood flow and causes further deterioration of. The source of bleeding should always be confirmed by endoscopy, because about 20% of bleeding from non- variceal lesions. All patients should receive prophylactic broad-spectrum antibiotics, such as oral ciprofloxacin or intravenous cephalosporin,because sepsis is common and antibiotics has been shown to improve outcomes. The
measures used to control bleeding include vasoactive medications (e.g. terlipressin), endoscopic therapy (banding or sclerotherapy), balloon tamponade, transjugular intrahepatic portosystemic stent
shunting (TIPSS) and, rarely, oesophageal transection.
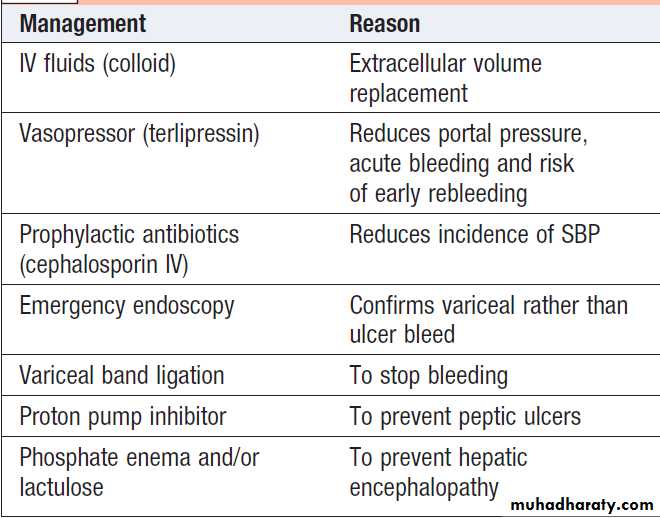
Emergency management of bleeding
Pharmacological reduction of portal venous pressure.Terlipressin is a synthetic vasopressin analogue that, in contrast to vasopressin, can be given by intermittent injection rather than continuous infusion, reduces portal blood flow and/or intrahepatic resistance and hence reduces portal pressure. It reduces mortality in the setting of acute variceal bleeding. The dose is 2 mg IV 4 times daily until bleeding stops, and then 1 mg 4 times daily for up to 72 hours.
Caution is needed in patients with severe ischaemic heart disease or peripheral vascular disease because of the drug’s vasoconstrictor properties.
Banding ligation and sclerotherapy. This is the most
widely used initial treatment. It stops variceal bleeding in 80% of patients and can be repeated if bleeding recurs.
Band ligation involves the varices being sucked into a cap placed, allowing them to be occluded with a tight rubber band. The occluded varix subsequently sloughs with variceal obliteration. Banding is repeated every 4–6 weeks until the varices are obliterated. Regular follow-up endoscopy is required to identify and treat any recurrence. Band ligation has fewer side effects than, and has largely replaced, sclerotherapy, a technique in which varices are injected with a sclerosing agent. Banding is associated with a lower risk of oesophageal perforation or stricturing.
Prophylactic acid suppression with PPI reduces the risk of secondary bleeding from banding-induced ulceration.
Protection of the patient’s airway with endotracheal
intubation may aid the endoscopist, facilitating therapy, and significantly reduce the risk of pulmonary aspiration.
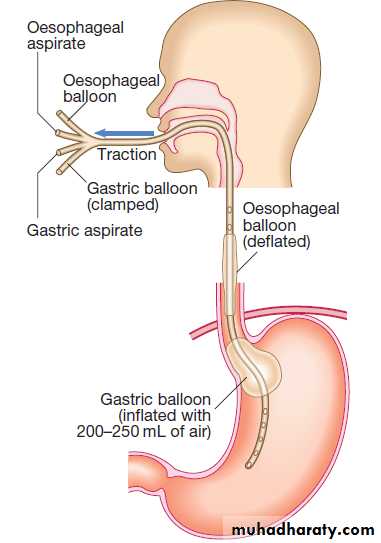
Sengstaken–Blakemore tube.
Balloon tamponade.Sengstaken–Blakemore tube possessing two balloons that exert pressure in the fundus of the stomach and in the lower oesophagus respectively . Additional lumens allow material to be aspirated from the stomach and from the oesophagus above the oesophageal balloon. This technique may be used in the event of life-threatening haemorrhage if early endoscopic therapy is not available or is unsuccessful.
The tube should be passed through the mouth and its presence in the stomach should be checked by auscultating the upper abdomen while injecting air and by radiology.
The safest technique is to inflate the balloon in the
stomach under direct endoscopic vision. Gentle traction is essential to maintain pressure on the varices. Initially, only the gastric balloon should be inflated, with 200– 250 mL of air, as this will usually control bleeding. Inflation of the gastric balloon must be stopped if the patient experiences pain because inadvertent inflation in the oesophagus can cause oesophageal rupture.
If the oesophageal balloon needs to be used because of continued bleeding, it should be deflated for about 10 minutes every 3 hours to avoid oesophageal mucosal damage. Pressure in the oesophageal balloon should be monitored with a sphygmomanometer, and should not exceed 40 mm Hg. Balloon tamponade will almost always stop variceal bleeding, but is only a bridge to more definitive therapy. Self-expanding removable oesophageal stents are a new alternative in patients with bleeding oesophageal, but not gastric, varices.
TIPSS. This technique uses a stent placed between the
portal vein and the hepatic vein within the liver to
provide a portosystemic shunt and portal pressure
It is carried out under radiological control via the internal jugular vein; prior patency of the portal vein must be determined angiographically, coagulation deficiencies may require correction with fresh frozen plasma, and antibiotic cover is provided. Successful shunt placement stops and prevents variceal bleeding.
Hepatic encephalopathy may occur following TIPSS and is managed by reducing the shunt diameter. Although associated with less rebleeding than endoscopic therapy, survival is not improved .

Portosystemic shunt surgery. prevents recurrent bleeding, carries high mortality and often leads to encephalopathy, now reserved for patients in whom other treatments have not been successful and those with good liver function.
Oesophageal transection. Rarely performed as a last resort when bleeding cannot be controlled by other means, mortality is high.
Secondary prevention of variceal bleeding
Beta-blockers are used as a secondary measure to
prevent recurrent variceal bleeding. Following successful
treatment by endoscopic therapy, patients should be
entered into an oesophageal banding programme with
repeated sessions of therapy at 1- to 2-week intervals
until the varices are obliterated. In selected individuals,
TIPSS may also be considered in this setting.
Congestive gastropathy
Long-standing portal hypertension causes chronic
gastric congestion recognisable at endoscopy as multiple
areas of punctate erythema (‘snake skin gastropathy’).
Rarely, similar lesions occur more distally in the gastrointestinal tract. These areas may become eroded,
causing bleeding from multiple sites. Acute bleeding
can occur but repeated minor bleeding causing iron deficiency anaemia is more common. Anaemia may be
prevented by oral iron supplements but repeated blood
transfusions can become necessary. Reduction of the
portal pressure using propranolol (80–160 mg/day) is
the best initial treatment. If this is ineffective, a TIPSS
procedure can be undertaken.
INFECTIONS AND THE LIVER
Viral hepatitisAll these viruses cause illnesses with similar clinical and pathological features and which are frequently anicteric
or even asymptomatic. They differ in their tendency
to cause acute and chronic infections. A non-specific prodromal illness characterised by headache, myalgia, arthralgia, nausea and abdominal discomfort usually precedes the development of jaundice by a few days to 2 weeks. Dark urine and pale stools may precede jaundice. The liver is often tender but only minimally enlarged. Occasionally, mild splenomegaly and cervical lymphadenopathy are seen. Symptoms rarely last longer than 3–6 weeks. Complications may occur but are rare.
Investigations
A hepatitic pattern of LFTs develops, with serum transaminases typically between 200 and 2000 U/L in an acute infection .The plasma bilirubin reflects the degree of liver damage. The ALP rarely exceeds twice the upperlimit of normal. Prolongation of the PT indicates the
severity of the hepatitis but rarely exceeds 25 seconds,
except in rare cases of acute liver failure. The white cell
count is usually normal with a relative lymphocytosis.
Serological tests confirm the aetiology of the infection.
Management
Most individuals do not need hospital care. Alcohol, sedatives and narcotics, which are metabolised in the
liver, should be avoided. No specific dietary modifications
are needed. Elective surgery should be avoided.
Causes of viral hepatitis

Complications of acute viral hepatitis
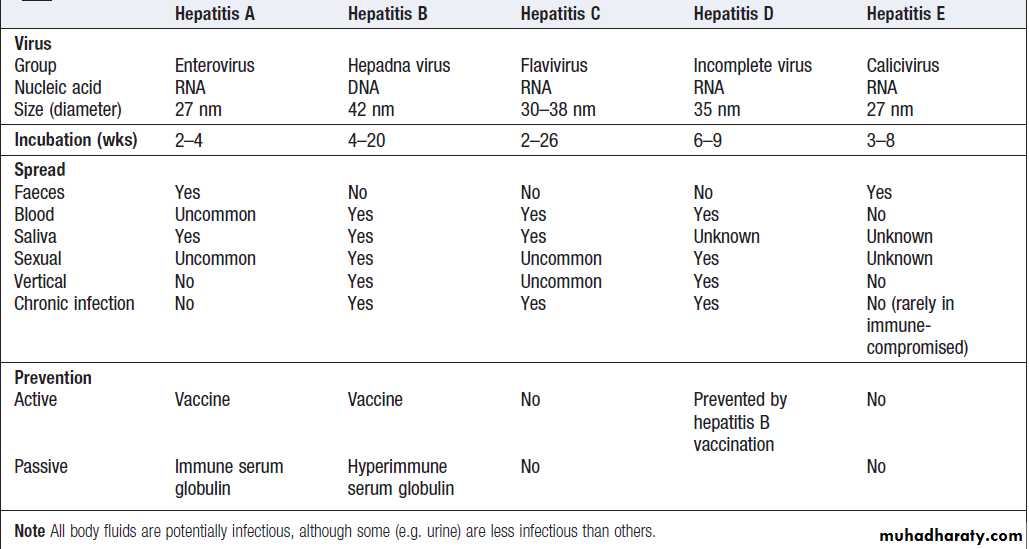
Features of the main hepatitis viruses
Hepatitis AHAV enterovirus is highly infectious , spread by the faecal–oral route. Infected, excrete the virus in faeces for about 2–3 weeks before the onset and further 2 weeks or so. 30% of adults will have serological evidence of past infection but give no history of jaundice. chronic carrier state does not occur. Acute liver failure is rare (0.1%) .In adults, a cholestatic phase may complicate infection
Investigations
One HAV antigen has been found , anti-HAV is important in diagnosis, as HAV present in the blood transiently during the incubation period. Excretion in the stools occurs for 7–14 days virus cannot be grown readily. Anti-HAV of the IgM and is diagnostic of an acute infection. Titres fall to low levels within about 3 months of recovery.
Anti-HAV of the IgG type is of no diagnostic value, persists for years , used as a marker of previous HAV infection. Its presence indicates immunity to HAV.
Management :
There is no role for antiviral drugs . Infection is best prevented by improving social conditions, especially overcrowding and poor sanitation. Active immunisation with an inactivated virus vaccine, should be considered for individuals with chronic hepatitis B or C, close contacts, the elderly, those with other major disease, People travelling to endemic areas and perhaps pregnant women. Immune serum globulin (Anti-HAV) give immediate protection if given soon after exposure to the virus. Can be effective in an outbreak of hepatitis, in a school or nursery, as injection of those at risk prevents secondary spread to families.
Hepatitis B
Consists of a core containing DNA and a DNA polymerase enzyme needed for virus replication. Humans are the only source of infection. Hepatitis B is one of the most common causes of chronic liver disease and hepatocellular carcinoma worldwide. Approximately one-third of the world’s population have serological evidence of past or current infection. Many individuals with acute and chronic hepatitis B asymptomatic. The risk of progression to chronic disease depends on the source and timing of infection. Vertical transmission from mother to child in the perinatal period is the most common cause of infection and carries the highest risk of ongoing chronic infection .
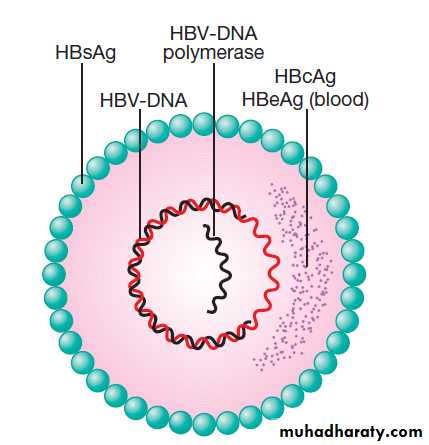
Schematic diagram of hepatitis B virus. Hepatitis B
surface antigen (HBsAg) is a protein that makes up part of the viral envelope. Hepatitis B core antigen (HBcAg) is a protein that makes upthe capsid or core part of the virus (found in the liver but not in blood).
Hepatitis B e antigen (HBeAg) is part of the HBcAg that can be found in the blood and indicates infectivity.

Source of hepatitis B infection and risk of chronic infection
Chronic hepatitis can lead to cirrhosis or HCC, usually after decades of infection . It should be remembered that the virus is not directly cytotoxic to cells; rather it is an immune response to viral antigens displayed on infected hepatocytes that initiates liver injury.Investigations
Hepatitis B surface antigen (HBsAg) is an indicator of active infection. In acute liver failure from hepatitis B, the liver damage is mediated by viral clearance and so HBsAg is negative, with evidence of recent infection shown by the presence of hepatitis B core IgM.
HBsAg appears in the blood late in the incubation period, usually lasts for 3–4 weeks and persist for up to 5 months.
The persistence >6 months indicates chronic infection.
Antibody to HBsAg (anti-HBs) usually appears after 3–6 months and persists for many years or permanently.
Anti-HBs implies either a previous infection, in which case anti-HBc is usually also present, or previous vaccination, in which case anti-HBc is not present.
Hepatitis B core antigen (HBcAg). HBcAg is not found
in the blood, but antibody to it (anti- HBc) appears early in the illness and rapidly reaches a high titre, which
subsides gradually but then persists. Anti-HBc is initially
of IgM type, with IgG antibody appearing later.
Anti-HBc (IgM) can sometimes reveal an acute HBV
infection when the HBsAg has disappeared and before
anti-HBs has developed .
Hepatitis B e antigen (HBeAg). HBeAg is an indicator
of viral replication, it may appear only transiently at the outset followed by the production of anti-HBe. The HBeAg reflects active replication of the virus in the liver.Chronic HBV infection is marked by the presence of HBsAg and anti-HBc (IgG) in the blood.
Viral load and genotype
HBV-DNA can be measured by PCR in the blood. Viral
loads are usually >105 copies/mL in the presence of active viral replication, as indicated by the presence of e antigen. In contrast, in those with low viral replication, HBsAg- and anti-HBe-positive, viral loads are less than 105 copies/ mL.
The exception is in patients who have a mutation in the pre-core protein, which means they cannot secrete e antigen into serum. Such individuals will be anti-HBe-positive but have a high viral load and often evidence of chronic hepatitis.
They respond differently to antiviral drugs from those with
classical e antigen-positive chronic hepatitis.
Measurement of viral load is important in monitoring
antiviral therapy and identifying patients with pre-core mutants. Specific HBV genotypes (A–H) can also be identified using PCR.
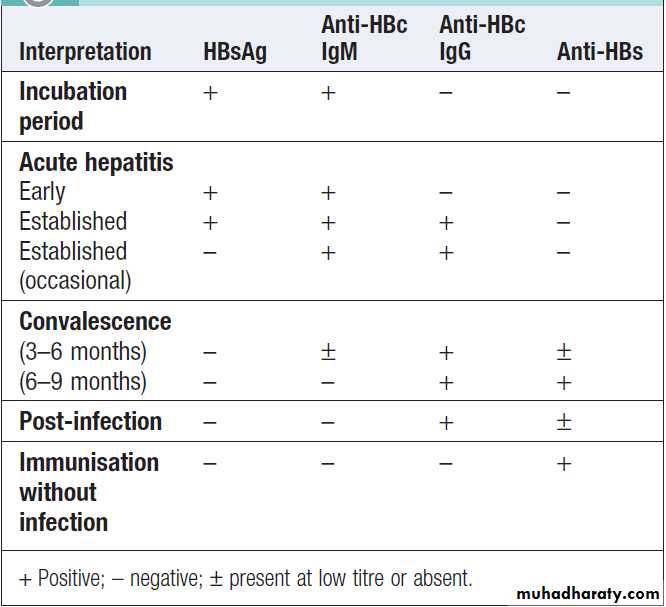
How to interpret the serological tests of
acute hepatitis B virus infectionManagement of acute hepatitis B
Treatment is supportive, monitoring for acute liver failure, which occurs in <1%. There is no definitive evidence that antiviral therapy reduces the severity or duration. Full recovery occurs in 90–95% of adults . 5–10% develop a chronic infection that usually continues for life, although later recovery occasionally occurs. Infection passing from mother to child at birth leads to chronic infection in the child in 90% of cases and recovery is rare. Chronic infection is also common in immunodeficient. Recovery from acute infection occurs within 6 months. Persistence of HBeAg beyond this time indicates chronic infection.Combined HBV and HDV causes more aggressive disease.
Management of chronic hepatitis B
Treatments are still limited, as no drug is consistentlyable to eradicate hepatitis B infection completely (i.e.
render the patient HBsAg-negative).
The goals of treatment are HBeAg seroconversion, reduction in HBV-DNA and normalisation of the LFTs.
The indication for treatment
High viral load in the presence of active hepatitis(elevated transaminases and/or histological evidence)
The oral antiviral agents are more effective in reducing
viral loads in patients with e antigen-negative than in those with e antigen-positive chronic hepatitis B, as the pre-treatment viral loads are lower.
Most patients with chronic hepatitis B are asymptomatic
and develop complications such as cirrhosis andhepatocellular carcinoma only after many years.
Cirrhosis develops in 15–20% of chronic HBV over 5–20 years. This proportion is higher in e antigen-positive.
Two different types of drug are used :
direct-acting nucleoside/nucleotide analogues and
pegylated interferon-alfa.
Direct-acting oral nucleoside/nucleotide are the mainstay of therapy. These act by inhibiting the reverse transcription of pre-genomic RNA to HBV-DNA by HBV-DNA polymerase , relapse is common if treatment is withdrawn.
Entecavir and tenofovir are potent with a high barrier to genetic resistance, and so are first-line agents.
Antiviral resistance mutations occur in only1–2% after 3 years of entecavir drug exposure. Both drugs have anti-HIV and so their use as monotherapy is contraindicated in HIV-positive, as it may lead to HIV antiviral resistance.
Lamivudine. Although effective, long-term therapy is
often complicated by the development of HBV-DNA
polymerase mutants characterised by a rise in viral load during treatment. Telbivudine is more potent but is also susceptible to viral resistance. Adefovir is associated with development of HBV-DNA mutants lower than lamivudine.

