
Hussien Mohammed Jumaah
CABMLecturer in internal medicine
Mosul College of Medicine
2016

learning-topics
Blood diseases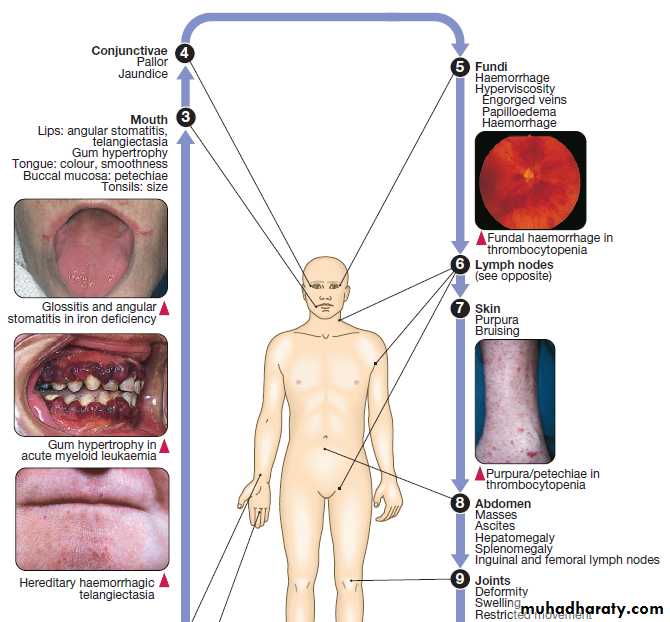
CLINICAL EXAMINATION IN BLOOD DISEASE
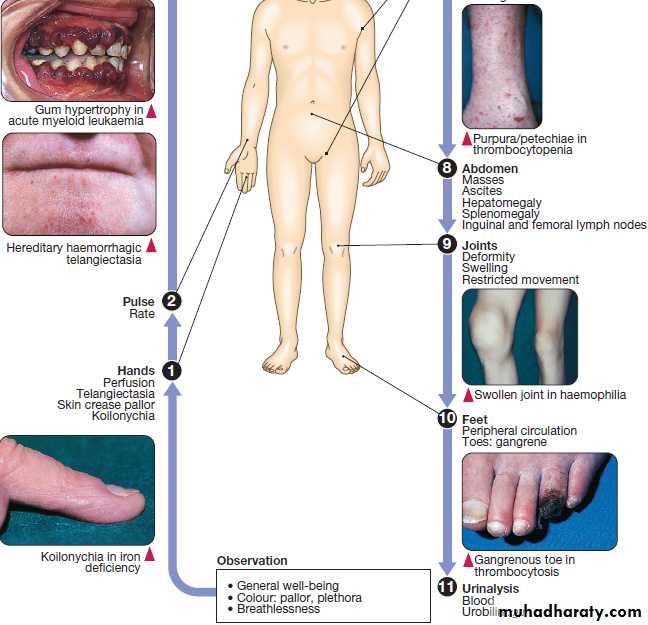

Anaemia
Symptoms and signs help to indicate the clinical severity of anaemia.A full history and examination is needed to
identify the underlying cause.
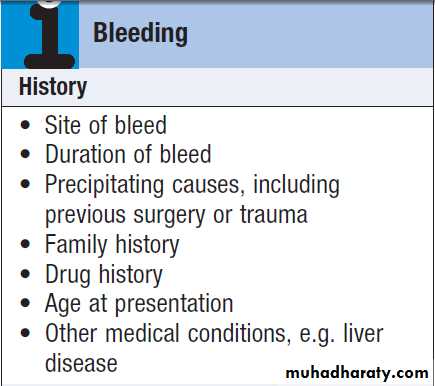

Bleeding can be due to congenital or acquired abnormalities in the clotting system. History and examination help to clarify the severity and underlying cause of the bleeding.
FUNCTIONAL ANATOMY AND PHYSIOLOGY
Blood flows throughout the body in the vascular system,and consists of:
• red cells, which transport oxygen from the lungs to
the tissues
• white cells, which defend against infection
• platelets, which interact with blood vessels and
clotting factors to maintain vascular integrity and
prevent bleeding
• plasma, which contains proteins with many functions, including antibodies and coagulation factors.
Haematopoiesis
Haematopoiesis describes the formation of blood cells, an active process that must maintain normal numbers of circulating cells and be able to respond rapidly to increased demands such as bleeding or infection.During development, haematopoiesis occurs in the liver and spleen, and subsequently in red bone marrow in the medullary cavity of all bones. In childhood, red marrow is progressively replaced by fat (yellow marrow), so that, in adults, normal haematopoiesis is restricted to the vertebrae, pelvis, sternum, ribs, clavicles, skull, upper humeri and proximal femora. However, red marrow can expand in response to increased demands for blood cells.
Bone marrow contains a range of immature haematopoietic precursor cells and a storage pool of mature cells for release at times of increased demand. In normal marrow, nests of red cell precursors cluster around a central macrophage, which provides iron and also phagocytoses nuclei from red cells prior to their release into the circulation.
Megakaryocytes are large cells which produce and release platelets into vascular sinuses.
White cell precursors are clustered next to the bone trabeculae; maturing cells migrate into the marrow spaces towards the vascular sinuses.
Plasma cells are antibody-secreting mature B cells which normally represent < 5% of the marrow population.
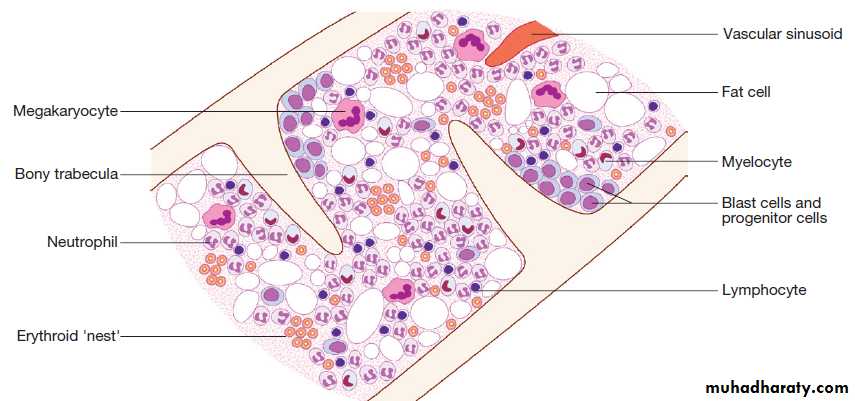
Structural organisation of normal bone marrow.
Stem cellsAll blood cells are derived from pluripotent haematopoietic stem cells. These comprise only 0.01% of the total marrow cells, but they can self-renew (i.e. make more stem cells) or differentiate to produce a hierarchy of lineage-committed stem cells. The resulting primitive
progenitor cells cannot be identified morphologically, so they are named according to the types of cell (or colony) they form during cell culture experiments.
CFU–GM (colony-forming unit – granulocyte, monocyte) are stem cells that produce granulocytic and monocytic lines, CFU–E produce erythroid cells, CFU–Meg produce megakaryocytes and ultimately platelets .
Growth factors, produced in bone marrow stromal cells and elsewhere, control the survival, proliferation, differentiation and function of stem cells and their progeny. Some, such as interleukin-3 (IL-3), stem cell
factor (SCF) and granulocyte, macrophage–colony stimulating factor (GM–CSF), act on a wide number of cell types at various stages of differentiation. Others, such as erythropoietin (Epo), granulocyte–colony stimulating
factor (G–CSF) and thrombopoietin (Tpo), are lineage-specific. Many of these growth factors are now synthesised by recombinant DNA technology and used as treatments:
for example, Epo to correct renal anaemia and G–CSF to hasten neutrophil recovery after chemotherapy.
The bone marrow also contains stem cells which can differentiate into
non- haematological cells, such as nerve, skeletal muscle, cardiac muscle, liver and blood vessel endothelium.This is termed stem-cell plasticity and may have exciting clinical applications in the future.