Entecavir and telbivudine in chronic hepatitis B infection

Tenofovir for chronic hepatitis B
Interferon-alfaThis is most effective in patients with a low viral load ,
in whom it acts by augmenting a native immune response. In HBeAg-positive chronic hepatitis, 33% lose e antigen after 4–6 months of treatment, compared to 12% of controls. Response rates are lower in HBeAg-negative chronic hepatitis, even with longer courses of treatment.
Interferon is contraindicated in the presence of cirrhosis, as it may cause a rise in serum transaminases and precipitate liver failure.Longer-acting pegylated interferons that can be given once weekly have been evaluated.
Side-effects are common , include fatigue, depression, irritability, bone marrow suppression and the triggering of autoimmune thyroid disease.
Liver transplantation
Historically, was contraindicated in hepatitis B because infection often recurred in the graft. However, the use of post-liver transplant prophylaxis with direct-acting antiviral agents and hepatitis B immunoglobulins has reduced the re-infection to 10% and increased 5-year survival to 80%, making transplantation an acceptable treatment option.
Pegylated interferons in chronic hepatitis B infection
Prevention
Individuals are most infectious when markers of continuingviral replication, such as HBeAg, and high levels
of HBV-DNA are present in the blood. HBV-DNA can
be found in saliva, urine, semen and vaginal secretions.
The virus is about ten times more infectious than hepatitis C, which in turn is about ten times more infectious than HIV.
A recombinant hepatitis B vaccine containing HBsAg
is available (Engerix) and is capable of producing active
immunisation in 95% of normal individuals.
The vaccine should be offered to those at special risk of infection who are not already immune, as evidenced by anti-HBs in the blood .
The vaccine is ineffective in those already infected by HBV.
Infection can also be prevented or minimised by the intramuscular injection of hyperimmune serum globulin prepared from blood containing anti-HBs. This should be given within 24 hours, or at most a week, of exposure to infected blood in circumstances likely to cause infection (e.g. needlestick injury, contamination of cuts or mucous membranes). Vaccine can be given together with hyperimmune globulin (active–passive immunisation).Neonates born to hepatitis B-infected mothers should
be immunised at birth and given immunoglobulin. Hepatitis B serology should then be checked at 12 months of age.

At-risk groups meriting hepatitis B
vaccination in low-endemic areas
Hepatitis D (Delta virus)
RNA-defective virus that has no independent existence; it requires HBV for replication and has the same sources and modes of spread. It can simultaneously infect individuals with HBV, or can superinfect those who are already chronic carriers of HBV. Simultaneous infections give rise to acute hepatitis, which is often severe but is limited by recovery from the HBV infection. Chronic infection with HBV and HDV can also occur, and this frequently causes rapidly progressive chronic hepatitis and cirrhosis. HDV has a worldwide distribution, transmission is mainly by close contact and occasionally by vertical transmission. In non-endemic areas, transmission is mainly a consequence of parenteral drug misuse.Investigations
HDV contains a single antigen to which infected individuals make an antibody (anti-HDV). Delta antigen appears in the blood only transiently, and in practice diagnosis depends on detecting anti-HDV. Simultaneous infection with HBV and HDV followed by full recovery is associated with the appearance of low titres of anti-HDV of IgM type within a few days of the onset of the illness. This antibody generally disappears within 2 months but persists in a few patients. Superinfection of patients with chronic HBV infection leads to the production of high titres of anti-HDV, initially IgM and later IgG. Such patients may then develop chronic infection with both viruses.Effective management of hepatitis B prevents hepatitis D.
Hepatitis C
This is caused by an RNA flavivirus. Acute symptomatic infection is rare. Most individuals identified when they develop chronic liver disease. 80% of individuals exposed to the virus become chronically infected and late spontaneous viral clearance is rare. Hepatitis C infection is usually identified in asymptomatic screened because they have risk factors for infection, such as previous injecting drug use , or have incidentally abnormal liver blood tests. InvestigationsSerology and virology
The HCV protein contains several antigens that give rise
to antibodies in an infected person. Active infection is confirmed by the presence of serum hepatitis C RNA.
Anti-HCV antibodies persist in serum even after viral clearance, whether spontaneous or post-treatment.
Molecular analysis
There are six common viral genotypes. Genotype 1 is less easy to eradicate than types 2 and 3 with traditional pegylated interferon alfa/ ribavirin-based treatments.
Liver function tests
Jaundice is rare , LFTs may be normal or show fluctuating serum transaminases between 50 and 200 U/L. and only usually appears in end-stage cirrhosis.
Liver histology
biopsy is often required to stage the degree of liver
damage. The degree of inflammation and fibrosis can be scored histologically. Metavir system, which scores fibrosis from 1 to 4, the latter equating to cirrhosis.

Risk factors for the acquisition of chronic
hepatitis C infectionManagement
The aim of treatment is to eradicate infection. Untilrecently, the treatment of choice was dual therapy with
pegylated interferon- alfa given as a weekly subcutaneous injection, together with oral ribavirin, a synthetic nucleotide analogue .The main side-effects of ribavirin are haemolytic anaemia and teratogenicity.
Side-effects of interferon are significant and include
flu-like symptoms, irritability and depression.
Virological relapse can occur in the first 3 months after stopping treatment, and cure is defined as loss of virus from serum 6 months after completing therapy (sustained virological response, or SVR).
The length of treatment and efficacy depend on viral genotype (12 months’ treatment for genotype
1 results in a 40% SVR, whereas 6 months’ treatment
for genotype 2 or 3 leads to an SVR in > 70%). Response to treatment is better in patients who have an early virological response, as defined by negativity of HCVRNA in serum 1 month after starting therapy.
The recent availability of triple therapy with the
addition of protease inhibitors such as telaprevir and
boceprevir to standard pegylated interferon/ribavirin
has provided a significant advance in therapy, with SVR
rates for genotype 1 individuals comparable to those
previously only achieved in genotypes 2 and 3.
Liver transplantation should be considered when
complications of cirrhosis occur, such as diuretic resistant
ascites. Unfortunately, hepatitis C almost always recurs in the transplanted liver and up to 15% will develop cirrhosis in the liver graft within 5 years of transplantation.
There is no active or passive protection against HCV.
Progression from chronic hepatitis to cirrhosis, 20% occurs
over 20–40 years. Risk factors for progression include
male gender, immunosuppression , prothrombotic states and heavy alcohol misuse.
Once cirrhosis is present, 2–5% per year will develop primary hepatocellular carcinoma. Once complications like ascites have arisen, the 5-year survival is around 50%.

Treatment of hepatitis C
Hepatitis EHepatitis E is caused by an RNA virus. The clinical presentation and management of hepatitis E are similar to that of hepatitis A. Disease is spread via the faecal–oral route; in most cases, it presents as a self-limiting acute hepatitis and does not usually cause chronic liver disease, although some cases have been described, usually in immunocompromised patients.
Hepatitis E differs from hepatitis A in that infection
during pregnancy is associated with the development of acute liver failure, which has a high mortality. In acute
infection, IgM antibodies to HEV are positive.
Other forms of viral hepatitis
Non-A, non-B, non-C (NANBNC) or non-A–E hepatitis .Other viruses that affect the liver, Cytomegalovirus and EBV infection. Herpes simplex is a rare cause in adults, and most of these patients are immunocompromised.
Abnormal LFTs are also common in chickenpox, measles, rubella and acute HIV infection.
Liver abscess
Classified as pyogenic, hydatid or amoebic.
Pyogenic liver abscess
Uncommon but important because they are potentially curable, carry significant morbidity and mortality if untreated, and are easily overlooked. The mortality of liver abscesses is 20–40%.
Causes of pyogenic liver abscesses
PathophysiologyMost common in older patients and usually result from ascending infection due to biliary obstruction (cholangitis) or contiguous spread from an empyema of the gallbladder. They can also complicate dental sepsis. Abscesses complicating suppurative appendicitis now rare. Immunocompromised are particularly likely to develop liver abscesses. Single lesions are more common in the right liver; multiple abscesses are usually due to infection secondary to biliary obstruction. E coli and various streptococci, are the most common organisms; anaerobes, including streptococci and Bacteroides, can often be found when infection has been transmitted from large bowel pathology via the portal vein, and multiple organisms are present in one-third of patients.
Clinical features
Patients are generally ill with fever, and sometimesrigors and weight loss. Abdominal pain is the most
common symptom and is usually in the right upper
quadrant, sometimes with radiation to the right shoulder.
The pain may be pleuritic in nature. Tender
hepatomegaly is found in more than 50% of patients.
Mild jaundice may be present, becoming severe if large
abscesses cause biliary obstruction. Atypical presentations are common and explain the frequency with which the diagnosis is made only at postmortem. This is a particular problem in patients with gradually developing illnesses or pyrexia of unknown origin without localising features.
Investigations
Liver imaging is the most revealing investigation and
shows 90% or more of symptomatic abscesses. aspiration under ultrasound guidance confirms the diagnosis and provides pus for culture. A leucocytosis is frequently found, plasma ALP activity is usually increased, and the serum albumin is often low.
The chest X-ray may show a raised right diaphragm and lung collapse, or an effusion at the base of the right lung. Blood cultures are positive in 50–80%. Abscesses caused by gut-derived organisms require active exclusion of significant colonic pathology, such as a colonoscopy to
exclude colorectal carcinoma.
Management
This includes prolonged antibiotic therapy and drainageof the abscess. Associated biliary obstruction and cholangitis require biliary drainage (endoscopically).
Pending the results of culture of blood and pus
from the abscess, treatment should be commenced with
a combination of antibiotics such as ampicillin, gentamicin and metronidazole. Aspiration or drainage with a catheter placed in the abscess under ultrasound guidance is required if the abscess is large or if it does not respond to antibiotics. Surgical drainage is rarely undertaken, although hepatic resection may be indicated for a chronic persistent abscess or ‘pseudotumour’.
Hydatid cysts
Caused by Echinococcus granulosus infection .They have an outer layer derived from the host, an intermediate laminated layer and an inner germinal layer. They can be single or multiple. Chronic cysts become calcified. The cysts may be asymptomatic but may present with abdominal pain or a mass. Peripheral blood eosinophilia is present in 20% of cases, whilst X-rays may show calcification of the rim of the abscess. CT reliably shows the cyst(s), and Echinococcus enzyme-linked immunosorbent assay (ELISA) has 90% sensitivity for hepatic hydatid cysts.Rupture or secondary infection of cysts can occur, and a
communication with the intrahepatic biliary tree canthen result, with associated biliary obstruction.
All patients should be treated medically with albendazole or mebendazole prior to definitive therapy. In the absence of communications with the biliary tree, treatment consists of percutaneous aspiration of the cyst followed by the injection of 100% ethanol into the cysts and then re-aspiration of the cyst contents (PAIR).
Where there is communication between the cyst and biliary system, surgical removal of the intact cyst is the preferred treatment.
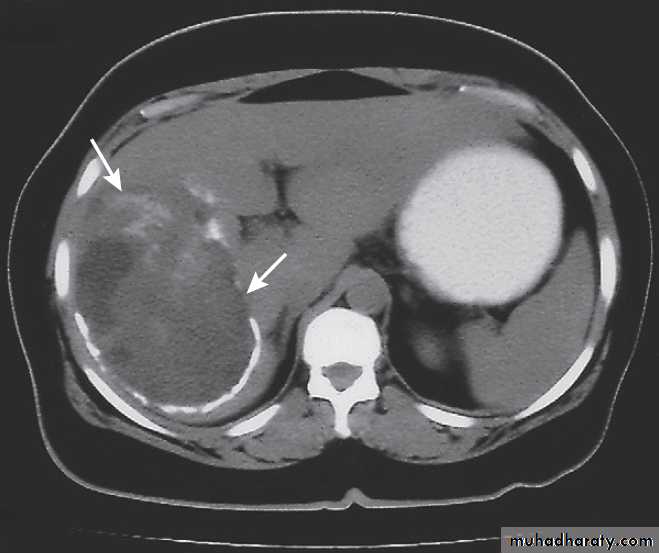
Hydatid cyst of the liver on CT (arrows).
Amoebic liver abscessesCaused by Entamoeba histolytica infection .Up to 50% of cases do not have a previous history of intestinal disease. Abscesses are usually large, single and located in the right lobe; multiple abscesses may occur in advanced disease. Fever and abdominal pain or swelling are the most common symptoms. Diagnosis may depend on cyst aspiration revealing the classic anchovy sauce appearance of the cyst fluid. Analysis of serum for Entamoeba antibodies by immunoassay carries 99% sensitivity and over 90% specificity, and is more accurate than stool analysis in amoebic liver disease.
ALCOHOLIC LIVER DISEASE (ALD)
Alcohol is one of the most common causes of chronicliver disease worldwide. Patients with ALD may also have risk factors for other liver diseases (e.g. coexisting NAFLD or chronic viral hepatitis infection), and these interact to increase disease severity. A threshold of 14 units/week in women and 21 units/week in men is generally considered safe. The risk threshold for developing ALD begins at 30 g/day. 1 unit = 8gm of ethanol .
Consumption of > 80 g /day, for >5 years, is required to confer significant risk of advanced liver disease.
The average alcohol consumption of a man with cirrhosis is 160 g/day for over 8 years.
Some of the risk factors for ALD are:
• Drinking pattern. Liver damage is more likely to occur in continuous rather than intermittent or ‘binge’ drinkers, as this pattern gives the liver a chance to recover. It is therefore recommended that people should have at least two alcohol-free days each week. The type of beveragedoes not affect risk.
• Gender. The incidence is increasing in women, who have higher blood ethanol levels than men after consuming the same amount of alcohol. This may be related to the reduced volume of distribution of alcohol.
• Genetics. Alcoholism is more concordant in monozygotic than dizygotic twins.
• Nutrition. Obesity increases the incidence of liver-related mortality by over fivefold in heavy drinkers.
Ethanol itself produces 7 kcal/g (29.3 kJ/g) and many alcoholic drinks also contain sugar, which further increases the calorific value and may contribute to weight gain. Excess alcohol consumption is frequently associated with nutritional deficiencies that contribute to morbidity.
Pathophysiology
Alcohol reaches peak blood concentrations after about
20 minutes, although this may be influenced by stomach
contents. It is metabolised almost exclusively by the liver via one of two pathways .
Approximately 80% of alcohol is metabolised to
acetaldehyde by the mitochondrial enzyme, alcohol
dehydrogenase (ADH).
Acetaldehyde is then metabolised to acetyl-CoA and acetate by aldehyde dehydrogenase. This generates NADH from NAD (nicotinamide adenine dinucleotide), which changes the redox potential of the cell. Acetaldehyde activate the immune system, contributing to cell injury. The remaining 20% of alcohol is metabolised by the microsomal ethanol- oxidising system (MEOS) pathway.
Cytochrome CYP2E1 is an enzyme that oxidises ethanol to acetate. It is induced by alcohol, and during metabolism of ethanol it releases oxygen free radicals, leading to lipid peroxidation and mitochondrial damage. The CYP2E1 enzyme also metabolises acetaminophen, and hence chronic alcoholics are more susceptible to hepatotoxicity from low doses of paracetamol.
It is thought that pro-inflammatory cytokines may also be involved in inducing hepatic damage in alcoholic hepatitis, since endotoxin is released into the blood because of increased gut permeability, leading to release of tumour necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-2 and IL-8 from immune cells. All of these cytokines have been implicated in the pathogenesis of liver fibrosis .
In about 80% of patients with severe alcoholic hepatitis, cirrhosis will coexist at presentation.
Iron deposition is common and does not necessarily indicate haemochromatosis.

Pathological features of alcoholic liver disease
Clinical features
ALD has a wide clinical spectrum ranging from mild abnormalities of LFTs on biochemical testing to advanced cirrhosis. The liver is often enlarged in ALD, even in the presence of cirrhosis.
Stigmata of chronic liver disease, such as palmar erythema, are more common in alcoholic cirrhosis than in cirrhosis of other aetiologies.
Alcohol misuse may also cause damage of other organs and this should be specifically looked for . Three types of ALD are recognised but these overlap considerably, as do the pathological changes seen in the liver.
Alcoholic fatty liver disease
Alcoholic fatty liver disease (AFLD) usually presents with elevated transaminases in the absence of hepatomegaly.It has a good prognosis and steatosis usually disappears after 3 months of abstinence.
Alcoholic hepatitis
This presents with jaundice and hepatomegaly; complications of portal hypertension may also be present. About one third of patients die in the acute episode, particularly those with hepatic encephalopathy or a prolonged PT.
Cirrhosis often coexists; if not present, it is the likely outcome if drinking continues. Even if they abstain, it may take up to 6 months for jaundice to resolve. In patients presenting with jaundice who subsequently abstain, the 3- and 5-year survival is 70%. In contrast, those who continue to drink have 3- and 5-year survival rates of
60% and 34% respectively.
Alcoholic cirrhosis
Alcoholic cirrhosis often presents with a serious complication, such as variceal haemorrhage or ascites, and only half of such patients will survive 5 years from presentation.
However, most who survive the initial illness and who become abstinent will survive beyond 5 years.
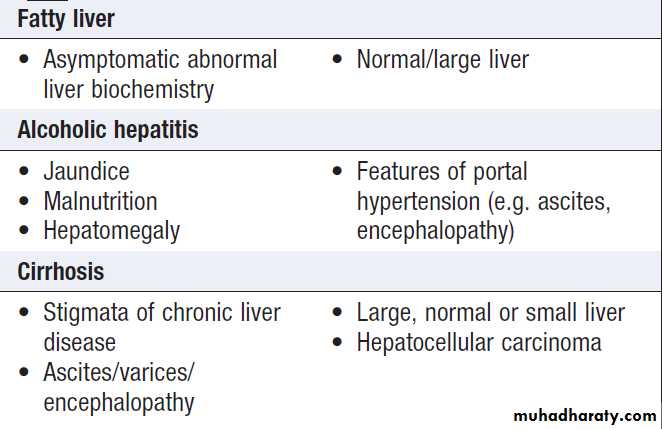
Clinical syndromes of alcoholic liver disease
Investigations
Investigations aim to establish alcohol misuse, to exclude alternative or additional coexistent causes of liver disease and to assess the severity of liver damage. Biological markers, particularly macrocytosis in the absence of anaemia, may suggest and support a history of alcohol misuse. A raised GGT is not specific for alcohol misuse and may also be elevated in the presence of other conditions, including NAFLD and GGT should not be relied on as an indicator of ongoing alcohol consumption.
The presence of jaundice may suggest alcoholic hepatitis. Determining the extent of liver damage often requires a liver biopsy.
In alcoholic hepatitis, PT and bilirubin are used to calculate a ‘discriminant function’ (DF), also known as the Maddrey score, which enables the clinician to assess prognosis (PT = prothrombin time; serum bilirubin in μmol/L is divided by 17 to convert to mg/ dL):
DF = [4.6X Increase PT in (sec)]+Bilirubin (mg/ dL) A value over 32 implies severe liver disease with a poor prognosis and is used to guide treatment decisions .
A second scoring system, the Glasgow score, uses the age, white cell count and renal function, in addition to PT and bilirubin, to assess prognosis with a cutoff of 9 .
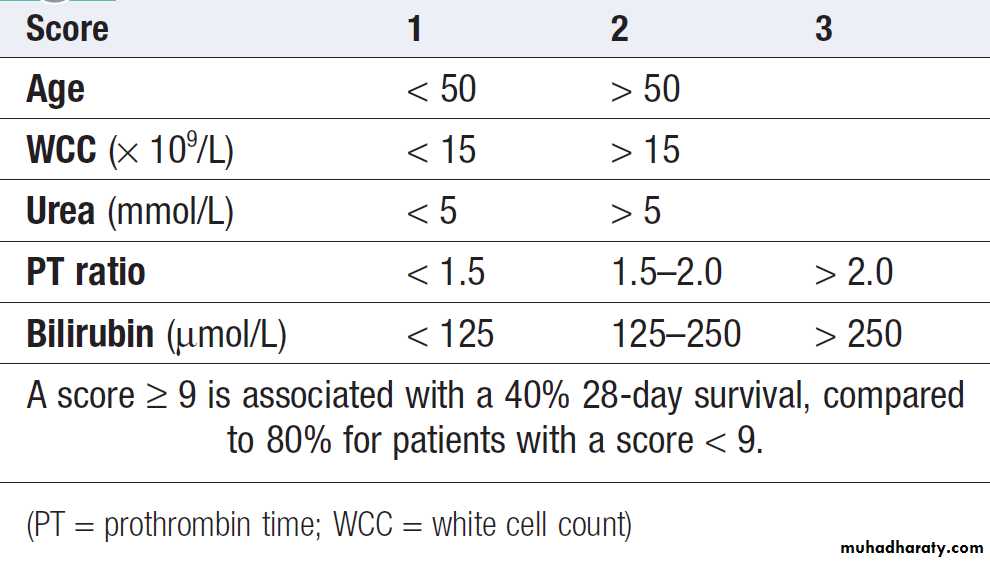
How to assess prognosis using the Glasgow alcoholic hepatitis score
ManagementCessation of alcohol consumption is the single most important. General health and life expectancy are improved when this occurs, irrespective of the stage of liver disease. In the acute presentation it is important to identify and anticipate alcohol withdrawal and Wernicke’s encephalopathy, which need treating in parallel with the liver disease and any complications of cirrhosis.
Nutrition
Good nutrition is very important, and enteral feeding via a nasogastric tube may be needed in severely ill patients.
Corticosteroids
These are of value in patients with severe alcoholic hepatitis (Maddrey’s discriminative score > 32) and increase survival .
A similar improvement in 28-day survival from 52% to 78% is seen when steroids are given to those with a Glasgow score of more than 9. Sepsis is the main side-effect of steroids, and existing sepsis and variceal haemorrhage are the main contraindications to their use. If the bilirubin has not fallen 7 days after starting steroids, the drugs are unlikely to reduce mortality and should be stopped.
Pentoxifylline , has a weak anti-TNF action, may be beneficial in severe hepatitis. It reduces the incidence of hepatorenal failure.
Liver transplantation
The role of liver transplantation in the management of ALD remains controversial. In many centres, ALD is a common indication for liver transplantation. The challenge is to identify patients with an unacceptable risk of returning to harmful alcohol consumption. Many programmes require a 6-month period of abstinence from alcohol before a patient is considered for transplantation. Although this relates poorly to the incidence of alcohol relapse after transplantation, liver function may improve to the extent that transplantation is no longer necessary. The outcome of transplantation for ALD is good and, if the patient remains abstinent, there is no risk of disease recurrence. Transplantation for alcoholic hepatitis has a poorer outcome and is seldom performed.


Pentoxifylline in alcoholic hepatitis
Corticosteroids in alcoholic hepatitisNON-ALCOHOLIC FATTY LIVER DISEASE (NAFLD)
NAFLD disease represents a spectrum of liver disease encompassing simple fatty infiltration (steatosis),fat and inflammation (nonalcoholic steatohepatitis, NASH) and cirrhosis, in the absence of excessive alcohol consumption (typically a threshold of < 20 g/day for women and < 30 g/day for men is adopted 1 unit = 8gm).
While simple steatosis has not been associated with liver-related morbidity, NASH is linked with progressive liver fibrosis, cirrhosis and liver cancer, as well as increased cardiovascular risk. in one study NASH was associated with a greater than tenfold increased risk of liver-related death and a doubling of cardiovascular risk.
NAFLD is strongly associated with obesity, dyslipidaemia, insulin resistance and type 2 diabetes, and so may be considered to be the hepatic manifestation of the ‘metabolic syndrome’.
Increasingly sedentary lifestyles and changing dietary patterns mean that the prevalence of obesity and insulin resistance has increased, making NAFLD the leading cause of liver dysfunction in the non-alcoholic, viral hepatitis-negative population, one large European study found NAFLD in 94% of obese (body mass index (BMI) > 30 kg/m2), 67% of overweight patients (BMI > 25 kg/m2) and 25% of normal-weight patients.
The overall prevalence of NAFLD in patients with type 2 diabetes ranges from 40% to 70%.
Pathophysiology
Whilst the majority of patients with the metabolic syndrome develop steatosis, only a minority exhibit NASH or fibrosis. The initiating events are obesity and insulin resistance, leading to increased hepatic free fatty acid flux. This may be an adaptive response through which hepatocytes store potentially toxic lipids as relatively inert triglyceride. A ‘two-hit’ hypothesis has been proposed to describe the pathogenesis of NAFLD, the ‘first hit’ causing steatosis then progresses to steatohepatitis if a ‘second hit’ occurs.Progression probably follows hepatocellular injury caused by a combination of several different ‘hits’, including:
• oxidative stress due to free radicals produced during fatty acid oxidation
• direct lipotoxicity
• gut-derived endotoxin
• cytokine release (TNF-α etc.)
• endoplasmic reticulum stress.
Cellular damage triggers a mixture of immune-mediated hepatocellular injury and cell death, which leads to stellate cell activation and hepatic fibrosis .
As with many other liver diseases, subtle inter-patient genetic variations and environmental factors interact to determine disease progression.
Clinical features
NAFLD is frequently asymptomatic, although it may be associated with fatigue and mild right upper quadrantdiscomfort. It is commonly identified as an incidental
biochemical abnormality. Alternatively, patients may present late with cirrhosis and portal hypertension, variceal haemorrhage, or HCC. The average age of NASH patients is 40–50 years, however. Most patients have insulin resistance and exhibit features of the metabolic syndrome. Recognised independent risk factors for disease progression are age over 45 years, presence of diabetes , obesity (BMI > 30 kg/m2) and hypertension. NAFLD is also associated with polycystic ovary syndrome, obstructive sleep apnoea and small-bowel bacterial overgrowth.
Investigations
Should be directed first towards exclusion of excess alcohol consumption and other liver diseases (including viral, autoimmune causes), and then at confirming the presence of NAFLD, discriminating simple steatosis from NASH and determining the extent of any hepatic fibrosis. Elevations of serum ALT and AST are modest, and usually less than twice the upper limit of normal. ALT levels fall as hepatic fibrosis increases and the characteristic AST : ALT ratio of <1 seen in NASH reverses (AST : ALT > 1) as progresses towards cirrhosis, meaning that advanced steatohepatitis with disease may be present even in those with normal-range ALT levels. Other abnormalities that may be present include non-specific elevations of GGT, low- titre ANA in 20–30% of patients and elevated ferritin levels.Imaging
Ultrasound provides a qualitative assessment of hepatic fat content. CT, MRI or MR spectroscopy, which offer greater sensitivity for detecting lesser degrees of steatosis. Currently, no routine imaging modality can distinguishsimple steatosis from steatohepatitis or accurately quantify hepatic fibrosis.
Liver biopsy
Remains the ‘gold standard’ investigation. The histological definition of NASH is based on a combination of three lesions (steatosis, cellular injury and inflammation).
There may be accompanied by Mallory– Denk bodies.
Perisinusoidal fibrosis is a characteristic feature of .
It is important to note that hepatic fat content tends to diminish as cirrhosis develops and so NASH is likely to be under-diagnosed in the setting of advanced liver disease, where it is thought to be the underlying cause of 30–75% of cases in which no specific aetiology is readily identified (so-called ‘cryptogenic cirrhosis’).
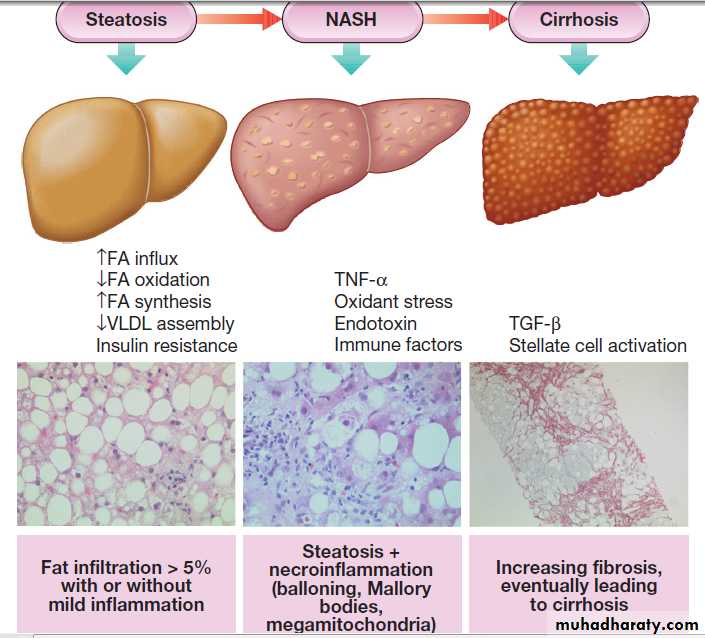
Features of non-alcoholic fatty liver disease and non-alcoholic
steatohepatitis. Rate of progression isdetermined by environmental (dietary) and
genetic factors. (FA = fatty acids; TGF-β =
transforming growth factor beta; TNF-α =
tumour necrosis factor alpha; VLDL = very
low-density lipoproteins)
Management
As a marker of the metabolic syndrome, identification ofNAFLD should prompt screening for and treatment of
cardiovascular risk factors.
Non-pharmacological treatment
Current treatment comprises lifestyle interventions to
promote weight loss and improve insulin sensitivity
through dietary changes and physical exercise. Sustained
weight reduction of 7–10% is associated with significant
improvement in histological and biochemical NASH severity. Pharmacological treatment
No pharmacological agents are currently licensed. Treatment directed at coexisting metabolic disorders, such as dyslipidaemia and hypertension, should be given.
Although use of statins does not ameliorate NAFLD, there does not appear to be any increased risk of hepatotoxicity, and so they may be used to treat dyslipidaemia.
Specific insulin- sensitising agents, in particular glitazones, may help selected patients, while positive results with high-dose vitamin E (800 U/day) have been tempered by evidence that high doses may be associated
with an increased risk of prostate cancer and all-cause
mortality, which has limited its use.
Autoimmune hepatitis
Immune-mediated liver injury characterised by the presence of serum antibodies and peripheral blood T lymphocytes reactive with self-proteins, a strong association with other autoimmune diseases and high serum immunoglobulins , in particular, IgG. Most commonly seen in women, in the second and third decades. Cross-reactivity with viruses such as HAV and EBV in immunogenetically susceptible individuals (human leucocyte antigen (HLA)-DR3 and DR4) has been suggested as a mechanism.
Conditions associated with autoimmune hepatitis
PathophysiologySeveral subtypes of this disorder have been proposed
that have differing immunological markers, histological patterns are similar and treatment (immunosuppressive drugs) is the same.
The most frequently seen autoantibody pattern is high titre of antinuclear and anti-smooth muscle antibodies, with IgG hyperglobulinaemia (type I autoimmune hepatitis in the old classification), seen in young females.
Disease characterised by the presence of anti-LKM (liver–kidney microsomal) antibodies typically seen in paediatric populations and can be more resistant to treatment. Adult onset of anti-LKM can be seen in chronic HCV infection.
This was classified as type II disease in the old system.
Clinical features
The onset is usually insidious, with fatigue, anorexiaand jaundice. In about one-quarter of patients, the onset is acute, resembling viral hepatitis, but resolution does not occur and can lead to extensive liver necrosis and liver failure. Other features fever, arthralgia, vitiligo and epistaxis. Amenorrhoea can occur. Jaundice is mild or absent, but signs of chronic liver disease, especially spider naevi and hepatosplenomegaly, can be present.
Associated autoimmune disease, such as Hashimoto’s
thyroiditis or rheumatoid arthritis, is often present and
can modulate the clinical presentation.
Investigations
Serological tests for autoantibodies are often positive
but low titres of these occur in some healthy people and in patients with other inflammatory liver diseases. Elevated IgG levels are an important diagnostic and treatment response feature of autoimmune hepatitis if present. If the diagnosis is suspected, liver biopsy should be performed.
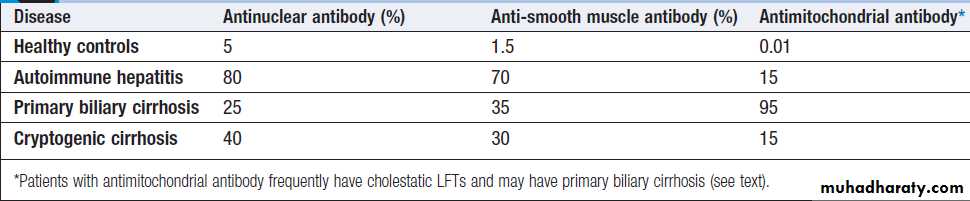
Frequency of autoantibodies in chronic non-viral liver diseases and in healthy people
Management of autoimmune hepatitisCorticosteroids is life-saving, particularly during exacerbations Initially, prednisolone 40 mg/day; gradually reduced as the patient and LFTs improve. Maintenance instituted once LFTs are normal (as well as IgG if elevated). Approaches to maintenance include reduced-dose prednisolone (ideally below 5–10 mg/day), usually in the context of azathioprine 1.0–1.5 mg/kg/day. Azathioprine can also be used as the sole maintenance agent in low-activity disease. Newer agents such as mycophenolate mofetil (MMF) are increasingly used. Patients should be monitored for acute exacerbations (LFT and IgG) and such exacerbations should be treated with corticosteroids. Although treatment can significantly reduce the rate of progression to cirrhosis, end-stage disease can be seen despite treatment.