Blood cells and their functions
Red cellsRed cell precursors formed in the bone marrow from the
erythroid (CFU–E) progenitor cells are called erythroblasts
or normoblasts .These divide and acquire haemoglobin, which turns the cytoplasm pink; the nucleus condenses and is extruded from the cell. The first non-nucleated red cell is a reticulocyte, which still contains ribosomal material in the cytoplasm, giving these large cells a faint blue tinge (‘polychromasia’). Reticulocytes lose their ribosomal material and mature over 3 days, during which time they are released into the circulation.
Increased numbers of circulating reticulocytes (reticulocytosis) reflect increased erythropoiesis.
Proliferation and differentiation of red cell precursors
is stimulated by erythropoietin, a polypeptide hormone
produced by renal interstitial peritubular cells in response to hypoxia. Normal mature red cells circulate for about 120 days. They are 8 μm biconcave discs lacking a nucleus but filled with haemoglobin, which delivers oxygen to the tissues. In order to pass through the smallest capillaries, the red cell membrane is deformable, with a lipid bilayer to which a ‘skeleton’ of filamentous proteins is attached via special linkage proteins . Inherited abnormalities of any of these proteins result in loss of membrane as cells pass through the spleen, and the formation of abnormally shaped red cells called spherocytes or elliptocytes .
Red cells are exposed to osmotic stress in the pulmonary and renal circulation; in order to maintain homeostasis, the membrane contains ion pumps, which control intracellular levels of sodium, potassium, chloride and bicarbonate. In the absence of mitochondria, the energy for these functions is provided by anaerobic glycolysis and the pentose phosphate pathway. Membrane glycoproteins inserted into the lipid bilayer also form the antigens recognised by blood grouping .The ABO and Rhesus systems are the most commonly recognised but over 400 blood group antigens have been described.
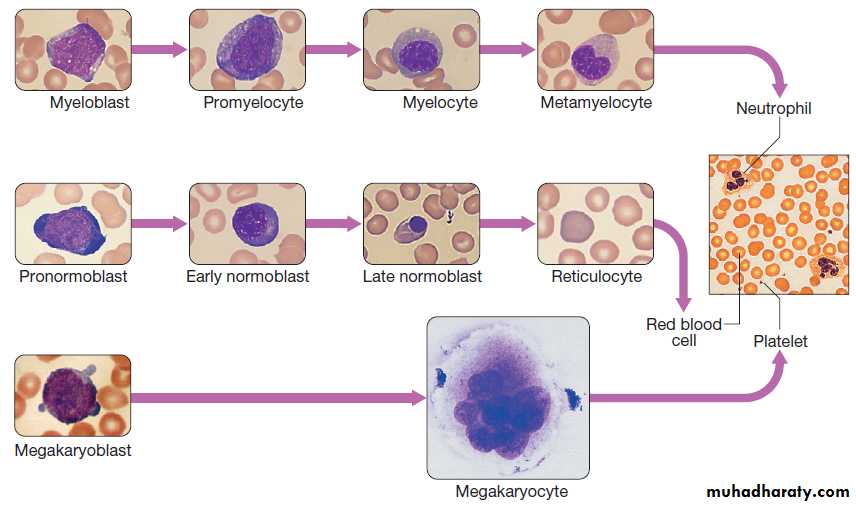
Maturation pathway of red cells, granulocytes and platelets.The image on the right is normal blood film.
Haemoglobin
Haemoglobin is a protein specially adapted for oxygentransport. It is composed of four globin chains, each surrounding an iron-containing porphyrin pigment molecule termed haem. Globin chains are a combination of two alpha and two non-alpha chains; haemoglobin A (αα/ββ) represents over 90% of adult Hb, whereas Hb F (αα/ γγ) is the predominant type in the fetus. Each haem molecule contains a ferrous ion (Fe2+), to which oxygen reversibly binds; the affinity for oxygen increases as successive oxygen molecules bind. In the ‘open’ deoxygenated state, 2,3 diphosphoglycerate (DPG), a product of red cell metabolism, binds to the haemoglobin molecule and lowers its oxygen affinity.
These complex interactions produce the sigmoid shape of the oxygen dissociation curve .
The position of this curve depends upon the concentrations of 2,3 DPG, H+ ions and CO2; increased levels shift the curve to the right and cause oxygen to be released more readily, e.g. when red cells reach hypoxic tissues. Haemoglobin F is unable to bind 2,3 DPG and has a left-shifted oxygen dissociation curve, which, together with the low pH of fetal blood, ensures fetal oxygenation. Genetic mutations affecting the haem-binding pockets of globin chains or the ‘hinge’ interactions between globin chains result in haemoglobinopathies or unstable haemoglobins.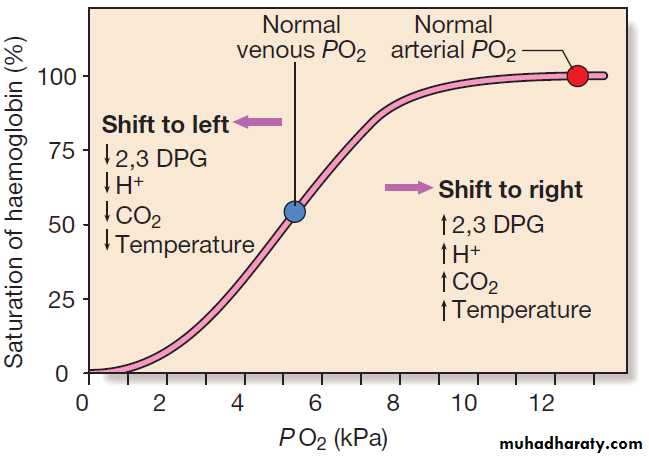
The haemoglobin oxygen dissociation curve. Factors are
listed which shift the curve to the right (more oxygen released from blood) and to the left (less oxygen released) at given PO2. (To convert kPa to mmHg, multiply by 7.5.)
Alpha globin chains are produced by two genes on chromosome 16, and beta globin chains by a single gene on chromosome 11; imbalance in the production of globin chains results in the thalassaemias.
Defects in haem synthesis cause the porphyrias.
Destruction
Red cells at the end of their lifespan of approximately120 days are phagocytosed by the reticulo-endothelial
system. Amino acids from globin chains are recycled
and iron is removed from haem for re-use in haemoglobin
synthesis. The remnant haem structure is degraded to bilirubin and conjugated with glucuronic acid before being excreted in bile. In the small bowel, bilirubin is converted to stercobilin; most of this is excreted, but a small amount is reabsorbed and excreted by the kidney as urobilinogen. Increased red cell destruction due to haemolysis or ineffective haematopoiesis results in jaundice and increased urinary urobilinogen. Free intravascular haemoglobin is toxic and is normally bound by haptoglobins, which are plasma proteins produced by the liver.
White cells
White cells or leucocytes in the blood consist of granulocytes (neutrophils, eosinophils and basophils), monocytes and lymphocytes .Granulocytes and monocytes are formed from bone
marrow CFU–GM progenitor cells during myelopoiesis.
The first recognisable granulocyte in the marrow is
the myeloblast, a large cell with a small amount of
basophilic cytoplasm and a primitive nucleus with
open chromatin and nucleoli. As the cells divide and
mature, the nucleus segments and the cytoplasm
acquires specific neutrophilic, eosinophilic or basophilic
granules . This takes about 14 days.
The cytokines G–CSF, GM–CSF and M–CSF are involved in the production of myeloid cells, and G–CSF can be used clinically to hasten recovery of neutrophil counts after chemotherapy.
Myelocytes or metamyelocytes are normally found
only in the marrow but may appear in the circulation in
infection or toxic states. The appearance of more primitive myeloid precursors in the blood is often associated with the presence of nucleated red cells and is termed a ‘leucoerythroblastic’ picture; this indicates a serious disturbance of marrow function.
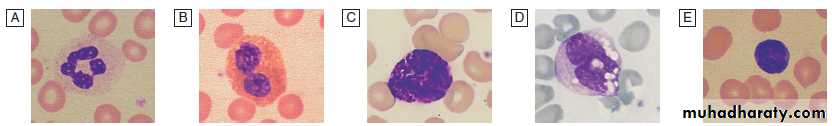
Neutrophils
10–14 μm in diameter, with a multilobular nucleus containing 2–5 segments and granules in their cytoplasm. Their function is to recognise, ingest and destroy foreign particles and microorganisms.
A large storage pool of mature neutrophils exists in the bone marrow. Every day, some 1011 neutrophils enter the circulation, where cells may be circulating freely or attached to endothelium in the marginating pool. These two pools are equal in size; factors such as exercise or catecholamines increase the number of cells flowing in the blood. Neutrophils spend 6–10 hours in the circulation before being removed, principally by the spleen. Alternatively, they pass into the tissues and either are consumed in the inflammatory process or undergo apoptotic cell death and phagocytosis by macrophages.
Eosinophils, represent 1–6% of the circulating white cells.They are a similar size to neutrophils but have a bilobed nucleus and prominent orange granules on Romanowsky staining. Eosinophils are phagocytic and their granules contain a peroxidase capable of generating reactive oxygen species and proteins involved in the intracellular killing of protozoa and helminths.They are also involved in allergic reactions (e.g. atopic asthma). Basophils less than 1% of circulating white cells. They contain dense black granules which obscure the nucleus. Mast cells resemble basophils ,are found only in the tissues. These cells are involved in hypersensitivity reactions.
Monocytes are the largest with a diameter of 12–20 μm and an irregular nucleus in abundant pale blue cytoplasm containing occasional cytoplasmic vacuoles. These cells circulate for a few hours and then migrate into tissue, where they become macrophages, Kupffer cells or antigen-presenting dendritic cells. The former phagocytose debris, apoptotic cells and microorganisms.
Lymphocytes
Lymphocytes are derived from pluripotent haematopoietic
stem cells in the bone marrow. There are two main
types: T cells (which mediate cellular immunity) and B
cells (which mediate humoral immunity) .Lymphoid
cells that migrate to the thymus develop into T cells, whereas B cells develop in the bone marrow.
The majority (about 80%) of lymphocytes in the circulation are T cells. Lymphocytes are heterogeneous, the
smallest being the size of red cells and the largest the
size of neutrophils. Small lymphocytes are circular with
scanty cytoplasm but larger cells are more irregular with
abundant blue cytoplasm. Lymphocyte subpopulations
have specific functions and lifespan can vary from a few
days to many years. Cell surface antigens (‘cluster of
differentiation’ (CD) antigens), which appear at different
points of lymphocyte maturation, are used to classify
lymphomas and lymphoid leukaemias.
Haemostasis
Blood must be maintained in a fluid state in order to
function as a transport system, but must be able to solidify
to form a clot following vascular injury in order to
prevent excessive bleeding, a process known as haemostasis.
Successful haemostasis is localised to the area of tissue damage and is followed by removal of the clot and
tissue repair. This is achieved by complex interactions
between the vascular endothelium, platelets, coagulation
factors, natural anticoagulants and fibrinolytic
enzymes . Dysfunction of any of these components
may result in haemorrhage or thrombosis.
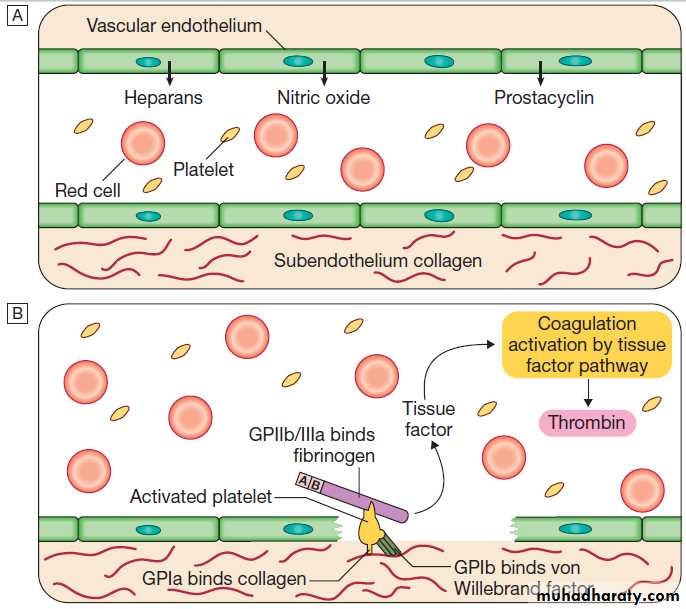


The stages of normal
haemostasis.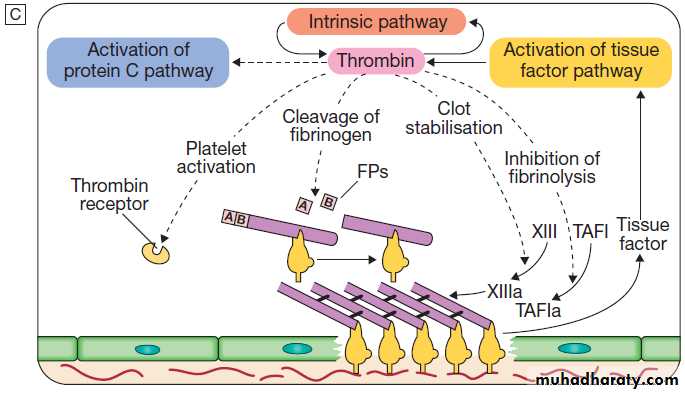
C Stage 3 Fibrin clot formation: platelets become activated and aggregate; fibrin formation is supported by the platelet membrane; stable fibrin clot forms. The adherent platelets are activated by many pathways, including binding of ADP, collagen, thrombin and adrenaline (epinephrine) to surface receptors. The cyclo-oxygenase pathway converts arachidonic acid from the platelet membrane into thromboxane A2, which causes aggregation of platelets. Activation of the platelets results in release of the platelet granule contents, enhancing coagulation further. Thrombin plays a key role in the control of coagulation: the small amount generated via the TF pathway massively amplifies its own production; the ‘intrinsic’ pathway becomes



Platelets
Platelets are formed in the bone marrow from megakaryocytes. Meg- stem cells (CFU–Meg) divide to form megakaryoblasts, which undergo a process called ‘endomitotic reduplication’, in which there is division of the nucleus but not the cell. This creates mature megak, large cells with several nuclei and cytoplasm containing platelet granules. Large numbers of platelets then fragment off from each megakaryocyte into the circulation. The formation and maturation of megakaryocytes are stimulated by thrombopoietin produced in the liver. Platelets circulate for 8–10 days before they are destroyed in the reticulo-endothelial system. Some 30% of peripheral platelets are normally pooled in the spleen and do not circulate.Under normal conditions platelets are discoid, with a
diameter of 2–4 μm . The surface membrane invaginates to form a tubular network, the canalicular system, which provides a conduit for the discharge of the granule content following platelet activation.
Drugs which inhibit platelet function and thrombosis include aspirin (cyclo-oxygenase inhibitor), clopidogrel (adenosine diphosphate (ADP)-mediated activation inhibitor), dipyridamole (phosphodiesterase inhibitor),
and the IIb/ IIIa inhibitors abciximab, tirofiban and eptifibatide (which prevent fibrinogen binding).