Immunosuppression in autoimmune hepatitis
Primary biliary cirrhosis (PBC)A chronic, progressive cholestatic liver disease that predominantly affects middle-aged women , female-to-
male 9 : 1; also more common amongst cigarette smokers.
Clustering of cases has been reported, suggesting an environmental trigger in susceptible individuals.
It is strongly associated with the presence of antimitochondrial antibodies (AMA), which are diagnostic, and is characterised by a granulomatous inflammation of the portal tracts, leading to progressive damage and eventually loss of the small and middle sized bile ducts. This in turn leads to fibrosis and cirrhosis of the liver. The condition typically presents with an insidious onset of itching and/or tiredness; it may also be found incidentally as the result of routine blood tests.
Pathophysiology
Immune mechanisms are clearly involved, associated with other autoimmune nonhepatic diseases, such as thyroid disease, and there is a genetic association with HLA-DR8. AMA directed at pyruvate dehydrogenase mitochondrial enzyme that plays a key role in cellular energy generation. PBC-specific ANA (such as those directed at the nuclear pore antigen gp210) have a characteristic staining pattern in immunofluorescence assays. Elevations in serum Ig levels are frequent but, unlike in autoimmune hepatitis, elevation is typically of IgM. Pathologically, chronic granulomatous inflammation destroys the interlobular bile ducts; progressive lymphocyte-mediated inflammatory damage causes fibrosis, which spreads from the portal tracts to the liver parenchyma and eventually leads to cirrhosis.
Clinical features
Systemic symptoms such as fatigue are common andmay precede diagnosis for years. Pruritus, which can
be a feature of any cholestatic disease, is a common
presenting complaint and may precede jaundice by
months or years. Jaundice is rarely a presenting feature.
The itching is usually worse on the limbs. Although there may be right upper abdominal discomfort, fever and rigors do not occur. Bone pain or fractures can rarely result from osteomalacia (fat-soluble vitamin malabsorption) or, more commonly, from osteoporosis (hepatic osteodystrophy). Initially, patients are well nourished but weight loss can occur as the disease progresses. Scratch marks may be found in patients with severe pruritus.
Jaundice is only prominent late in the disease and can
become intense. Xanthomatous deposits occur in aminority, especially around the eyes. Mild hepatomegaly
is common and splenomegaly becomes increasingly
common as portal hypertension develops. Liver failure
may supervene.
Associated diseases
Autoimmune and connective tissue diseases occur with
increased frequency in PBC, particularly the sicca
syndrome ,systemic sclerosis, coeliac disease and thyroid diseases. Hypothyroidism should always be considered in patients with fatigue.
Diagnosis and investigations
The LFTs show a pattern of cholestasis. Hypercholesterolaemia is common and worsens as disease progresses; however, it is of no diagnostic value. AMA is present in over 95% of patients, and when it is absent the diagnosis should not be made without obtaining histological evidence and considering cholangiography (MRCP or ERCP) to exclude other biliary disease. ANA and anti-smooth muscle antibodies are present in around 15% of patients . US examination shows no sign of biliary obstruction. Liver biopsy is only necessary if there is diagnostic uncertainty. The histological features correlate poorly with the clinical features; portal hypertension can develop before the histological onset of cirrhosis.
Management
The hydrophilic bile acid, ursodeoxycholic acid (UDCA),at a dose of 13–15 mg/kg/day improves bile flow,
replaces toxic hydrophobic bile acids in the bile acid
pool, and reduces apoptosis of the biliary epithelium.
Clinically, UDCA improves LFTs, may slow down histological progression and has few side-effects . it is therefore should be regarded as the optimal first-line treatment.
A significant minority of patients either fail to normalise their LFTs with UDCA or show an inadequate response, and such patients have an increased risk of
developing end-stage liver disease compared to patients
showing a full response.
There is currently no consensus as to how to treat such patients. Immunosuppressants, such as corticosteroids, azathioprine, penicillamine and ciclosporin, have all been trialled . None shows overall benefit.
Whether they offer benefit to the specific subgroup of UDCA non-responding patients requiring second-line
approaches to treatment is not clear.
Liver transplantation should be considered once liver
failure has developed and may be indicated in patients
with intractable pruritus. Serum bilirubin remains the
most reliable marker of declining liver function. Transplantation is associated with an excellent 5-year survival of over 80%, although the disease will recur in over one third of patients at 10 years.

Ursodeoxycholic acid (UDCA) in primary biliary cirrhosis
PruritusThis is the main symptom. The cause is unknown but up-regulation of opioid receptors and increased levels of endogenous opioids may play a role. First-line treatment is with the anion-binding resin colestyramine, which probably acts by binding potential pruritogens in the intestine and increasing their excretion in the stool. A dose of 4–16 g/day orally is used. The powder is mixed in orange juice and the main dose (8 g) taken before and after breakfast, when maximal duodenal bile acid concentrations occur. Colestyramine may bind other drugs in the gut (most obviously UDCA), and adequate spacing should be used between drugs. Colestyramine is sometimes ineffective, especially in complete biliary obstruction, and can be difficult for some to tolerate.
Alternative treatments include rifampicin (150 mg/day, titrated up to a maximum of 600 mg/day as required and contingent on there being no deterioration in LFTs),
naltrexone (an opioid antagonist; 25 mg/day initially,
increasing up to 300 mg/day), plasmapheresis and a
liver support device (e.g. a molecular adsorbent recirculating system (MARS)).
Fatigue
Fatigue affects about one-third of patients with PBC.
The cause is unknown but it may reflect intracerebral
changes due to cholestasis. Unfortunately, once depression,
hypothyroidism and coeliac disease have been excluded, there is currently no specific treatment. The impact on patients’ lives can be substantial.
Malabsorption
Prolonged cholestasis is associated with steatorrhoeaand malabsorption of fat-soluble vitamins, which should
be replaced as necessary. Coeliac disease should be
excluded since its incidence is increased in PBC.
Bone disease
Osteopenia and osteoporosis are common, and normal
post-menopausal bone loss is accelerated. Baseline bone
density should be measured and treatment
started with replacement calcium and vitamin D3.
Bisphosphonates should be used if there is evidence of
osteoporosis. Osteomalacia is rare.
Primary sclerosing cholangitis (PSC)
PSC is a cholestatic liver disease caused by diffuse inflammation and fibrosis; it can involve the entire biliary tree and leads to the gradual obliteration of intrahepatic and extrahepatic bile ducts, and ultimately biliary cirrhosis, portal hypertension and hepatic failure.
Although considered as an autoimmune disease, evidence for an autoimmune pathophysiology is weaker than is the case for PBC and autoimmune hepatitis. Cholangiocarcinoma develops in about 10–30%.
PSC is twice as common in young men.
Most patients present at age 25–40 years, although the condition may be diagnosed at any age and is an important cause of chronic liver disease in children
The generally accepted diagnostic criteria are:
• generalised beading and stenosis of the biliarysystem on cholangiography
• absence of choledocholithiasis (or history of bile
duct surgery)
• exclusion of bile duct cancer, by prolonged follow-up.

Causes of secondary sclerosing cholangitis
PathophysiologyThe cause is unknown but there is a close association with IBD, particularly ulcerative colitis . About two-thirds have coexisting ulcerative colitis, and PSC is the most common form of CLD in UC. Between 3% and 10% of patients with ulcerative colitis develop PSC, particularly those with extensive colitis or pancolitis. The prevalence of is lower in patients with Crohn’s colitis (about 1%). Patients with PSC and UC are at greater risk of colorectal neoplasia than those with UC alone, and those who develop colorectal neoplasia are at greater risk of cholangiocarcinoma. It is currently believed that PSC is an immunologically mediated disease, triggered in genetically susceptible individuals by toxic or infectious agents, which may gain access to the biliary tract through a leaky, diseased colon.
A close link with HLA haplotype A1 B8 DR3 DRW52A has been identified. This haplotype is commonly found in association with other organ-specific autoimmune diseases (e.g. autoimmune hepatitis). Perinuclear antineutrophil cytoplasmic antibodies (ANCA) have been detected in the sera of 60–80% of patients with PSC with or without ulcerative colitis, and in 30–40% of patients with ulcerative colitis alone.
The antibody is not specific for PSC and is found in other chronic liver diseases (e.g. 50% of patients with autoimmune hepatitis).
Diseases associated with primary sclerosing cholangitis
Clinical featuresThe diagnosis is often made incidentally when persistently
raised serum ALP is discovered in an individual with UC. Common symptoms ,fatigue, intermittent jaundice, weight loss, right upper quadrant pain and pruritus. Physical examination is abnormal in 50% of symptomatic; the most common findings are hepato/splenomegaly and jaundice. The key investigation is now MRCP, which is usually diagnostic, revealing multiple irregular stricturing and dilatation . ERCP should be reserved for patients in whom therapeutic intervention is likely to be necessary. On liver biopsy the characteristic early features of are periductal ‘onion-skin’ fibrosis and inflammation, with portal oedema and bile ductular proliferation resulting in expansion of the portal tracts . Later, fibrosis spreads, progressing to biliary cirrhosis; obliterative cholangitis ‘vanishing bile duct syndrome’.
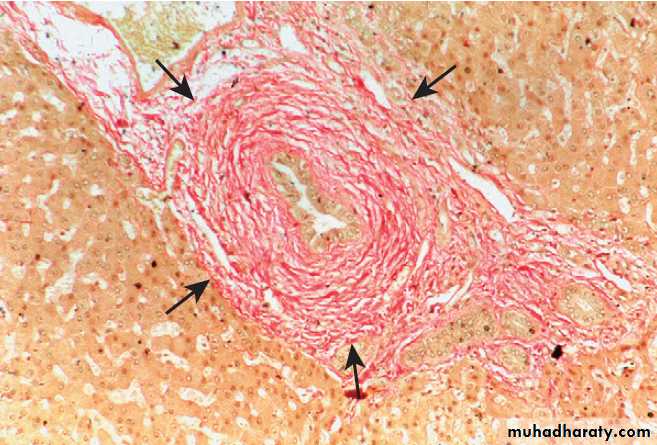
Primary sclerosing cholangitis. Note onion-skin scarring (arrows) surrounding a bile duct.
ManagementThere is no cure for PSC. UDCA is widely used, although the evidence to support this is limited. UDCA may have benefit in terms of reducing colon carcinoma risk.
The course is variable. In symptomatic, median survival from presentation to death or liver transplantation is about 12 years. About 75% of asymptomatic patients survive 15 years or more. Most patients die from liver failure, about 30% die from bile duct carcinoma, and the remainder die from colonic cancer or complications of colitis.
Immunosuppressive agents, including prednisolone, azathioprine, methotrexate and ciclosporin, have been tried; generally, results have been disappointing.
Management of pruritus is as for PBC. Fatigue appears to be less prominent than in PBC.
Management of complications
Broad-spectrum antibiotics (e.g. ciprofloxacin) should begiven for acute attacks of cholangitis. If cholangiography shows a well-defined obstruction to the extrahepatic bile ducts (‘dominant stricture’), mechanical relief can be obtained by placement of a plastic stent or by balloon dilatation performed at ERCP. It is important, in this situation, actively to consider the possibility of cholangiocarcinoma (the differential diagnosis for dominant extrahepatic stricture). Fat-soluble vitamin replacement is necessary in jaundiced patients. osteoporosis requires treatment .
Surgical treatment
Surgical resection of the extrahepatic bile duct and biliary reconstruction have a limited role in the management of non-cirrhotic patients with dominant extrahepatic disease. Orthotopic transplantation is the only surgical option in patients with advanced liver disease; 5-year survival is 80–90% in most centres. Unfortunately, the condition may recur in the graft and there are no identified therapies able to prevent this. Cholangiocarcinoma is a contraindication to transplantation.
Colon carcinoma risk can be increased following transplant because of the effects of immune suppression.
LIVER TUMOURS AND OTHER FOCAL LIVER LESIONS
Identification of a hepatic mass lesion is common .The critical steps to be taken in diagnosing hepatic mass lesions are:
• determining the presence, nature and severity of any underlying chronic liver disease(CLD), as the differential diagnosis is very different in patients with and without CLD
• use of optimal (usually multiple) imaging modalities. Primary malignant tumours
Hepatocellular carcinoma (HCC)
Is the most common primary liver tumour, and the sixth most common cause of cancer worldwide. Cirrhosis is present in 75–90% of individuals with HCC and is an important risk factor for the disease. The risk is between 1% and 5% in cirrhosis caused by hepatitis B and C.
Chronic hepatitis B infection increases the risk of HCC
100-fold and is the major risk factor worldwide. The risk is four times higher in HBeAg-positive individuals than in those who are HBeAg-negative.Hepatitis B vaccination has led to a fall in HCC in.
The risk is higher in men and rises with age. Macroscopically, appears as a single mass in the absence of cirrhosis, or as a single nodule or multiple nodules in the presence of cirrhosis. It takes its blood supply from the hepatic artery and tends to spread by invasion into the portal vein and its radicals. Lymph node metastases are common, while lung and bone metastases are rare.
Well-differentiated can resemble normal hepatocytes and can be difficult to distinguish from normal liver.
Clinical features
Patients typically present with HCC in one of two ways.Commonly, those with underlying cirrhosis develop a
deterioration in their liver function, with worsening ascites and/or jaundice or variceal haemorrhage.
Other symptoms weight loss, anorexia and abdominal pain. Examination may reveal hepatomegaly or a right
hypochondrial mass. Tumour vascularity can lead to an abdominal bruit, and hepatic rupture with intraabdominal bleeding may occur. The advanced nature of the disease that presents in this way makes curative therapy unlikely.
The second presentation is through screening of patients at risk of HCC.
Investigations
Serum markers
Alpha-fetoprotein (AFP) is produced by 60% of HCCs.
Levels increase with the size of the tumour. Serum AFP can also rise in the presence of active hepatitis B and C viral
replication; very high levels are seen in acute hepatic
necrosis, such as that following paracetamol toxicity. Nevertheless, in the absence of a marked hepatic flare of disease, a progressively rising AFP, or AFP over 400 ng/mL (normal is < 10 ng/mL), warrants an aggressive search for HCC. In HCC patients with elevated AFP levels, serial measurement scan be a useful biomarker of disease progression/response to treatment.
Imaging
Ultrasound will detect focal liver lesions as small as 2–3 cm. The use of ultrasound contrast agents has increased sensitivity and specificity, may also show evidence of portal vein involvement and features of coexistent cirrhosis.Multidetector row CT, following intravenous
contrast, identifies HCC by its classical hypervascular
appearance . Small lesions of less than 2 cm
can be difficult to differentiate from hyperplastic nodules
in cirrhosis. MRI can be used instead of CT. Angiography
is now seldom performed. Combination of imaging
modalities more accurately diagnoses and stages the
extent of disease, and using at least two modalities (typically, CT or MRI following initial screening ultrasound
identification of a mass lesion) is recommended.
Liver biopsy
Histological confirmation is advisable in patients withlarge tumours who do not have cirrhosis or hepatitis B,
in order to confirm the diagnosis and exclude metastatic
tumour. Biopsy should be avoided in patients who may
be eligible for transplantation or surgical resection
because there is a small (< 2%) risk of tumour seeding
along the needle tract.
Role of screening by ultrasound scanning and AFP
measurements at 6-month intervals, is indicated in high risk patients, such as those with cirrhosis due to hepatitis B and C, haemochromatosis, alcohol, NASH and α1-antitrypsin deficiency. Screening identifies smaller tumours, often ,3 cm in size, which are more likely to be cured by surgical resection, local ablative or transplantation .
Guidelines for screening and management
of hepatocellular carcinomaManagement
This is different for patients with cirrhosis and those without. In the presence of cirrhosis, tumour size, extent of liver disease (Child–Pugh score). Prognosis depends on tumour size, the presence of vascular invasion, and liver function in those with cirrhosis. Screening has improved the outlook.Hepatic resection : This is the treatment of choice for non-cirrhotic patients. The 5-year survival in this group is about 50%. However, there is a 50% recurrence rate at 5 years. Few patients with cirrhosis are suitable for hepatic resection because of the high risk of hepatic failure; nevertheless, surgery is offered, particularly in the Far East, to some cirrhotic patients with small tumours and good liver function (Child–Pugh A with no portal hypertension).
Liver transplantation
Has the benefit of curing underlying cirrhosis and removing the risk of a second, de novo tumour. The requirement for immunosuppression creates its own risks of reactivation. 5-year survival following transplantation is 75% for single tumour< 5 cm in size or three tumours < 3 cm (the Milan criteria). Unfortunately, the underlying liver disease, in particular hepatitis C, may recur and can result in recurrent cirrhosis giving rise to de novo HCC risk, now complicated by the presence of immunosuppression.Percutaneous therapy
Percutaneous ethanol injection into the tumour under ultrasound guidance is efficacious (80% cure rate) for tumours of 3 cm or smaller. Recurrence rates (50% at 3 years) are similar to those following surgical resection.
Radiofrequency ablation, using a single electrode inserted into the tumour under radiological guidance, is an alternative that takes longer to perform but may cause more complete tumour necrosis. Their role in primary therapy as an alternative to curative resection or transplantation remains to be established.
Trans-arterial chemo- embolisation
Hepatocellular cancers are not radiosensitive and the response rate to chemotherapy such as doxorubicin is only around 30%. In contrast, hepatic artery embolisation with Gelfoam and doxorubicin is more effective, with survival rates of 60% in cirrhotic patients with unresectable HCC and good liver function (compared with 20% in untreated patients) at 2 years.
Trans-arterial chemo-embolisation (TACE) is contraindicated in decompensated cirrhosis and multifocal HCC. Chemotherapy
Sorafenib improves survival from 7.9 to 10.7 months in cirrhotic. The drug is a multikinase inhibitor with activity against Raf, vascular endothelial growth. factor (VEGF) and platelet-derived growth factor (PDGF) signalling, and is the first systemic therapy to prolong survival in HCC.
The ultimate role of sorafenib in HCC – in particular, when and how best to use it – remains to be established.
Other primary malignant tumours
Sarcomas and Cholangiocarcinoma (bile duct cancer) typically presents with bile duct obstruction rather than as a hepatic mass lesion, although the latter occasionally occurs.
Secondary malignant tumours
These are common and usually originate from carcinomas
in the lung, breast, abdomen or pelvis. They may be single or multiple. Peritoneal dissemination frequently results in ascites.
Clinical features
The primary neoplasm is asymptomatic in 50% of patients, being detected on either radiological, endoscopic or blood biochemistry screening. There is liver enlargement and weight loss; jaundice may be present.
Investigations
A raised ALP activity is the most common biochemical abnormality but LFTs may be normal. Ascitic fluid, if present, has a high protein content and may be bloodstained; cytology sometimes reveals malignant cells. Imaging shows filling defects , laparoscopy
may reveal the tumour and facilitates liver biopsy.
Management
Hepatic resection can improve survival for slow-growingtumours such as colonic carcinomas, and is an approach
that should be actively explored in patients who are fit
for liver resection, have had the primary tumour resected
and in whom extrahepatic disease has been excluded.
Patients with neuro-endocrine tumours, such as gastrinomas, insulinomas and glucagonomas, and those with lymphomas may benefit from surgery, hormonal treatment or chemotherapy.
Unfortunately, palliative treatment to relieve pain is all that is available for most patients; this may include arterial embolisation of the tumour masses.
Benign tumours
Hepatic adenomas, rare vascular tumours present as an abdominal mass, abdominal pain or intraperitoneal bleeding, more common in women and may be caused by oral contraceptives, androgens and anabolic steroids. Resection is indicated for the relief of symptoms. It can increase in size during pregnancy. Rapidly growing adenomas can, rupture, causing intraperitoneal bleeding.Haemangiomas
These are the most common benign liver tumours. Most are smaller than 5 cm and rarely cause symptoms . The diagnosis is usually made by ultrasound, but CT may show a low-density lesion with delayed arterial filling. Surgery is only needed for very large symptomatic lesions or where the diagnosis is in doubt.
Cystic liver disease and liver abscess
Isolated or multiple simple cysts are common in the liver and are a relatively frequent finding on ultrasound screening. They can be associated with polycystic renal disease .
They are intrinsically benign and require no therapy, other than in rare cases where the mass effect of very large or multiple cysts causes abdominal discomfort. In such cases, percutaneous or surgical debulking can be attempted but recurrence is typical.
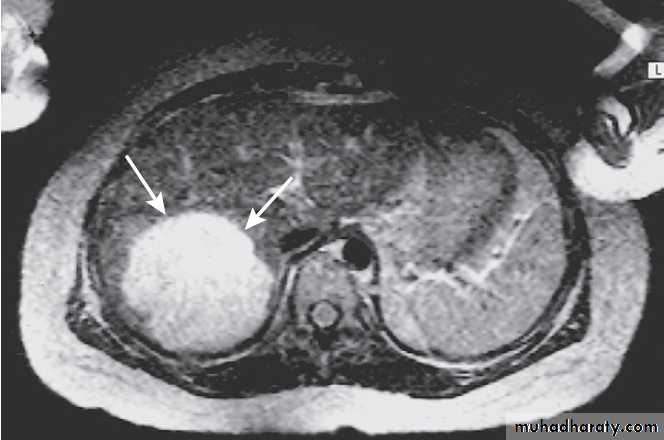
MRI showing a haemangioma (arrows) in the liver.
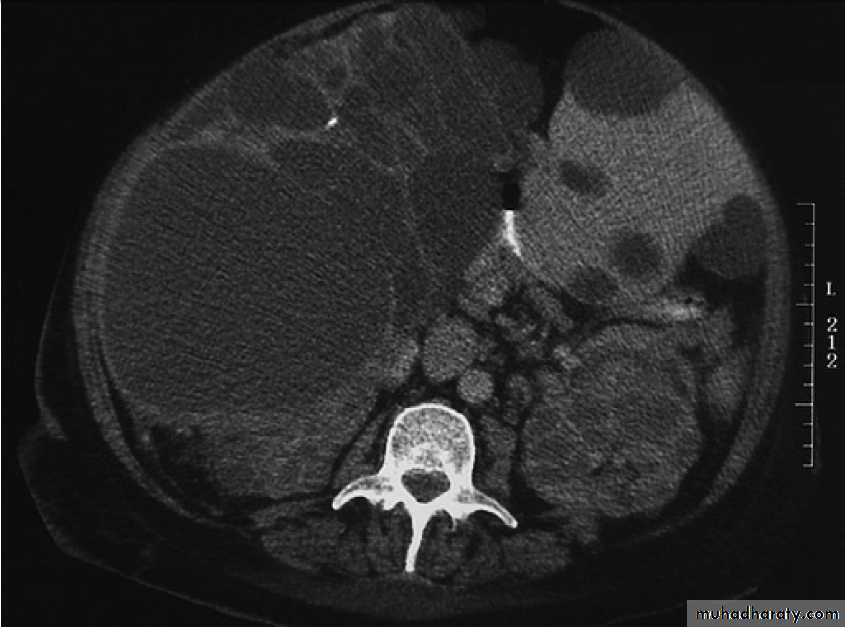
CT : multiple cysts in the liver and kidneys in polycystic disease.
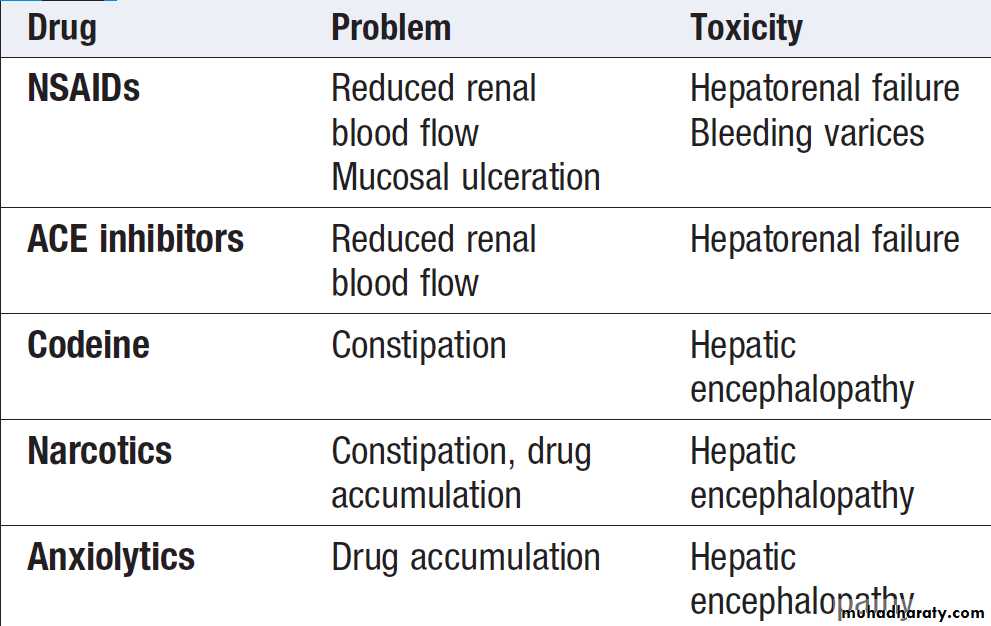
Drugs to be avoided in cirrhosis
The liver is the primary site of drug metabolism and an important target for drug-induced injury. Pre-existing liver disease may affect the capacity of the liver to metabolise drugs, and unexpected toxicity may occur when patients with liver disease are given drugs in normal doses.Drug-induced liver injury
Should always be considered in the differential diagnosis of acute liver failure, jaundice or abnormal LFT. Jaundice indicates more severe damage. Although acute liver failure can occur, most drug reactions are self-limiting and chronic liver damage is rare. Abnormal LFTs often take weeks to normalise. Occasionally, permanent bile duct loss (ductopenia) follows a cholestatic drug reaction, such as to co- amoxiclav. looking for temporal relationships between drug exposure and onset of liver abnormality (bearing in mind the fact that liver injury can frequently take weeks or even months to develop following exposure). A liver biopsy considered if there is suspicion of pre-existing liver disease Where drug-induced injury is suspected, the potential culprit drug should be discontinued.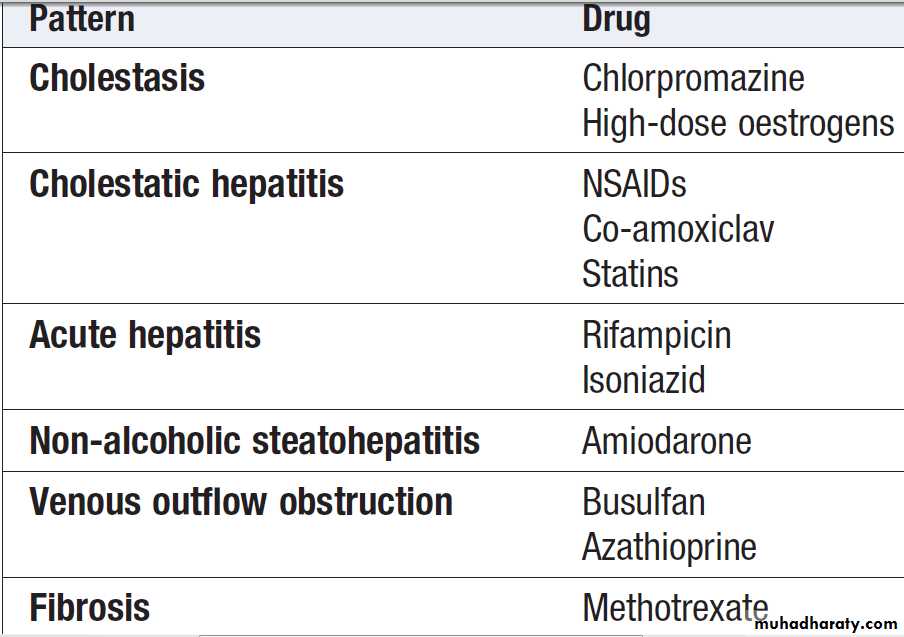
Examples of common causes of drug-induced hepatotoxicity
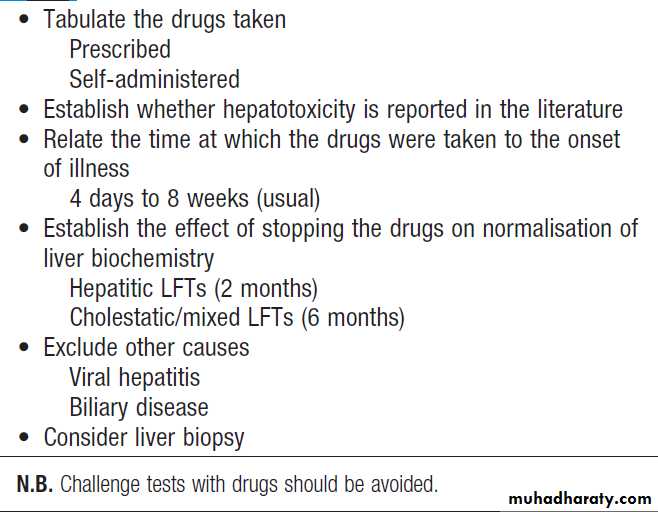
The diagnosis of acute drug-induced liver disease
Types of liver injuryCholestasis
Pure cholestasis can occur with oestrogens (50 μg/day) were used as contraceptives. Chlorpromazine and antibiotics such as flucloxacillin can cause cholestatic hepatitis. Co- amoxiclav is the most common antibiotic to cause abnormal LFTs but, unlike other, it may not produce symptoms until 10–42 days after it is stopped.
Anabolic steroids used by body-builders may also cause a cholestatic hepatitis. In some cases (NSAIDs and (COX)-2 inhibitors) there is overlap with acute hepatocellular injury.
Hepatocyte necrosis
Paracetamol. Diclofenac and isoniazid. Herbal remedies, Recreational drugs, including cocaine. Granulomas may be seen in liver injury following allopurinol.
Steatosis
can follow exposure to tetracyclines and sodium valproate , tamoxifen, and amiodarone.Vascular/sinusoidal lesions
Alkylating agents used in oncology.
Chronic overdose of vitamin A.
Hepatic fibrosis
Most drugs cause reversible liver injury and hepatic fibrosis is very uncommon. Methotrexate, however, as well as causing acute liver injury when it is started, can lead to cirrhosis when used in high doses over a long period of time. Risk factors for include pre-existing liver disease and a high alcohol intake.
INHERITED LIVER DISEASES
Haemochromatosis
A condition in which the amount of total body iron is increased; the excess iron causes damage to several organs. It may be primary or secondary .
Hereditary haemochromatosis (HHC)
Iron is deposited throughout the body and total body iron may reach 20–60 g (normally 4 g). The important organs involved are the liver, pancreatic islets, endocrine glands and heart. The gradual development of fibrous septa leads to the formation of irregular nodules, and finally regeneration results in macronodular cirrhosis.
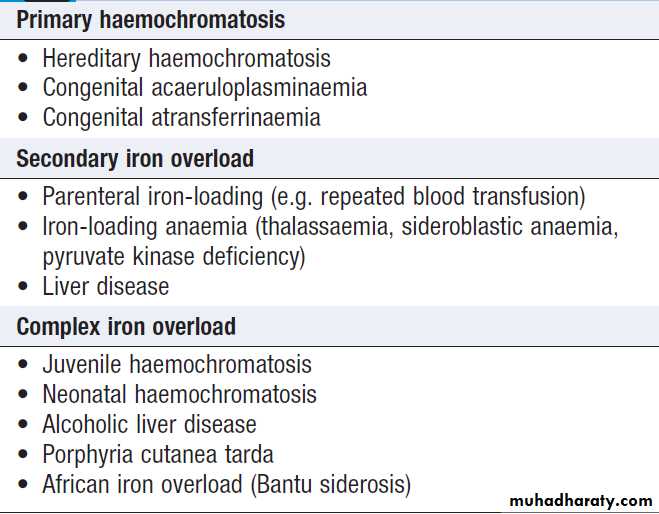
Causes of haemochromatosis
PathophysiologyCaused by increased absorption of dietary iron and is inherited as an autosomal recessive trait.
The mechanisms by which HFE regulates iron absorption are unclear. However, it is thought that a defect in uptake of transferrin-associated iron, leading to up-regulation of enterocyte iron-specific divalent metal transporters and excessive iron absorption.
HHC may promote accelerated liver disease with alcohol excess or hepatitis C infection.
Iron loss in menstruation and pregnancy can delay the onset of HHC in females.
Clinical features
Symptomatic disease usually presents in men over 40 years with features of liver disease (often with hepatomegaly), diabetes mellitus or heart failure. Fatigue and arthropathy are early symptoms but are frequently absent.Leaden-grey skin pigmentation due to excess melanin occurs, especially in exposed parts, axillae, groins and genitalia: hence the term ‘bronzed diabetes’. Once again, absence of this feature does not preclude the diagnosis. Impotence, loss of libido, testicular atrophy and arthritis with chondrocalcinosis secondary to calcium pyrophosphate deposition are also common. Cardiac failure or cardiac dysrhythmia may occur due to iron deposition in the heart.
Investigations
Serum iron studies show a greatly increased ferritin, a raised plasma iron and saturated plasma iron-binding capacity. Transferrin saturation of > 45% is suggestive of iron overload.
The differential diagnoses for elevated ferritin are inflammatory disease or excess ethanol consumption
for modest elevations (< 1000 μg/L). Very significant ferritin elevation can be seen in adult Still’s disease. MRI has high specificity for iron overload, but poor sensitivity.
Liver biopsy allows assessment of fibrosis and distribution of iron (hepatocyte iron characteristic).The Hepatic Iron Index (HII) provides quantification of liver iron. HII of more than 1.9 suggests genetic haemochromatosis . C282Y and the H63D mutations can be identified by genetic testing.
Management
Treatment consists of weekly venesection of 500 mLblood (250 mg iron) until the serum iron is normal; this may take 2 years or more. The aim is to reduce
ferritin to < 50 μg/L. Thereafter, venesection is
continued as required to keep the serum ferritin
normal. Liver and cardiac problems improve after iron
removal, but joint pain is less predictable and can
improve or worsen after iron removal.
Diabetes mellitus does not resolve after venesection.
cirrhotic patients have a good prognosis compared with other forms of cirrhosis.
Venesection reduces but does not abolish the risk of hepatocellular carcinoma in the presence of cirrhosis.
Other therapy includes that for cirrhosis and diabetes.
First degree family members should be investigated, preferably by genetic screening and also by checking the plasma ferritin and iron-binding saturation. Liver biopsy is only indicated in asymptomatic relatives if the LFTs are abnormal and/or the serum ferritin is greater than 1000 μg/L because these features are associated with significant fibrosis or cirrhosis.
Asymptomatic disease should also be treated by venesection until the serum ferritin is normal.
Pre-cirrhotic patients with HHC have a normal life
expectancy, and even cirrhotic patients have a good
prognosis compared with other forms of cirrhosis
(three-quarters of patients are alive 5 years after diagnosis).
This is probably because liver function is well
preserved at diagnosis and improves with therapy.
Screening for hepatocellular carcinoma is mandatory
because this is the main cause of death, affecting
one-third of patients with cirrhosis irrespective of
therapy.
Venesection reduces but does not abolish the risk of hepatocellular carcinoma in the presence of cirrhosis.
Secondary haemochromatosis
Many conditions, including chronic haemolytic disorders,sideroblastic anaemia, other conditions requiring
multiple blood transfusion (generally over 50 L),
porphyria cutanea tarda, dietary iron overload and
occasionally alcoholic cirrhosis, are associated with
widespread secondary siderosis. The features are similar
to primary haemochromatosis, but the history and clinical
findings point to the true diagnosis. Some patients
are heterozygotes for the HFE gene and this may contribute to the development of iron overload.
Wilson’s disease
Wilson’s disease (hepatolenticular degeneration) is a rare autosomal recessive disorder of copper metabolism caused by a variety of mutations in the ATP7B gene on chromosome 13. Total body copper is increased, with excess copper deposited in, and causing damage to, several organs.
Pathophysiology
Normally, dietary copper is absorbed from the stomach
and proximal small intestine, rapidly taken into the liver, where it is stored and incorporated into caeruloplasmin, which is secreted into the blood. The accumulation of excessive copper in the body is ultimately prevented by its excretion, the most important route being via bile.
In Wilson’s disease, there is almost always a failure of synthesis of caeruloplasmin; however, some 5% of patients have a normal circulating caeruloplasmin concentration and this is not the primary pathogenic defect. The organs most affected are the liver, basal ganglia, eyes, kidneys and skeleton.
The large number of culprit mutations means that, in contrast to haemochromatosis, genetic diagnosis is not routine in Wilson’s disease, although it may have a role in screening families following identification of the Genotype in an index patient.
Clinical features
Symptoms usually arise between the ages of 5 and 45years. Hepatic disease occurs predominantly in childhood
and early adolescence. Neurological damage causes basal ganglion syndromes and dementia, which tends to
present in later adolescence. Other manifestations include
renal tubular damage and osteoporosis.
Liver disease
Episodes of acute hepatitis, sometimes recurrent, can
occur, especially in children, and may progress to
fulminant liver failure. The latter is characterised by
the liberation of free copper into the blood stream,
causing massive haemolysis and renal tubulopathy.
Chronic hepatitis can also develop insidiously and eventually present with established cirrhosis; liver failure and portal hypertension may supervene. The possibility of Wilson’s disease should be considered in any patient under the age of 40 presenting with recurrent acute hepatitis or CLD of unknown cause, especially when accompanied by haemolysis.
Neurological disease
Clinical features include a variety of extrapyramidal
features, particularly tremor, choreoathetosis, dystonia,
parkinsonism and dementia . Unusual clumsiness
for age may be an early symptom.
Neurological disease typically develops after the onset of liver disease and can be prevented by effective treatment started following diagnosis in the liver disease phase. This increases the importance of diagnosis in the liver phase.
Kayser–Fleischer rings
These constitute the most important single clinical clue
to the diagnosis and can be seen in 60% of adults with Wilson’s disease (less often in children but almost
always in neurological Wilson’s disease), albeit sometimes only by slit-lamp examination. Kayser–Fleischer rings are characterised by greenish-brown discoloration of the corneal margin appearing first at the upper periphery. They disappear with treatment.
Investigations
A low serum caeruloplasmin is the best single laboratory clue to the diagnosis. However, advanced liver failure from any cause can reduce the serum caeruloplasmin, and occasionally it is normal in Wilson’s disease. Other features of disordered copper metabolism should thereforebe sought; these include a high free serum copper
concentration, a high urine copper excretion of greater
than 0.6 μmol/24 hrs (38 μg/24 hrs) and a very high
hepatic copper content.
Measuring 24-hour urinary copper excretion whilst giving D- penicillamine is a useful confirmatory test; more than 25 μmol/24 hrs is considered diagnostic of Wilson’s .
Management
The copper-binding agent, penicillamine, is the drug of
choice. The dose given must be sufficient to produce
cupriuresis and most patients require 1.5 g/day (range
1–4 g). The dose can be reduced once the disease is in
remission but treatment must continue for life, even
through pregnancy. Care must be taken to ensure that
re-accumulation of copper does not occur. Abrupt discontinuation of treatment must be avoided because this may precipitate acute liver failure. Toxic effects occur in one-third and include rashes, protein-losing nephropathy, lupus-like syndrome and bone marrow depression. If these do occur, trientine dihydrochloride (1.2–2.4 g/day) and zinc (50 mg 3 times daily) are potential alternatives.
Liver transplantation is indicated for fulminant liver failure or for advanced cirrhosis with liver failure. The value of liver transplantation in severe neurological Wilson’s disease is unclear. Prognosis is excellent, provided treatment is started before there is irreversible damage.
Siblings and children of patients with Wilson’s disease must be investigated and treatment should be given to all affected individuals, even if they are asymptomatic.
Alpha1-antitrypsin deficiency
Alpha1-antitrypsin (α1-AT) is a serine protease inhibitor(Pi) produced by the liver. The mutated form of α1-AT (PiZ) cannot be secreted into the blood by liver cells because it is retained within the endoplasmic reticulum of the hepatocyte. Homozygous individuals (PiZZ) have low plasma α1-AT concentrations, although globules containing α1-AT are found in the liver, and these people
may develop hepatic and emphysema.
Liver manifestations include cholestatic jaundice in the neonatal period (neonatal hepatitis), which can resolve spontaneously, chronic hepatitis and cirrhosis in adults, and, in the long term, HCC.
Alpha1-AT deficiency is a not uncommon exacerbating factor for liver disease of other aetiologies, and the possibility of dual pathology
should be considered in patients in whom severity of disease, such as ALD, appears disproportionate to the level of underlying insult.
There are no clinical features distinguishing liver
disease due to α1-AT deficiency from liver disease due
to other causes, and the diagnosis is made from the low plasma α1-AT concentration and genotyping for the
presence of the mutation.
There is no specific treatment; the risk of severe and early-onset emphysema means that all patients should be advised to stop smoking.
Gilbert’s syndrome
Gilbert’s syndrome is by far the most common inherited disorder of bilirubin metabolism .
It is an autosomal dominant trait caused by a mutation in the promoter region of the UDP- glucuronyl transferase enzyme, which leads to reduced enzyme expression.
This results in decreased conjugation of bilirubin,
which accumulates as unconjugated bilirubin in the
blood. The levels of unconjugated bilirubin increase
during fasting, as fasting reduces levels of UDPglucuronyl transferase.
Clinical features
The typical presentation is with isolated elevation ofbilirubin, typically, although not exclusively, in the
setting of physical stress or illness. There are no stigmata of chronic liver disease other than jaundice. Increased excretion of bilirubin and hence stercobilinogen leads to normal- coloured or dark stools, and increased urobilinogen excretion causes the urine to turn dark on standing as urobilin is formed.
In the presence of haemolysis, pallor due to anaemia and splenomegaly due to excessive reticulo-endothelial activity are usually present.
Investigations
The plasma bilirubin is usually less than 100 μmol/L(~6 mg/ dL) and the LFTs are otherwise normal. There
is no bilirubinuria because the hyperbilirubinaemia is
predominantly unconjugated. Hepatic histology is
normal and liver biopsy is not recommended for the
investigation of patients with possible Gilbert’s syndrome.
The condition is not associated with liver injury
and thus has an excellent prognosis, needs no treatment, and is clinically important only because it may be mistaken for more serious liver disease.
VASCULAR LIVER DISEASE
Metabolically, the liver is highly active and has large
oxygen requirements. This places it at risk of ischaemic
injury in settings of impaired perfusion. The risk is mitigated, however, by the dual perfusion of the liver (via portal vein as well as hepatic artery), with the former
representing a low-pressure perfusion system that offers
protection against the potential effects of arterial hypotension.
The single outflow through the hepatic vein and the low-pressure perfusion system of the portal vein make the liver vulnerable to venous thrombotic ischaemia
in the context of Budd– Chiari syndrome and portal vein thrombosis, respectively.
Portal vein thrombosis
Portal venous thrombosis as a primary event is rare, butcan occur in any condition predisposing to thrombosis.
It may also complicate intra-abdominal inflammatory or
neoplastic disease, and is a recognised cause of portal
hypertension. Acute portal venous thrombosis causes
abdominal pain and diarrhoea, and may rarely lead to
bowel infarction, requiring surgery. Treatment is otherwise based on anticoagulation, although there are no randomised data on efficacy. An underlying thrombophilia needs to be excluded.
Subacute thrombosis can be asymptomatic but may subsequently lead to extrahepatic portal hypertension . Ascites is unusual in non-cirrhotic portal hypertension, unless the albumin is particularly low.
Portal vein thrombosis can arise as a secondary event
in patients with cirrhosis and portal hypertension, and
is a recognised cause of decompensation in patients with
previously stable cirrhosis. In patients showing such
decompensation, portal vein patency should be assessed
by ultrasound with Doppler flow studies.
Hepatopulmonary syndrome
This condition is characterised by resistant hypoxaemia
(PaO2 < 9.3 kPa or 70 mm Hg), intrapulmonary vascular dilatation in patients with cirrhosis, and portal hypertension.
Clinical features include finger clubbing, cyanosis,
spider naevi and a characteristic reduction in arterial
oxygen saturation on standing. The hypoxia is due to
intrapulmonary shunting through direct arteriovenous communications. Nitric oxide (NO) overproduction may
be important in pathogenesis. The hepatopulmonary
syndrome can be treated by liver transplantation but, if
severe (PaO2< 6.7 kPa or 50 mm Hg), is associated with an increased operative risk.
Portopulmonary hypertension
This unusual complication of portal hypertension issimilar to ‘primary pulmonary hypertension’ .It
is defined as pulmonary hypertension with increased
pulmonary vascular resistance and a normal pulmonary
artery wedge pressure in a patient with portal hypertension.
The condition is caused by vasoconstriction and
obliteration of the pulmonary arterial system, and leads
to breathlessness and fatigue.
Budd– Chiari syndrome
This uncommon caused by thrombosis of the larger hepatic veins and sometimes the inferior vena cava. Many patients have haematological disorders such as myelofibrosis, primary proliferative polycythaemia, paroxysmal nocturnal haemoglobinuria, or antithrombin III, protein C or protein S deficiencies. Pregnancy and oral contraceptive use, obstruction due to tumours (particularly carcinomas of the liver, kidneys or adrenals), congenital venous webs and occasionally inferior vena caval stenosis. The underlying cause cannot be found in about 50% of patients.
Hepatic congestion affecting the centrilobular areas is followed by centrilobular fibrosis, and eventually cirrhosis supervenes in those who survive long enough.
Clinical features
Acute venous occlusion causes rapid development of
upper abdominal pain, marked ascites and occasionally acute liver failure. More gradual occlusion causes gross ascites and, often, upper abdominal discomfort.
Hepatomegaly, frequently with tenderness over the
liver, is almost always present. Peripheral oedema
occurs only when there is inferior vena cava obstruction.
Features of cirrhosis and portal hypertension develop in those who survive the acute event.
Investigations
The LFTs can show the features of acute hepatitis.Ascitic fluid analysis shows a protein concentration
above 25 g/L (exudate) in the early stages; however, this
often falls later in the disease. Doppler ultrasound may
reveal obliteration of the hepatic veins and reversed flow
or associated thrombosis in the portal vein.
CT and MRI may also demonstrate occlusion
of the hepatic veins and inferior vena cava. Liver
biopsy demonstrates centrilobular congestion with
fibrosis, depending on the duration of the illness. Venography is only needed if CT and MRI are unable to
demonstrate the hepatic venous anatomy clearly.
Management
Predisposing causes should be treated as far as possible;
where recent thrombosis is suspected, thrombolysis
with streptokinase, followed by heparin and oral anticoagulation, should be considered. Ascites is initially
treated medically but often with only limited success.
Short hepatic venous strictures can be treated with angioplasty. In the case of more extensive hepatic vein occlusion, many patients can be managed successfully by
insertion of a covered TIPSS, followed by anticoagulation.
Surgical shunts, such as portacaval shunts, are less
commonly performed now that TIPSS is available. Occasionally, a web can be resected or an inferior vena caval stenosis dilated.
Progressive liver failure is an indication for liver transplantation and life-long anticoagulation.
Prognosis without transplantation or shunting is
poor, particularly following an acute presentation with
liver failure. A 3-year survival of 50% is reported in
those who survive the initial event. The 1- and 10-year
survival following liver transplantation is 85% and 69%
respectively, and this compares with a 5- and 10-year
survival of 87% and 37% following surgical shunting.
Nodular regenerative hyperplasia of the liver
This is the most common cause of non-cirrhotic portalhypertension , characterised by small hepatocyte nodules throughout the liver without fibrosis. It is believed to be due to damage to small hepatic arterioles and portal venules. It occurs in older people and is associated with connective tissue disease, haematological diseases and immunosuppressive drugs, such as azathioprine. The condition is usually asymptomatic, but occasionally presents with portal hypertension or with an abdominal mass. The diagnosis is made by liver biopsy. Liver function is good and the prognosis is very favourable. Management is based on treatment of the portal hypertension.
PREGNANCY AND THE LIVER
The inter-relationship between liver disease and pregnancy can be a complex one. Three possibilities need to be borne in mind when faced with a pregnant woman with a liver abnormality:
• that this represents a worsening of pre-existing chronic liver or biliary disease
• that this represents a genuine first presentation of liver disease that is not intrinsically related to pregnancy
• that this represents a genuine pregnancy-associated liver injury process. Joint management between hepatologists and obstetricians is essential.