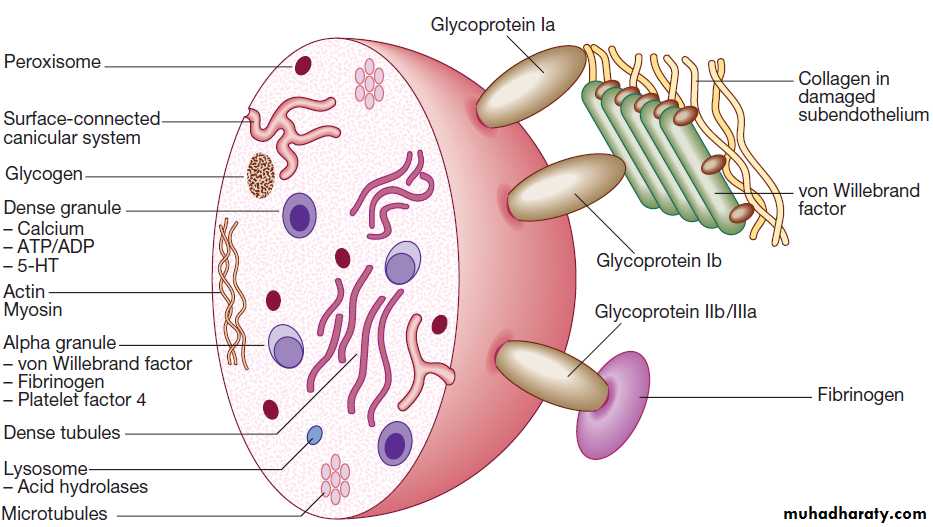
Normal platelet structure. (5-HT= 5-hydroxytryptamine, serotonin; ADP = adenosine diphosphate; ATP = adenosine triphosphate)
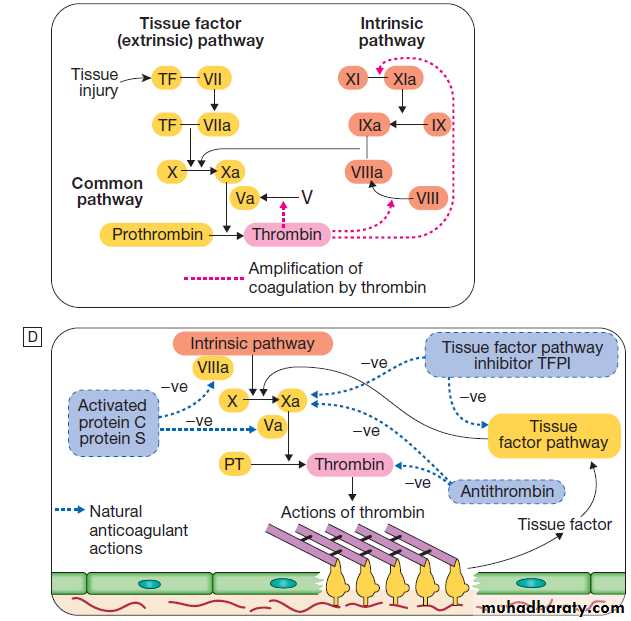
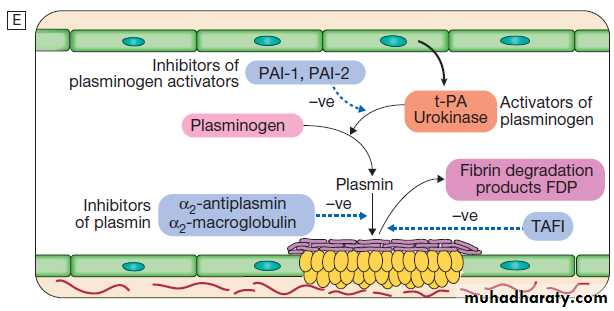
Clotting factors
The coagulation system consists of a cascade of solubleinactive zymogen proteins designated by Roman numerals.
When proteolytically cleaved and activated, each is
capable of activating one or more components of the
cascade. Activated factors are designated by the
suffix‘a’. Some of these reactions require phospholipid and
calcium. Coagulation occurs by two pathways: it is initiated
by the extrinsic (or tissue factor) pathway and amplified by the ‘intrinsic pathway’ .
Clotting factors are synthesised by the liver, although
factor V is also produced by platelets and endothelial
cells. Factors II, VII, IX and X require post-translational
carboxylation to allow them to participate in coagulation.
The carboxylase enzyme responsible for this in the
liver is vitamin K-dependent.
Vitamin K is converted to an epoxide in this reaction and must be reduced to its active form by a reductase enzyme. This reductase is inhibited by warfarin, and this is the basis of the anticoagulant effect of coumarins .
Congenital (e.g. haemophilia) and acquired (e.g. liver failure) causes of coagulation factor deficiency are associated with bleeding.
INVESTIGATION OF DISEASES OF THE BLOOD
The full blood count (FBC)To obtain a FBC anticoagulated blood is processed through automated blood analysers to measure the haematological parameters. These include numbers of circulating cells, the proportion of whole blood volume occupied by red cells (the haematocrit, Hct), and the red cell indices ,size of
red cells (mean cell volume, MCV) and the amount of
Haemoglobin present in the red cells (mean cell haemoglobin, MCH). Blood analysers can differentiate types of white blood cell and give automated counts of neutrophils, lymphocytes, monocytes, eosinophils and basophils. It is important to appreciate, however, that a
number of conditions can lead to spurious results
Blood film examination
Although technical advances in full blood count analysershave resulted in fewer blood samples requiring
manual examination, scrutiny of blood components prepared on a microscope slide (the ‘blood film’) can often yield valuable information .
Analysers cannot identify abnormalities of red cell
shape and content (e.g. Howell–Jolly bodies, basophilic
stippling, malaria parasites) or 1blasts.
Bone marrow examination
In adults, is usually obtained from the posterior iliac crest. After a local anaesthetic, marrow can be sucked out from the medullary space, stained and examined under the microscope (bone marrow aspirate).
In addition, a core of bone may be removed (trephine biopsy), fixed and decalcified before sections are cut for staining . A bone marrow aspirate is used to assess the composition and morphology of haematopoietic cells or abnormal infiltrates.
Further investigations may be performed, such as cell surface marker analysis (immunophenotyping), chromosome and molecular studies to assess malignant disease, or marrow culture for suspected tuberculosis.
A trephine biopsy is superior for assessing marrow cellularity, marrow fibrosis, and infiltration by abnormal cells such as metastatic carcinoma.
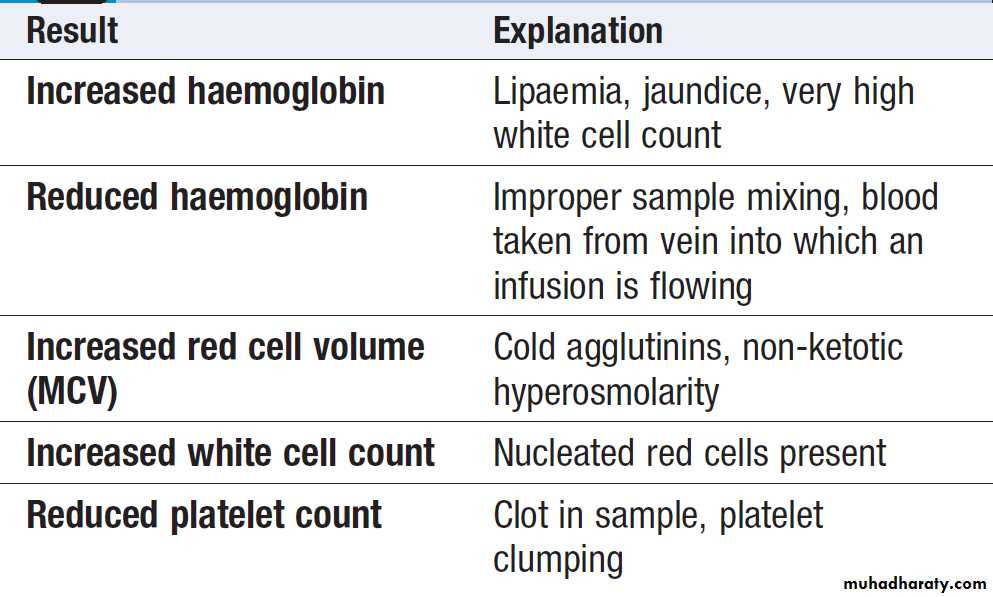
Spurious FBC results from autoanalysers
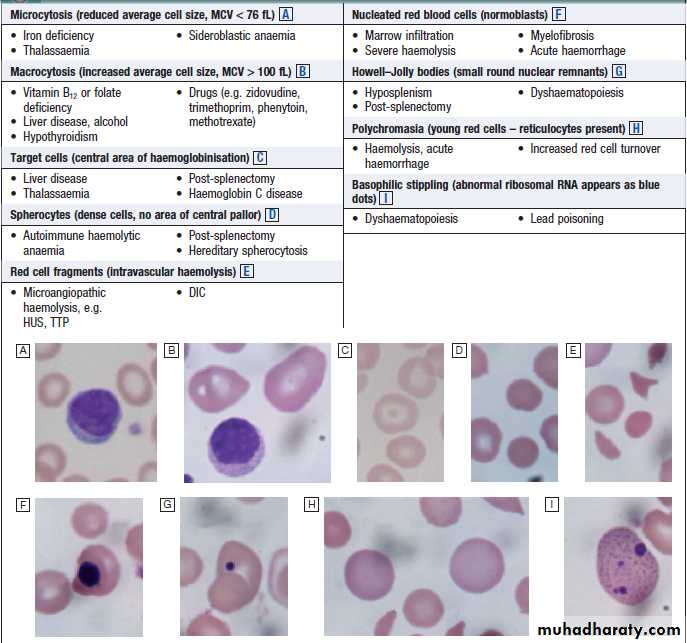
How to interpret red cell appearances
Appearance of red blood cells.A Microcytosis.