Abnormal liver function tests in pregnancy
Intercurrent and pre-existing liver diseaseAcute hepatitis A can occur during pregnancy but has
no effect on the fetus.
Chronic hepatitis B requires identification in pregnancy because of long-term health implications for the mother and the effectiveness of perinatal vaccination (with or without pre-delivery maternal antiviral therapy) in reducing neonatal acquisition of chronic hepatitis B. Maternal transmission of hepatitis C occurs in 1% of cases, and there is no convincing evidence that the mode of delivery affects this. Hepatitis E is reported to progress to acute liver failure much more commonly in pregnancy, with a 20% maternal mortality.
Pregnancy may be associated with either worsening or improvement of autoimmune hepatitis, although improvement during pregnancy and rebound postpartum is the most common pattern seen. Complications of portal hypertension may be a particular issue in the second and third trimesters.
Gallstones are more common during pregnancy, and may present with cholecystitis or biliary obstruction. In biliary obstruction due to gallstones, therapeutic ERCP can be safely performed, but lead protection for the fetus is essential and X-ray screening must be kept to a minimum.
Pregnancy-associated liver disease
The following conditions occur only during pregnancy,may recur in subsequent pregnancies and resolve after
delivery of the baby.
Acute cholestasis of pregnancy
This accounts for 20% of cases of jaundice in pregnancy;
it usually occurs in the third trimester of pregnancy but
can arise earlier. It may be linked with intrauterine
growth retardation and premature birth, but is most characteristically associated with intrauterine fetal death
if the pregnancy goes beyond 36 weeks of gestation. The condition characteristically presents with itching and cholestatic LFTs. A normal bilirubin and hepatitic LFTs do not, however, preclude the diagnosis.
Bile salts are elevated in the serum and this represents a useful clinical test. Delivery leads to resolution and should be considered from 36 weeks onwards.
Pregnancy should not be allowed to continue beyond term because of a steep rise in the risk of intrauterine death. UDCA (15 mg/kg daily) effectively controls itching and probably prevents premature birth. The major issue with UDCA in acute cholestasis of pregnancy is the relatively long time required to achieve effective levels within the bile pool, which renders it of little use in patients presenting in the very end stages of pregnancy. Cholestasis recurs in 60% of future pregnancies and the opportunities available for screening based on previous risk make UDCA a much more viable therapy in this setting.
Acute fatty liver of pregnancy
This is more common in twin and first pregnancies, and may arise more frequently when the fetus is male. It typically presents between 31 and 38 weeks of pregnancy with vomiting and abdominal pain followed by jaundice. In severe cases, this may be followed by lactic acidosis, coagulopathy, encephalopathy and renal failure. Hypoglycaemia can also occur. The features are characteristic of a defect in beta-oxidation of fatty acids in the mitochondria that leads to the formation of small fat droplets in liver cells (known as microvesicular fatty liver). Some women are heterozygous for loss-of-function mutations in the long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) gene.Other causes of microvesicular steatosis that have a similar clinical presentation outside pregnancy are shown in Box
Differentiation from toxaemia of pregnancy
(which is more common) can be achieved by the finding
of high serum uric acid levels and the absence of haemolysis.
Overlap between acute fatty liver of pregnancy,
HELLP (see below) and toxaemia of pregnancy can
occur. Early diagnosis, specialist care and delivery of the fetus have led to a fall in maternal and perinatal mortality to 1% and 7% respectively.
Toxaemia of pregnancy and HELLP
The HELLP syndrome (haemolysis, elevated liver enzymes and low platelets) is a variant of pre- eclampsia that tends to affect multiparous women. It usually presents at 27–36 weeks of pregnancy with hypertension, proteinuria and fluid retention. Jaundice only occurs in 5%. Blood tests may show low haemoglobin, with fragmented red cells, markedly elevated serum transaminases and raised D-dimers. The condition can be complicated by hepatic infarction and rupture. Maternal complications also include disseminated intravascular coagulation and placental abruption. Maternal mortality is 1% and perinatal mortality can be up to 30%. Delivery usually leads to prompt resolution, and disease recurs in fewer than 5% of subsequent pregnancies.LIVER TRANSPLANTATION
The outcome has improved significantly over the last decade, so in low-risk individuals now has a 1-year survival rate of more than 90%.Indications and contraindications
Around 9500 liver transplants are undertaken in Europe and the USA annually. About 10% for acute liver failure, 6% for metabolic diseases, 71% for cirrhosis and 11% for hepatocellular carcinoma. In North America the most common indication is hepatitis C cirrhosis, about 10–20% of transplants being for alcoholic cirrhosis . Patients with alcoholic liver disease need to show a capacity for abstinence.
The main contraindications are sepsis, extrahepatic malignancy, active alcohol or other substance misuse, and marked cardiorespiratory dysfunction.
Patients are matched for ABO group, but do not require HLA-matching with donors, as the liver is a relatively immune-privileged organ compared with heart or kidney.
MELD score is used to identify and prioritise patients for transplantation. In the UK, a similar score that also incorporates serum sodium, the United Kingdom End- Stage Liver Disease score (UKELD), is used.
Two types of transplant are increasingly used because
of insufficient cadaveric donors:
• Split liver transplantation. A cadaveric donor liver can be split into two, with the larger right lobe used in an adult and the smaller left lobe used in a child.
• Living donor transplantation. This is normally performed using the left lateral segment or the right lobe. The donor mortality is significant at 0.5–1%.
To be listed for elective (non- superurgent) transplantation in the UK, patients must have >50% projected post-transplant 5-year survival and fall into one of three categories:
• Category 1: estimated 1-year mortality without
transplantation of more than 9% (equivalent to a
UKELD score of more than 49 points)
• Category 2: HCC , based on CT, a single lesion of < 5 cm maximum diameter or fewer than three lesions each <3 cm in diameter without macrovascular invasion or metastases.
• Category 3: ‘variant syndromes’, including resistant ascites, hepatopulmonary syndrome, chronic encephalopathy, intractable pruritus, amyloidosis, primary hyperlipidaemia, polycystic liver disease and recurrent cholangitis.
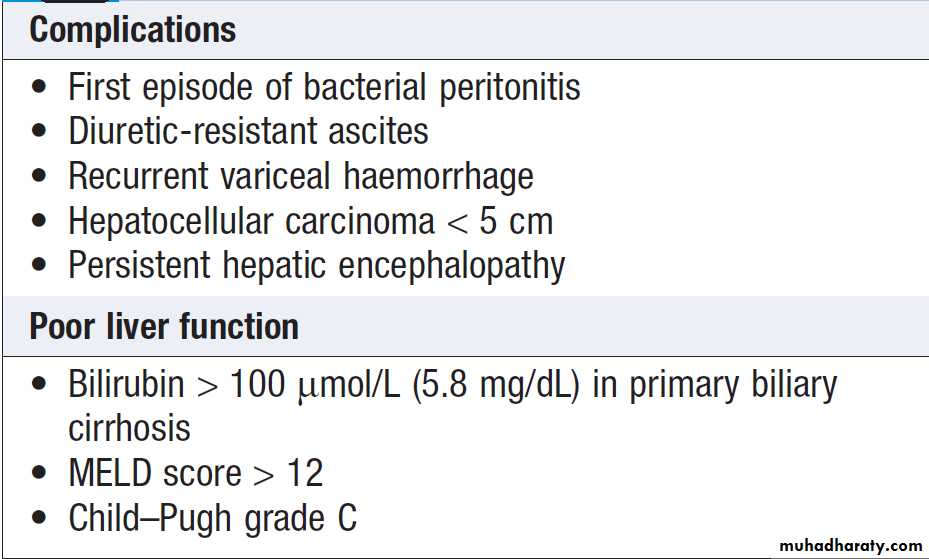
Indications for liver transplant assessment
for cirrhosis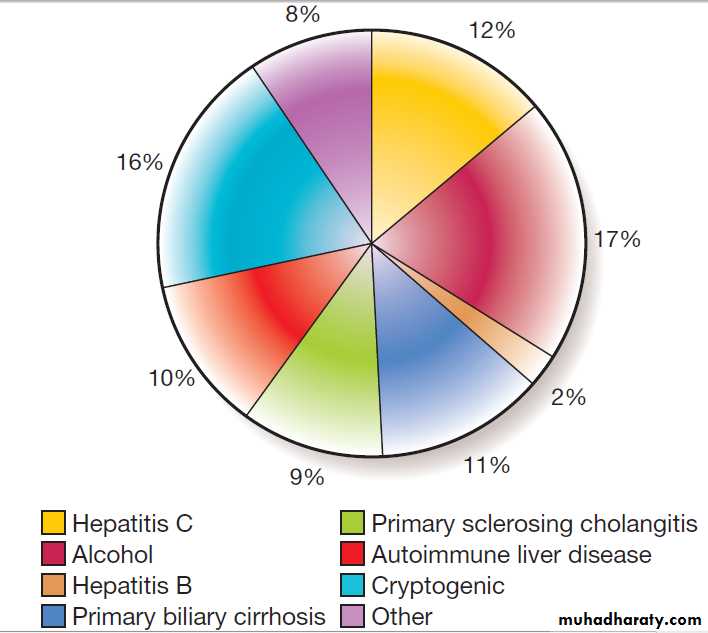
Indications for liver transplantation in the UK between
2001 and 2005.Complications
Early complicationsPrimary graft non-function
Results from ischaemia following removal from the donor and prior to reperfusion in the recipient. Factors that increase the likelihood of primary non-function include increasing donor age, degree of steatosis, and the length of ischaemia. Treatment is supportive. Occasionally, recovery is not seen and re-transplant is necessary. Technical complications
These include hepatic artery thrombosis, which may necessitate re-transplantation. Anastomotic biliary strictures can also occur; these may respond to endoscopic balloon dilatation and stenting, or require surgical reconstruction. Portal vein thrombosis is rare.
Rejection Less immunosuppression is needed following transplantation than with kidney or heart/lung grafting. Initial immunosuppression is usually with tacrolimus or ciclosporin,prednisolone and azathioprine or mycophenolate. Acute cellular rejection occurs in 60–80% of patients, commonly at 5–10 days post transplant. This normally responds to 3 days of high-dose IV methylprednisolone. Infections
Bacterial infections, such as pneumonia and wound infections, Cytomegalovirus (primary infection or reactivation) in the 3 months after transplantation and can cause hepatitis. Patients who have never had cytomegalovirus infection but who receive a liver from a donor who has been exposed are at greatest risk of infection, and are usually given prophylactic antiviral therapy such as valganciclovir.
Herpes simplex virus infection may occur. Prophylaxis is given to recipients who have had previous exposure to tuberculosis for the first 6 months after transplantation.
Late complications
These include recurrence of the initial disease and complications due to the immunosuppressive therapy.
Metabolic syndrome described in about 50% of transplant recipients within 6 months. Chronic vascular rejection is rare, occurring in only 5%.
Prognosis
The outcome following transplantation for acute liver failure is worse than for CLD because most patients have multi-organ failure at the time of transplantation. The 1-year survival is 65% ,59% at 5 years. The 1-year survival for cirrhosis is over 90%, falling to 70–75% at 5 years.