B Macrocytosis.
C Target cells.
D Spherocytes.
E Red cell fragments.
F Nucleated red blood cells.
G Howell–Jolly bodies.
H Polychromasia.
I Basophilic stippling.
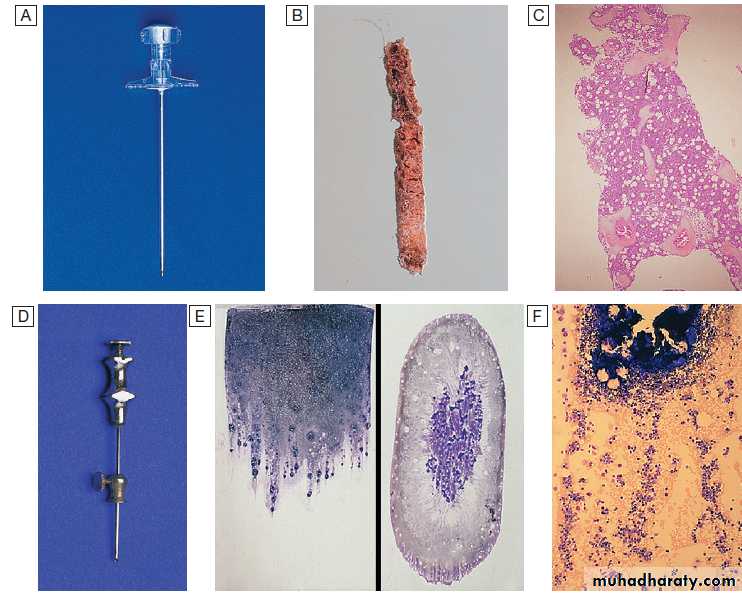
Bone marrow aspirate and trephine. A Trephine biopsy needle. B Macroscopic appearance of a trephine biopsy. C Microscopic appearance of stained section of trephine. D Bone marrow aspirate needle. E Stained macroscopic appearance of marrow aspirate: smear (left) and squash (right). F Microscopic appearance of stained marrow particles and trails of haematopoietic cells.
Investigation of coagulation
Bleeding disordersIn patients with clinical evidence of a bleeding disorder
there are recommended screening tests .
Coagulation tests measure the time to clot formation
in vitro in a plasma sample after the clotting process is
initiated by activators and calcium. The result of the test
sample is compared with normal controls. The tissue factor (‘extrinsic’) pathway is assessed by the prothrombin time (PT), and the ‘intrinsic’ pathway by the activated partial thromboplastin time (APTT), sometimes known as the partial thromboplastin time with kaolin (PTTK).
Coagulation is delayed by deficiencies of coagulation factors and by the presence of inhibitors of coagulation, such as heparin. If both the PT and APTT are prolonged, this indicates either deficiency or inhibition of the final
common pathway (which includes factors X, V, prothrombin and fibrinogen) or global coagulation factor deficiency involving more than one factor, as occurs in disseminated intravascular coagulation. Further specific tests may be performed based on interpretation of the clinical scenario and results of these screening tests.
A mixing test with normal plasma allows differentiation between a coagulation factor deficiency (the prolonged time corrects) and the presence of an inhibitor of coagulation (the prolonged time does not correct); the latter may be chemical (heparins) or an antibody (most often a lupus anticoagulant but occasionally a specific inhibitor of one of the coagulation factors, typically factor VIII). Von Willebrand disease may present with a normal APTT.
Platelet function has historically been assessed by the bleeding time, measured as the time to stop bleeding after a standardised incision. However, most centres have abandoned the use of this test.
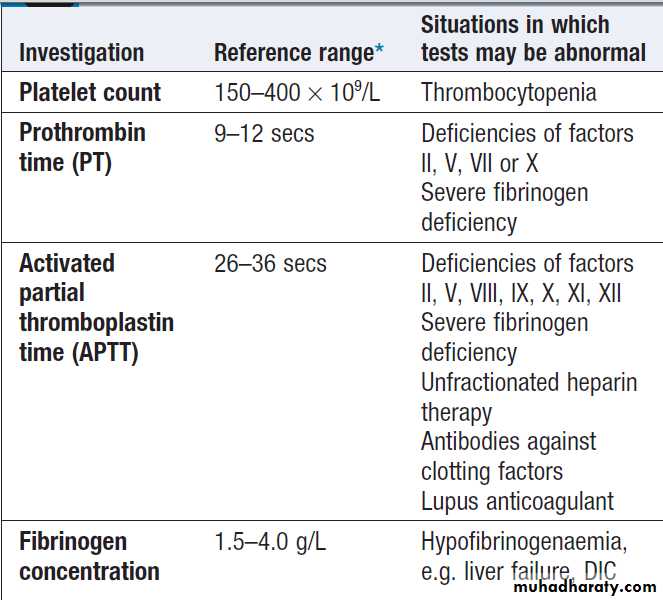
Coagulation screening tests
N.B. International normalised ratio (INR) is used only to monitor coumarintherapy and is not a coagulation screening test.
Platelet function can be assessed in vitro by measuring aggregation in response to various agonists, such as adrenaline (epinephrine), collagen, thrombin and ADP, or by measuring the constituents of the intracellular granules, e.g. adenosine triphosphate (ATP)/ADP.
Coagulation screening tests are also performed in
patients with suspected DIC, when clotting factors and
platelets are consumed, resulting in thrombocytopenia
and prolonged PT and APTT. In addition, there is evidence
of active coagulation with consumption of fibrinogen
and generation of fibrin degradation products (D-dimers). Note, however, that fibrinogen is an acute phase protein which may also be elevated in inflammatory disease
Monitoring anticoagulant therapy
The international normalised ratio (INR) is validated only to assess the therapeutic effect of coumarin anticoagulants, including warfarin. INR is the ratio of the patient’s PT to that of a normal control. Monitoring of heparin is, on the whole, only required with unfractionated heparins. Therapeutic anticoagulation prolongs the APTT relative to a control sample by a ratio of approximately 1.5–2.5.LMWH have such a predictable dose response that monitoring of the anticoagulant effect is not required, except in patients with renal impairment (GFR <30 mL/min). When monitoring is indicated,
an anti-Xa activity assay should be used.
Thrombotic disorders
Measurement of D-dimers derived from fibrin degradation is useful in excluding the diagnosis of active VTE. A variety of tests exist which may help to explain an underlying propensity to thrombosis, especially VTE. In most patients, the results do not affect clinical management but they may influence the duration of anticoagulation (e.g. antiphospholipid antibodies, justify family screening in inherited thrombophilias or suggest additional management strategies to reduce thrombosis risk (e.g. in PNH and myeloproliferative disease). Anticoagulants can interfere with some of these assays; warfarin reduces protein C and S levels and affects measurement of lupus anticoagulant, while heparin interferes with antithrombin and lupus anticoagulant assays. Therefore these tests, should be performed when the patient is not taking anticoagulants.
Investigation of possible thrombophilia
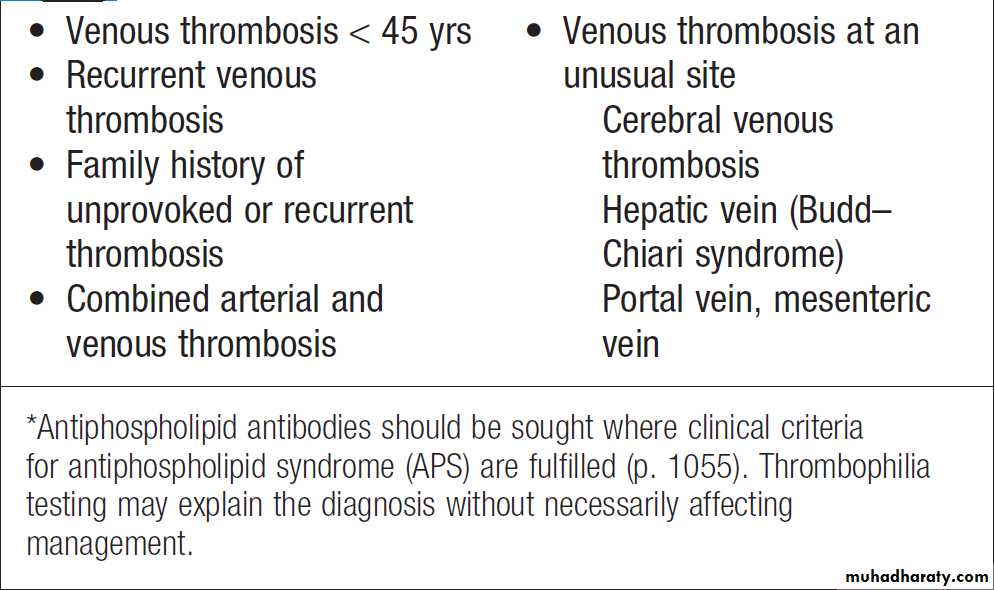
Indications for thrombophilia testing*

Haematological investigations in old age
PRESENTING PROBLEMS IN BLOOD DISEASEAnaemia
Anaemia refers to a state in which the level of haemoglobin in the blood is below the reference range appropriate for age and sex. Other factors, including pregnancy and altitude, also affect haemoglobin levels and must be taken into account when considering whether an individual is anaemic. The clinical features of anaemia reflect diminished oxygen supply to the tissues . A rapid onset of anaemia (e.g. due to blood loss) causes more profound symptoms than a gradually developing
anaemia. Individuals with cardiorespiratory disease are
more susceptible to symptoms of anaemia. The clinical assessment and investigation of anaemia should gauge its severity and define the underlying cause .

Causes of anaemia
Clinical assessment• Iron deficiency anaemia is the most common type worldwide.
A thorough GI history is important, looking in particular for symptoms of blood loss. Menorrhagiais a common cause in pre-menopausal.
• A dietary history should assess the intake of iron and
folate, which may become deficient in comparison to needs (e.g. in pregnancy or during periods of rapid growth).
• Past medical history may reveal a disease which is known to be associated with anaemia, such as rheumatoid (anaemia of chronic disease), or previous surgery (e.g. resection of the stomach or small bowel, which lead to malabsorption of iron and/or vitamin B12).
• Family history and ethnic background may raise suspicion of haemolytic anaemias, such as the haemoglobinopathies and hereditary spherocytosis. Pernicious anaemia may also be familial.
• A drug history may reveal the ingestion of drugs which cause blood loss (e.g. aspirin and anti-inflammatory drugs), haemolysis (e.g. sulphonamides) or aplasia (e.g. chloramphenicol).
On examination, as well as the general physical findings of anaemia, there may be specific findings related to the aetiology of the anaemia; for example, a patient may be found to have a right iliac fossa mass due to an underlying caecal carcinoma.
Haemolytic anaemias can cause jaundice. Vitamin B12
deficiency may be associated with neurological signs,
including peripheral neuropathy, dementia and signs of
subacute combined degeneration of the cord .
Sickle-cell anaemia may result in leg ulcers, stroke or features of pulmonary hypertension.
Investigations
Schemes for the investigation of anaemias are often
based on the size of the red cells, which is most accurately
indicated by the MCV in the FBC. Commonly, in
the presence of anaemia:
• A normal MCV (normocytic anaemia) suggests either acute blood loss or the anaemia of chronic disease (ACD) .
• A low MCV (microcytic anaemia) suggests iron
deficiency or thalassaemia .
• A high MCV (macrocytic anaemia) suggests
vitamin B12 or folate deficiency or myelodysplasia.
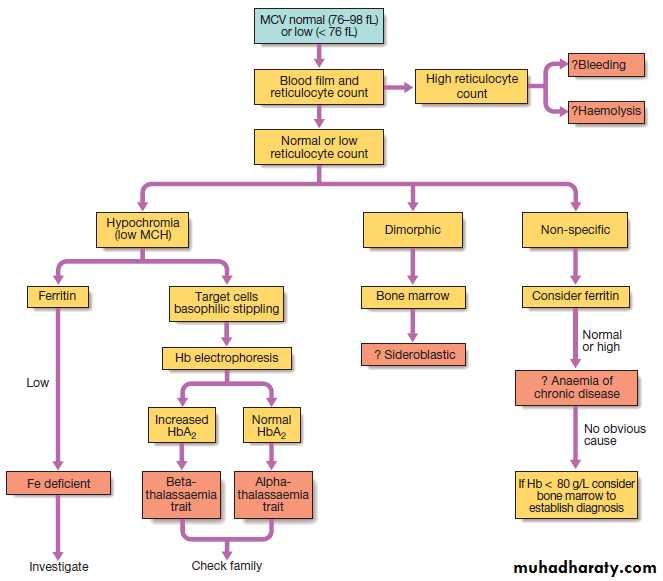
Investigation of anaemia with normal or low MCV.
High haemoglobinPatients with a persistently raised haematocrit (Hct)
(> 0.52 males, > 0.48 females) for more than 2 months
should be investigated. ‘True’ polycythaemia (or absolute
erythrocytosis) indicates an excess of red cells, while
‘relative’ (or ‘low-volume’) polycythaemia is due to a
decreased plasma volume. Causes are shown in Box.
Clinical assessment and investigations
Males and females with Hct values of over 0.60 and over
0.56, respectively, can be assumed to have an absolute
erythrocytosis. A clinical history and examination will identify most patients with polycythaemia secondary to
hypoxia. The presence of hypertension, smoking, excess
alcohol consumption and/or diuretic use is consistent
with low-volume polycythaemia (Gaisbock’s syndrome).
In polycythaemia rubra vera (PRV), a mutation in a
kinase, JAK-2 V617F, is found in over 90% of cases.
Patients with PRV have an increased risk of arterial thromboses, particularly stroke, and VTE.
They may also have aquagenic pruritus (worse after a hot bath), hepatosplenomegaly and gout (due to high red cell turnover). If the JAK-2 mutation is absent and there is no obvious secondary cause, a measurement of red cell mass is
required to confirm an absolute erythrocytosis, followed by further investigations to exclude hypoxia, and causes
of inappropriate erythropoietin secretion.
Red cell mass measurement is performed by radiolabelling an aliquot of the patient’s red cells, re-injecting them and measuring the dilution of the isotope.