Liver disease in old age
CHOLESTATIC AND BILIARY DISEASE‘Cholestasis’ relates to a biochemical abnormality
(elevation of ALP and, if the process is significant, elevation in serum bile acid levels and bilirubin) resulting from an abnormality in bile flow.
The cause can range from inherited or acquired dysfunction
of transporter molecules responsible for the production
of canalicular bile to physical obstruction of the
extrahepatic bile duct.
‘Biliary disease’ relates to pathology at any level from the small intrahepatic bile ducts to the sphincter of Oddi. Although there is very significant overlap, there are scenarios where cholestasis can exist without biliary disease (transporter disease or pure drug-induced cholestasis) and where biliary disease can exist without cholestasis
(when disease of the bile duct does not impact on bile flow).
Benign recurrent intrahepatic cholestasis
This rare condition usually presents in adolescence andis characterised by recurrent episodes of cholestasis,
lasting 1–6 months. It is now known to be mediated by mutations in the ATP8B1 gene, which lies on chromosome 18 and encodes FIC1.
Episodes start with pruritus, while painless jaundice
develops later.
LFTs show a cholestatic pattern.
Liver biopsy shows cholestasis during an episode but is
normal between episodes.
Treatment is required to relieve the symptoms, such as pruritus and the long-term prognosis is good.
Cystic fibrosis
associated with a biliary cirrhosis in about 5%. Splenomegaly and an elevated ALP are characteristic. UDCA improves liver blood tests but it is not known whether the drug can prevent progression.
Deficiency of fat-soluble vitamins (A, D, E and K) need to be treated in view of both biliary and pancreatic disease.
Extrahepatic biliary disease
Choledochal cysts
Cysts anywhere in the biliary tree. In the neonate, they may present with jaundice or biliary peritonitis. Recurrent jaundice, abdominal pain and cholangitis may arise in the adult. Liver abscess and biliary cirrhosis may develop, and increased incidence of cholangiocarcinoma. Excision of the cyst with hepatico-jejunostomy is of choice.
Secondary biliary cirrhosis
Secondary biliary cirrhosis develops after prolonged large duct biliary obstruction due to gallstones, benign bile duct strictures or sclerosing cholangitis .Carcinomas rarely cause secondary biliary cirrhosis because few patients survive long enough. The clinical features are those of chronic cholestasis with episodes of ascending cholangitis or even liver abscess .
Cirrhosis, ascites and portal hypertension are late features.
Relief of biliary obstruction may require endoscopic or surgical intervention.
Cholangitis requires treatment with antibiotics, which can be given continuously if attacks recur frequently.
Gallstones
Is the most common disorder. In developed countries, gallstones occur in 7% of males and 15% of females18–65 years, in the elderly the sex ratio is about equal. Increase in dietary cholesterol, fat, and refined carbohydrate or lack of dietary fibre has been implicated.
Pathophysiology
Gallstones are conveniently classified into cholesterol or pigment stones, although the majority are of mixed. Cholesterol stones are most common in developed countries, whereas pigment stones are more frequent in developing countries. Gallstones contain varying quantities of calcium salts, including calcium bilirubinate, carbonate, phosphate and palmitate, which are radio-opaque.
Cholesterol gallstones
Cholesterol is held in solution in bile by its association with bile acids and phospholipids in the form of micelles. In gallstone disease the liver produces bile that contains an excess of cholesterol, because there is either a relative deficiency of bile salts or a relative excess of cholesterol (‘lithogenic’ bile). Factors favouring nucleation (mucus, calcium, fatty acids, other proteins) and antinucleating factors (apolipoproteins) have been described.Pigment stones
Are almost always the consequence of bacterial or parasitic biliary infection. Haemolysis is important, as these stones occur in chronic haemolytic disease.

Pathogenic factors leading to the
production of lithogenic bile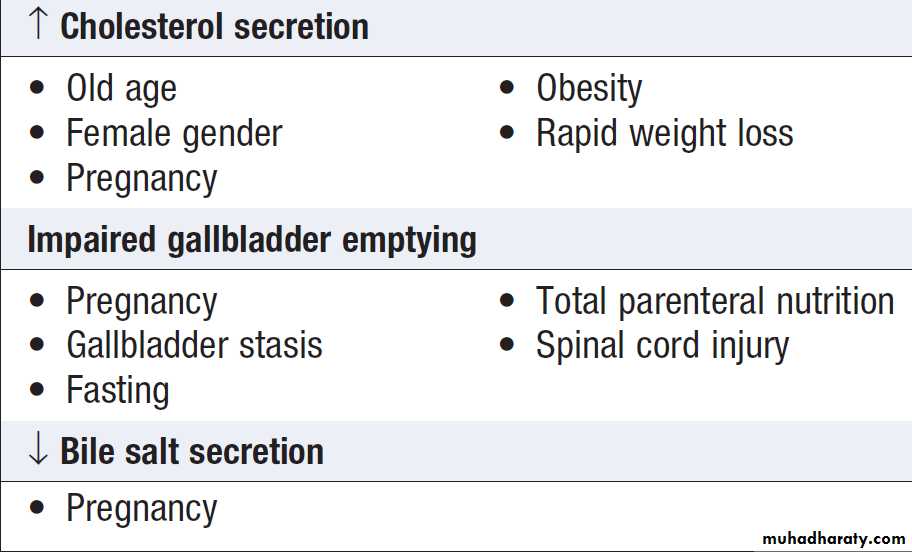
Risk factors and mechanisms for cholesterol gallstones
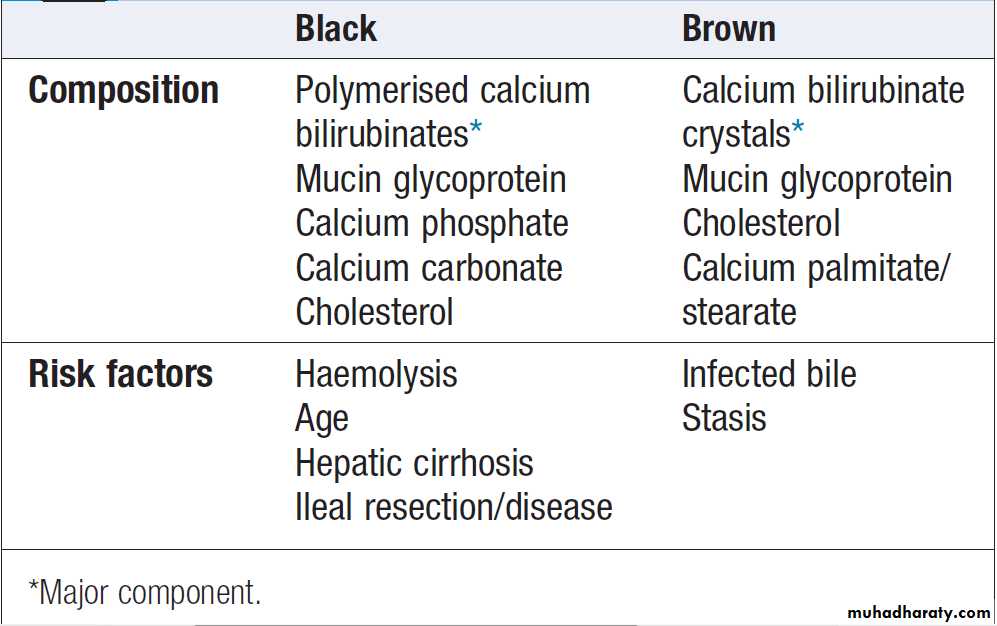
Composition of and risk factors for pigment stones
Biliary sludgeThis describes gelatinous bile that contains numerous
microspheroliths of calcium bilirubinate granules and
cholesterol crystals, as well as glycoproteins; it is an
important precursor to the formation of gallstones. Biliary sludge is frequently formed under normal conditions, but then either dissolves or is cleared by the gallbladder; in 15% it persist to form cholesterol stones. Fasting, parenteral nutrition and pregnancy are also associated with sludge formation.
Clinical features
Only 10% with gallstones develop clinical evidence of gallstone disease. Symptomatic stones manifest as either biliary pain (‘biliary colic’ stone acutely impacted in the cystic duct) or cholecystitis .The term ‘biliary colic’ is a misnomer because the pain does not rhythmically increase and decrease in intensity, the pain occurs suddenly and persists for about 2 hours; if it continues for > 6 hours, a complication such as cholecystitis or pancreatitis may be present. Pain is usually felt in the epigastrium 70% or right upper quadrant 20%, and radiates to the interscapular region or the tip of the right scapula, but other sites include the left upper quadrant and the lower chest. The pain can mimic oesophagitis, myocardial infarction or dissecting aneurysm.
Combinations of fatty food intolerance, dyspepsia and flatulence not attributable to other causes have been referred to as ‘gallstone dyspepsia’. These symptoms are not now recognised as being caused by gallstones and are best regarded as functional dyspepsia.
A mucocele may develop if there is slow distension of the gallbladder from continuous secretion of mucus; if this material becomes infected, an empyema supervenes.
Calcium may be secreted into the lumen of the hydropic gallbladder, causing ‘limey’ bile, and if calcium salts are precipitated in the gallbladder wall, the radiological
appearance of ‘porcelain’ gallbladder results.
Gallstones in the gallbladder (cholecystolithiasis) migrate to the common bile duct (choledocholithiasis,) in approximately 15% of patients and cause biliary colic. Rarely, fistulae develop between the gallbladder and the duodenum, colon or stomach. If this occurs, air will be seen in the biliary tree on plain abdominal X-rays. If a stone > 2.5 cm in diameter has migrated into the gut, it may impact either at the terminal ileum or occasionally in the duodenum or sigmoid colon. The resultant intestinal obstruction may be followed by ‘gallstone ileus’. Gallstones impacted in the cystic duct may cause stricturing of the common hepatic duct and the clinical picture of extrahepatic biliary diseases (with its important differential of malignant bile duct stricture).
Jaundice due to gallstones is a stone passing from the cystic duct into the common bile duct (choledocholithiasis), which may also result in cholangitis or acute pancreatitis.
It is usually very small stones that precipitate acute pancreatitis, due (it is thought) to oedema at the ampulla as the stone passes into the duodenum (no stone is seen within the bile duct in 80% of cases of presumed gallstone pancreatitis, suggesting stone passage). Previous stone passage is also the likely cause of most cases of benign papillary fibrosis, it may present with jaundice, obstructive LFTs with biliary dilatation, post-cholecystectomy pain or acute pancreatitis). Cancer of the gallbladder is uncommon but in over 95% of cases is associated with gallstones.
The diagnosis is usually made as an incidental histological finding following cholecystectomy for gallstone disease.
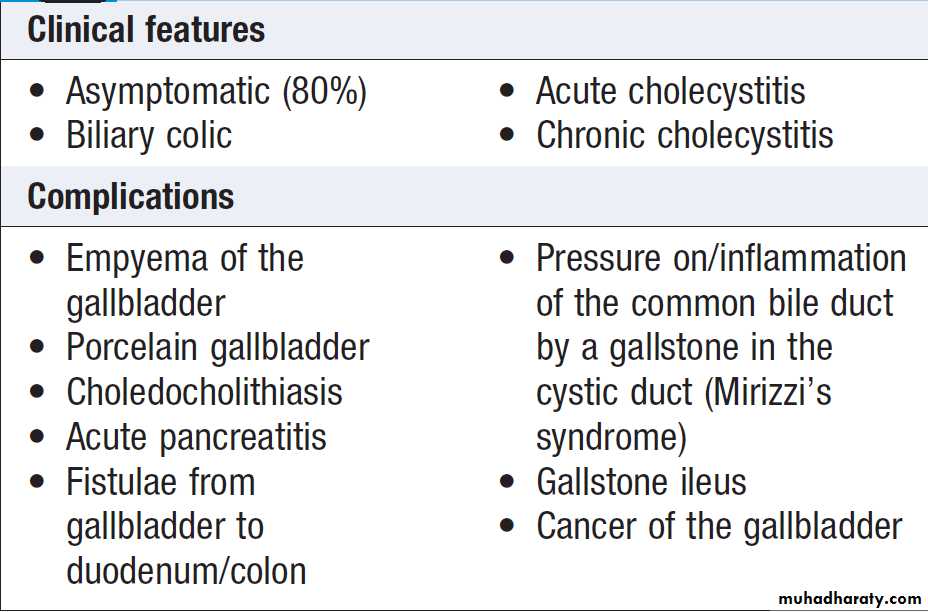
Clinical features and complications of gallstones
InvestigationsUltrasound is the investigation of choice for diagnosing gallstones. Most stones are diagnosed by transabdominal ultrasound, which has a greater than 92% sensitivity and 99% specificity for gallbladder stones . CT and MRCP are excellent modalities for detecting complications of gallstones (distal bile duct stone or gallbladder empyema), but are inferior to ultrasound in defining their presence in the gallbladder.
When recurrent attacks of otherwise unexplained acute pancreatitis occur, this may result from ‘microlithiasis’ in the gallbladder or common bile duct, and this is best assessed by endoscopic ultrasound (EUS).
Management
Asymptomatic gallstones found incidentally should not be treated because the majority will never cause symptoms. Symptomatic gallstones are best treated surgically by laparoscopic cholecystectomyy .

Treatment of gallstones
Acute cholecystitisAcute cholecystitis is almost always associated with obstruction of the gallbladder neck or cystic duct by a gallstone. Occasionally, obstruction may be by mucus, parasitic worms or a bile tumour, or may follow endoscopic bile duct stenting. The pathogenesis is unclear but the initial inflammation is possibly chemically induced. This leads to mucosal damage, which releases phospholipase, converting biliary lecithin to lysolecithin, a recognised mucosal toxin.
At the time of surgery, 50% of cultures of the gallbladder contents are sterile. Infection occurs eventually, and in elderly patients or those with diabetes mellitus a severe infection with gas-forming organisms can cause emphysematous cholecystitis.
Acalculous cholecystitis can occur in the intensive care setting and in association with parenteral nutrition, sickle cell disease and diabetes.
Clinical features
The cardinal feature is pain in the right upper quadrant but also in the epigastrium, the right shoulder tip or the interscapular region. Differentiation between biliary colic and acute cholecystitis may be difficult;
features suggesting cholecystitis include severe and prolonged pain, fever and leucocytosis.
Examination shows right hypochondrial tenderness, rigidity worse on inspiration (Murphy’s sign) and occasionally a gallbladder mass (30%). Fever but rigors are unusual.
Jaundice occurs in less than 10% and is usually due to passage of stones into the common bile duct, or compression or even stricturing of the common bile duct following stone impaction in the cystic duct (Mirizzi’s syndrome). Gallbladder perforation occurs in 10–15% of cases, and gallbladder empyema may arise.
Investigations
leucocytosis is common. Minor increase transaminases and amylase. Only when the amylase is higher than 1000 U/L can pain be confidently attributed to acute pancreatitis which may be a potentially serious complication of gallstones, since moderately elevated levels of amylase can occur in many other causes of abdominal pain
Plain X-rays of the abdomen and chest may show radio-opaque gallstones, and rarely intrabiliary gas due to fistulation of a gallstone into the intestine; they are important in excluding lower lobe pneumonia and a perforated viscus.
US detects gallstones and gallbladder thickening due to cholecystitis, but gallbladder empyema or perforation is best assessed by CT.
Management
Medical
This consists of bed rest, pain relief, antibiotics and IV fluids. Moderate pain can be treated with NSAIDs but more severe pain should be managed with opiates.
A cephalosporin (such as cefuroxime) or piperacillin/ tazobactam is the antibiotic of choice, but metronidazole is normally added in severely ill patients.
Nasogastric aspiration is only needed for persistent vomiting. Cholecystitis usually resolves with medical treatment, but the inflammation may progress to an empyema or perforation and peritonitis.
Surgical
Urgent surgery is the optimal treatment when cholecystitisprogresses in spite of medical therapy and when
complications such as empyema or perforation develop.
Operation should be carried out within 5 days of the
onset of symptoms. Delayed surgery after 2–3 months is
no longer favoured.
When cholecystectomy may be difficult due to extensive inflammatory change, percutaneous gallbladder drainage can be performed, with subsequent cholecystectomy 4–6 weeks later. Recurrent biliary colic or cholecystitis is frequent if the gallbladder is not removed.
Chronic cholecystitis
Chronic inflammation of the gallbladder is almost invariablyassociated with gallstones. The usual symptoms
are those of recurrent attacks of upper abdominal
pain, often at night and following a heavy meal. The
clinical features are similar to those of acute calculous cholecystitis but milder.
The patient may recover spontaneously or following analgesia and antibiotics. Patients are usually advised to undergo elective laparoscopic cholecystectomy.
Acute cholangitis
Acute cholangitis is caused by bacterial infection of bile ducts and occurs in patients with other biliary problems, such as choledocholithiasis , biliary strictures or tumours, or after ERCP. Jaundice, fever (with or without rigors) and right upper quadrant pain are the main presenting features (‘Charcot’s triad’).
Treatment
is with antibiotics, relief of biliary obstruction and removal (if possible) of the underlying cause.
Choledocholithiasis
Stones in the common bile duct (choledocholithiasis) occur in 10–15% of patients with gallstones , which have usually migrated from the gallbladder.Primary bile duct stones are rare but can develop within
the common bile duct many years after a cholecystectomy,
and are sometimes related to biliary sludge arising from dysfunction of the sphincter of Oddi. In Far Eastern countries, primary common bile duct stones are thought to follow bacterial infection secondary to parasitic infections with Clonorchis sinensis, Ascaris lumbricoides or Fasciola hepatica .Common bile duct stones can cause bile duct obstruction and may be complicated by cholangitis due to secondary bacterial infection, septicaemia, liver abscess and biliary stricture.
Clinical features
Choledocholithiasis may be asymptomatic, may be found incidentally by operative cholangiography at cholecystectomy, or may manifest as recurrent abdominal pain with or without jaundice. The pain is usually in the right upper quadrant, and fever, pruritus and dark urine may be present. Rigors may be a feature; jaundice is common and usually associated with pain.Physical examination may show the scar of a previous cholecystectomy; if the gallbladder is present, it is usually small, fibrotic and impalpable.
Investigations
The LFTs show a cholestatic pattern. If cholangitis is present, the patient usually has a leucocytosis. The most convenient method of demonstrating obstruction to the common bile duct is by transabdominal ultrasound. This shows dilated extrahepatic and intrahepatic bile ducts, together with gallbladder stones .50% of bile duct stones are missed on ultrasound, particularly those in the distal common bile duct. EUS is extremely accurate at identifying bile duct stones. MRCP is non-invasive, and is indicated when intervention is not necessarily mandatory (e.g. the patient with possible bile duct stones but no jaundice or sepsis).
Management
Cholangitis should be treated with analgesia, intravenous fluids and broad-spectrum antibiotics, such as cefuroxime and metronidazole .Blood cultures should be taken before the antibiotics are administered. Patients also require urgent decompression of the biliary tree and stone removal. ERCP with biliary sphincterotomy and stone extraction is the treatment of choice and is successful in about 90% of patients. If ERCP fails, other approaches include percutaneous transhepatic drainage and combined (‘rendezvous’) endoscopic procedures, extracorporeal shock wave lithotripsy (ESWL) and surgery.Surgical treatment of choledocholithiasis is performed less frequently than ERCP because it carries higher morbidity and mortality. Before the common bile duct is explored, the diagnosis of choledocholithiasis should be confirmed by intraoperative cholangiography.
If gallstones are found, the bile duct is explored, all stones are removed, stone clearance is checked by cholangiography or choledochoscopy, and a T-tube is inserted into the common bile duct. It is now possible to achieve these goals laparoscopically in specialist centres.
Tumours of the gallbladder and bile duct
Carcinoma of the gallbladderThis is an uncommon tumour, occurring more often in females and usually in those over the age of 70 years.
More than 90% are adenocarcinomas; the remainder are
anaplastic or, rarely, squamous tumours. Gallstones are
present in 70–80% of cases and are thought to be important in the aetiology of the tumour. Individuals with a calcified gallbladder (‘porcelain gallbladder’) are at high risk of malignant change, and gallbladder polyps over 1 cm in size are associated with increased risk of malignancy; preventative cholecystectomy should be considered in such patients.
Chronic infection with Salmonella, especially in areas where typhoid is endemic, is also a risk factor.
Carcinoma of the gallbladder may be diagnosed incidentally and is found in 1–3% of gallbladders removed at cholecystectomy for gallstone disease. It may manifest as repeated attacks of biliary pain and later persistent jaundice and weight loss. A gallbladder mass may be palpable in the right hypochondrium. LFTs show cholestasis, and porcelain gallbladder may be found on X-ray. The tumour can be diagnosed by ultrasonography and staged by CT. The treatment is surgical excision, but local extension of the tumour beyond the wall of the gallbladder into the liver, lymph nodes and surrounding tissues is invariable and palliative management is usually all that can be offered.
Survival is generally short, death typically occurring within 1 year in patients presenting with symptoms.
Cholangiocarcinoma
Cholangiocarcinoma (CCA) is an uncommon tumourthat can arise anywhere in the biliary tree, from the
intrahepatic bile ducts (20–25% of cases) and the confluence of the right and left hepatic ducts at the liver hilum (50–60%) to the distal common bile duct (20%).
It accounts for only 1.5% of all cancers but the incidence
is increasing.
The cause is unknown but the tumour is associated with gallstones, primary and secondary sclerosing cholangitis, Caroli’s disease and choledochal cysts . In the Far East, particularly northern Thailand, chronic liver fluke infection (Clonorchis sinensis) is a major risk factor for the development of CCA in men.
Primary sclerosing cholangitis carries a lifetime risk of CCA of approximately 20%, although only 5% of CCAs relate to primary sclerosing cholangitis.
Chronic biliary inflammation appears to be a common factor in the development of biliary dysplasia and cancer that is shared by all the predisposing causes.
Tumours typically invade the lymphatics and adjacent vessels, with a predilection for spread within perineural sheaths. The presentation is usually with obstructive jaundice. About 50% of patients also have
upper abdominal pain and weight loss. The diagnosis is made by a combination of CT and MRI but can be difficult to confirm in patients with sclerosing cholangitis.
Serum levels of the tumour marker CA19–9 are elevated in up to 80% of cases, although this may occur in biliary obstruction of any cause. In the setting of biliary obstruction, ERCP may result in positive biliary cytology.
Endoscopic ultrasound-fine needle aspiration (EUS-FNA) of bile duct masses is sometimes possible, and in specialist centres single-operator cholangioscopy with biopsy is now established.
CCAs can be treated surgically in about 20% of patients, which improves 5-year survival from < 5% to 20–40%.
Surgery involves excision of the extrahepatic biliary tree with or without a liver resection and a Roux loop reconstruction. However, most patients are treated by stent insertion across the malignant biliary stricture, using endoscopic or transhepatic techniques .
Combination chemotherapy is increasingly used and palliation with endoscopic photodynamic therapy has provided encouraging results.
Carcinoma at the ampulla of Vater
Nearly 40% of all adenocarcinomas of the small intestinearise in relationship to the ampulla of Vater, and present
with pain, anaemia, vomiting and weight loss. Jaundice
may be intermittent or persistent. The diagnosis is made
by duodenal endoscopy and biopsy of the tumour, but
staging by CT/MRI is essential. Ampullary carcinoma
must be differentiated from carcinoma of the head of the
pancreas and a cholangiocarcinoma because these latter
conditions both have a worse prognosis.
Imaging may show a ‘double duct sign’ with stricturing of both the common bile duct and pancreatic duct at the ampulla and upstream dilatation of the ducts. EUS is the most sensitive method of assessing and staging ampullary or periampullary tumours.
Curative surgical treatment can be undertaken by
pancreaticoduodenectomy, and the 5-year survival may be as high as 50%. If resection is impossible, palliative surgical bypass or stenting may be necessary.
Benign gallbladder tumours
These are uncommon, often asymptomatic and usually found incidentally at operation or postmortem. Cholesterol polyps, sometimes associated with cholesterolosis, papillomas and adenomas, are the main types.
Post-cholecystectomy syndrome
Dyspeptic symptoms following cholecystectomy (postcholecystectomy syndrome) occur in about 30% ofpatients. The syndrome occurs most frequently in women, in patients who have had symptoms for more than 5 years before cholecystectomy, and when the operation was undertaken for non- calculous gallbladder disease. An increase in bowel habit resulting from bile acid diarrhoea occurs in about 5–10% of patients and often responds to colestyramine 4–8 g daily. Severe post-cholecystectomy syndrome occurs in only 2–5%.
The usual symptoms include right upper quadrant pain, flatulence, fatty food intolerance, and occasionally jaundice and cholangitis. The LFTs may be abnormal and sometimes show cholestasis. Ultrasonography is used to detect biliary obstruction, and EUS or MRCP is used to seek common bile duct stones. If retained bile duct stones are excluded, sphincter of Oddi dysfunction should be considered (see below).
Other investigations that may be required include upper gastrointestinal endoscopy, small bowel radiology and pancreatic function tests. The possibility of a functional illness should also be considered.
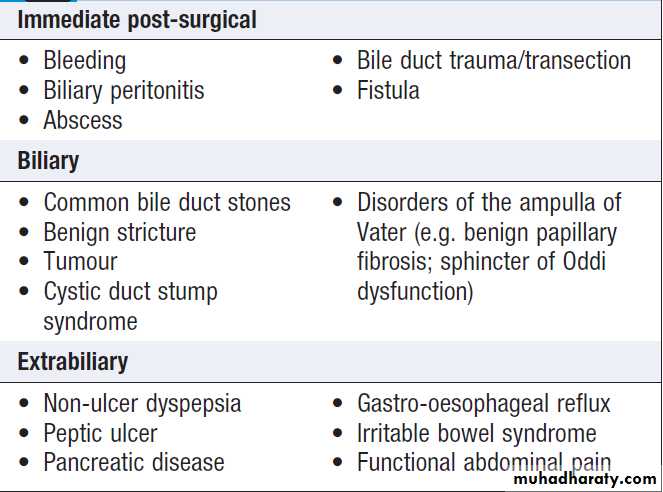
Causes of post-cholecystectomy symptoms
Sphincter of Oddi dysfunction (SOD)SO is a small smooth muscle sphincter situated at the junction of the bile duct and pancreatic duct in the duodenum, characterised by an increase in contractility that produces a benign non-calculous obstruction to the flow of bile or pancreatic juice. This may cause pain, deranged LFTs or recurrent pancreatitis.
Clinical features
Patients with SOD, who are predominantly female.
• Patients with biliary-type SOD experience recurrent, episodic biliary-type pain. They have often had a cholecystectomy but the gallbladder may be intact.
• Patients with pancreatic SOD usually present with unexplained recurrent attacks of pancreatitis.
Investigations
The diagnosis is established by excluding gallstones, and demonstrating a dilated or slowly draining bile duct. The gold standard for diagnosis is sphincter of Oddi manometry. This is associated with a high rate of procedure-related pancreatitis.
Management
Biliary-type Treated with endoscopic sphincterotomy. The results are good but patients should be warned that there is a high risk of complications, particularly acute pancreatitis. Nifedipine and/or low-dose amitriptyline may be tried.
The role of botulinum toxin (‘botox’) injection into the sphincter to improve sphincter function remains unclear.
Pancreatic SOD can be treated with pancreatic stenting followed by pancreatic sphincterotomy, carried out in specialised units. Emerging evidence suggests that all patients being investigated by ERCP for suspected SOD should also undergo prophylactic pancreatic duct stenting, because this significantly reduces the rate of procedure-related acute pancreatitis.
Classification of biliary sphincter of Oddi dysfunction
Classification of pancreatic sphincter of Oddi dysfunction
Cholesterolosis of the gallbladderlipid deposits in the submucosa and epithelium appear as multiple yellow spots on the pink mucosa, giving rise to the description ‘strawberry gallbladder’.is usually asymptomatic but may occasionally present with right upper quadrant pain. Small, fixed filling defects may be visible on ultrasonography; the radiologist can usually differentiate between gallstones and cholesterolosis.
The condition is usually diagnosed at cholecystectomy; if the diagnosis is made radiologically, cholecystectomy may be indicated, depending on symptoms.