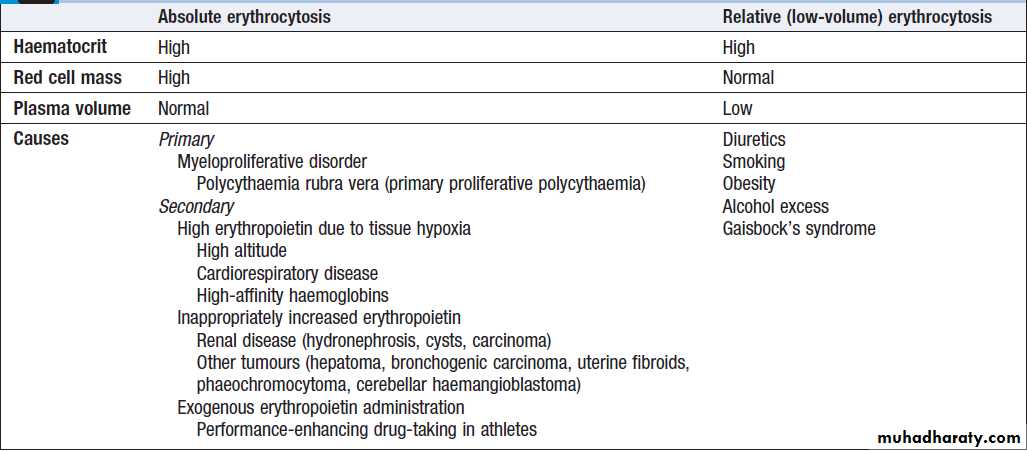
Classification and causes of erythrocytosis
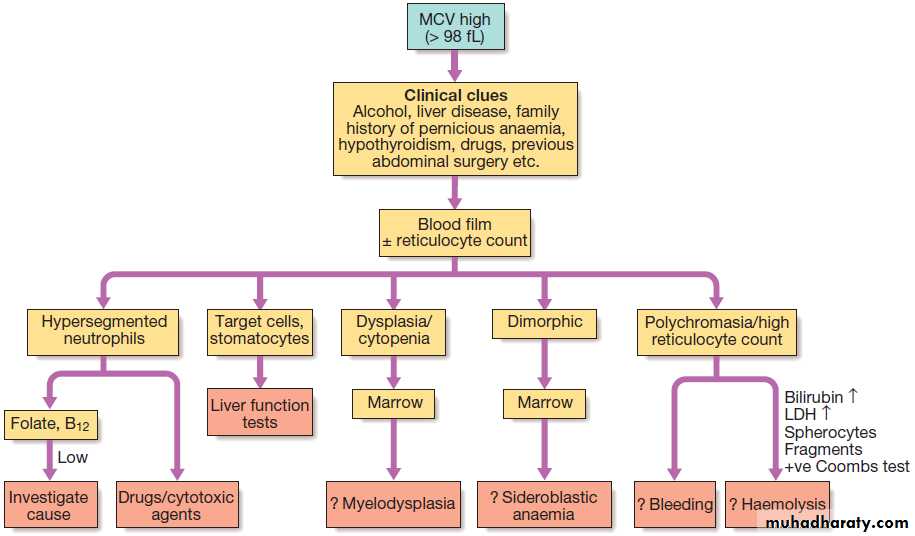
Investigation of anaemia with high MCV.
(LDH = lactate dehydrogenase)Leucopenia (low white cell count)
A reduction in the total numbers of circulating whitecells is called leucopenia. This may be due to a reduction
in all types of white cell or in individual cell types
(usually neutrophils or lymphocytes).
Neutropenia
A reduction in neutrophil count (< 1.5 × 109/L, but dependent on age and race) is called neutropenia. Drug-induced neutropenia is not uncommon . Clinical manifestations range from no symptoms to overwhelming sepsis. The risk of bacterial infection is related to the degree of neutropenia, with counts < 0.5 × 109/L considered to be critically low.
Fever is the first and often only manifestation of infection.
A sore throat, perianal pain or skin inflammation
may be present. The lack of neutrophils allows the
patient to become septicaemic and shocked within hours
if immediate antibiotic therapy is not commenced.
Lymphopenia
This is an absolute lymphocyte count of <1 × 109/L. Although minor reductions may be asymptomatic, deficiencies in cell-mediated immunity may result in infections (with organisms such as fungi, viruses and mycobacteria) and a propensity to lymphoid and other malignancies (particularly those associated with viral infections such as Epstein–Barr virus (EBV), human papillomavirus (HPV) and human herpesvirus 8 (HHV-8)).
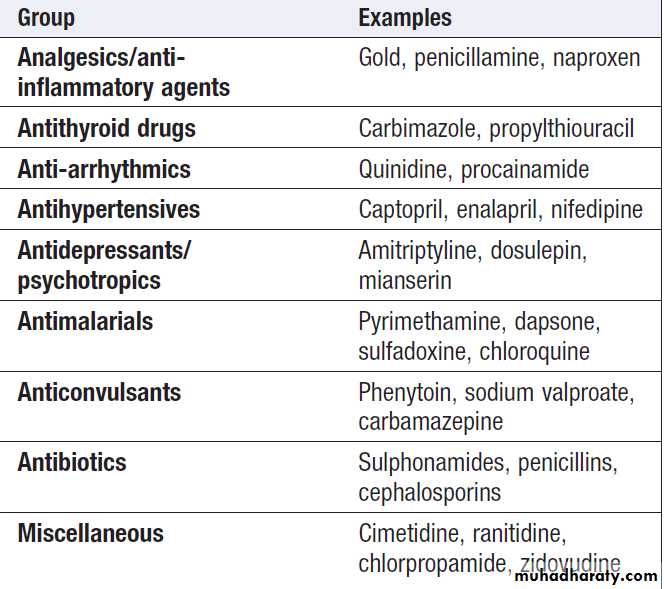
Drugs that can induce neutropenia
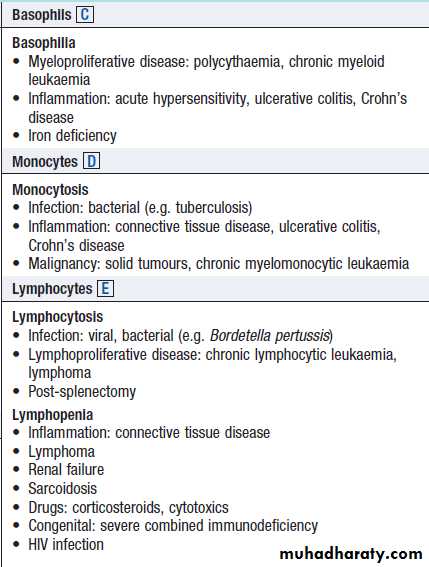
Neutrophils ANeutrophilia
• Infection: bacterial, fungal
• Trauma: surgery, burns
• Infarction: myocardial infarct, pulmonary embolus, sickle-cell crisis
• Inflammation: gout, rheumatoid arthritis, ulcerative colitis, Crohn’s
• Malignancy: solid tumours, Hodgkin lymphoma
• Myeloproliferative disease: polycythaemia, CML
• Physiological: exercise, pregnancy
Neutropenia
• Infection: viral, bacterial (e.g. Salmonella), protozoal (e.g. malaria)
• Drugs
• Autoimmune: connective tissue disease
• Alcohol
• Bone marrow infiltration: leukaemia, myelodysplasia
• Congenital: Kostmann’s syndrome
• Constitutional: Afro-Caribbean and Middle Eastern descent

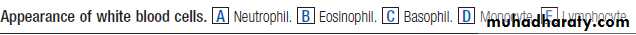
Leucocytosis (high white cell count)
An increase in the total numbers of circulating whitecells is called leucocytosis. This is usually due to an
increase in a specific type of cell .
It is important to realise that an increase in a single type of white cell (e.g. eosinophils or monocytes) may not
increase the total white cell count (WCC) above the
upper limit of normal and will only be apparent if the
‘differential’ of the white count is examined.
Neutrophilia
An increase in the number of circulating neutrophils iscalled a neutrophilia or a neutrophil leucocytosis. It can
result from an increased production of cells from the bone marrow or redistribution from the marginated pool. The normal neutrophil count depends upon age, race and certain physiological parameters.
During pregnancy, not only is there an increase in neutrophils but also earlier forms such as metamyelocytes can be found in the blood.
Eosinophilia
A high eosinophil count of >0.5 × 109/L is usually secondary to infection (especially parasites), allergy (e.g. eczema, asthma, reactions to drugs), immunological disorders (e.g. polyarteritis, sarcoidosis) or malignancy (e.g. lymphomas) .Usually, such eosinophilia is short-lived. In the rarer primary disorders, there is a persistently raised, often clonal: for example, in myeloproliferative disorders, subtypes of idiopathic hypereosinophilic syndrome (HES) and AML. Recently, specific mutations in receptor tyrosine kinase genes have been found in some primary eosinophilias (e.g. causing re-arrangements of platelet-derived growth factor receptors α and β or c-kit), which allow diagnosis and, in some cases, specific therapy with tyrosine kinase inhibitors such as imatinib.
Eosinophil infiltration can damage many organs (e.g.
heart, lungs, gastrointestinal tract, skin, musculoskeletalsystem); therefore evaluation of eosinophilia includes
not only the identification of any underlying cause and
its appropriate treatment, but also assessment of any
related organ damage.
Lymphocytosis
A lymphocytosis is an increase in circulating lymphocytes
above that expected for the patient’s age. In
adults, this is greater than 3.5 × 109/L. Infants and children have higher counts; age-related reference ranges
should be consulted, the most common is viral infection.
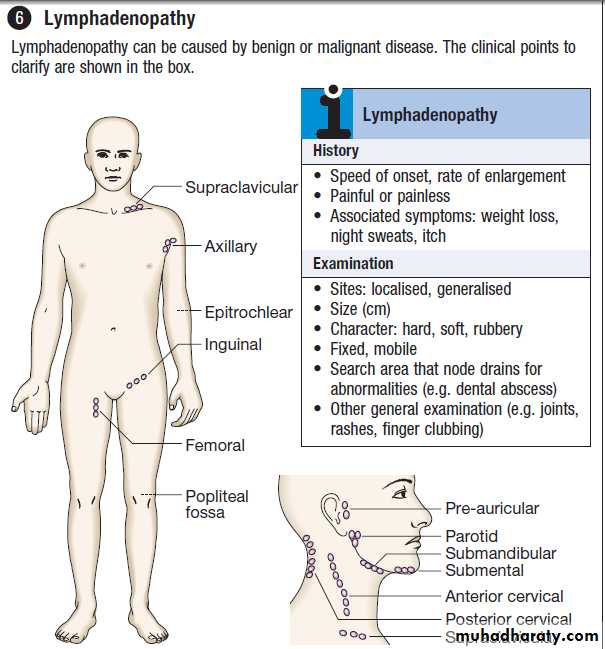
Lymphadenopathy
Enlarged lymph glands may be an important indicatorof haematological disease but they are not uncommon
in reaction to infection or inflammation.The sites of lymph node groups, symptoms and signs help elucidate the underlying cause. Nodes which enlarge in response to local
infection or inflammation (‘reactive nodes’) usually
expand rapidly and are painful, whereas those due to
haematological disease are more frequently painless.
Localised lymphadenopathy should elicit a search for a
source of inflammation in the appropriate drainage area:
• the scalp, ear, mouth, face or teeth for neck nodes
• the breast for axillary nodes
• the perineum or external genitalia for inguinal nodes.
Generalised lymphadenopathy may be secondary to
infection, connective tissue disease or extensive skin
disease, but is more likely to signify underlying haematological malignancy. Weight loss and drenching night sweats that may require a change of night clothes are associated with haematological malignancies, particularly lymphoma.
Initial investigations in lymphadenopathy include an
FBC (to detect neutrophilia in infection or evidence of
haematological disease), an ESR and a chest X-ray (to
detect mediastinal lymphadenopathy). If the findings
suggest malignancy, a formal cutting needle or excision biopsy of a representative node is indicated to obtain a histological diagnosis.

Causes of lymphadenopathy
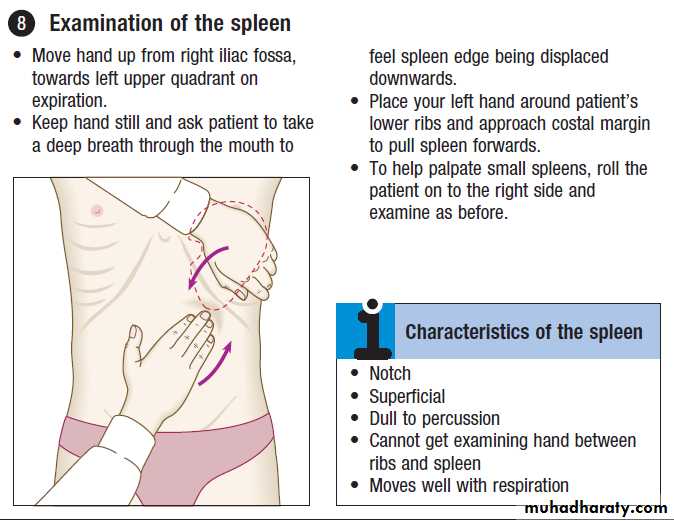
SplenomegalyThe spleen may be enlarged due to involvement by
lymphoproliferative disease, the resumption of extramedullary haematopoiesis in myeloproliferative disease, enhanced reticulo-endothelial activity in autoimmune haemolysis, expansion of the lymphoid tissue in response to infections, or vascular congestion as a result of portal hypertension . Associated lymphadenopathy is suggestive of lymphoproliferative disease. An enlarged spleen may cause abdominal discomfort, accompanied by back pain and abdominal bloating. Splenic infarction produces severe abdominal pain radiating to the left shoulder tip, associated with a splenic rub on auscultation.
Investigation should focus on the suspected cause.
US or CT will detect variations in the spleen, imaging of the liver and abdominal lymph nodes. Biopsy of enlarged abdominal or superficial lymph nodes may provide the diagnosis. A chest X-ray or CT of the thorax will detect mediastinal lymphadenopathy. An FBC may show pancytopenia secondary to hypersplenism, when the enlarged spleen has become overactive, destroying blood cells prematurely. If other abnormalities are present, such as abnormal lymphocytes or a leucoerythroblastic blood film, a bone marrow examination is indicated. Screening for infectious or liver disease may be appropriate. If all investigations are unhelpful, splenectomy may be diagnostic but is rarely carried out in these circumstances.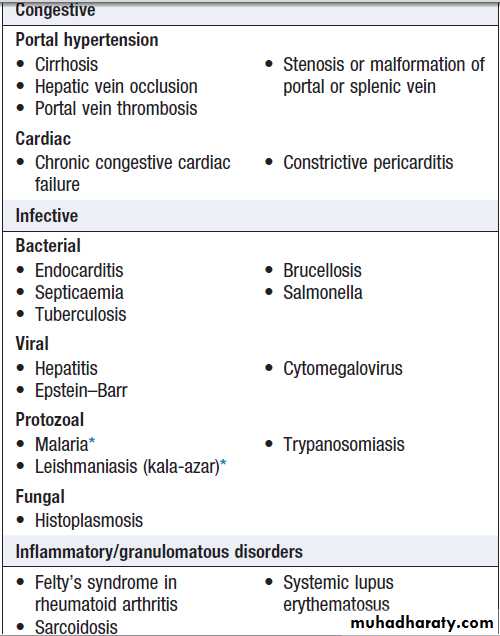
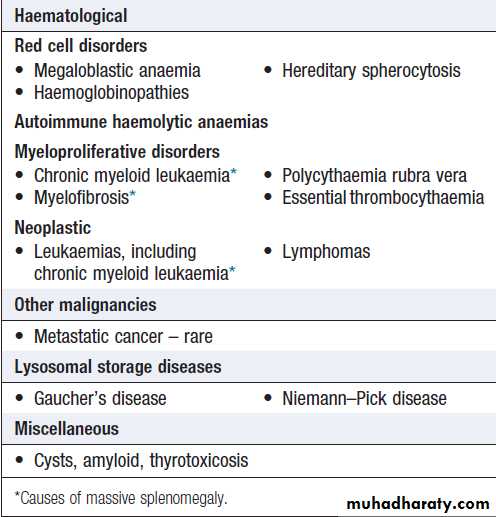
Causes of splenomegaly
BleedingNormal bleeding is seen following surgery and trauma.
Pathological bleeding occurs when structurally abnormal vessels rupture or when a vessel is breached in the presence of a defect in haemostasis. This may be due to a deficiency or dysfunction of platelets, to the coagulation factors, or occasionally to excessive fibrinolysis.
Clinical assessment
‘Screening’ blood tests do not reliably detect all causes of pathological bleeding (e.g. von Willebrand disease, scurvy, certain anticoagulant drugs and the causes of purpura) and should not be used indiscriminately.
A careful clinical evaluation is the key to diagnosis .
It is important to consider the following:
• Site of bleeding. Bleeding into muscle and joints, alongwith retroperitoneal and intracranial haemorrhage,
indicates a likely defect in coagulation factors.
Purpura, prolonged bleeding from superficial cuts,
epistaxis, gastrointestinal haemorrhage or menorrhagia is more likely to be due to thrombocytopenia, a platelet function disorder or VWD . Recurrent bleeds at a single site suggest a local structural abnormality.
• Duration of history. It may be possible to assess
whether the disorder is congenital or acquired.
• Precipitating causes. Bleeding arising spontaneously
indicates a more severe defect than bleeding that
occurs only after trauma.
• Surgery. Ask about operations. Dental extractions,
tonsillectomy and circumcision are stressful tests of the haemostatic system. Immediate post-surgical bleeding suggests defective platelet plug formation and primary haemostasis; delayed haemorrhage is more suggestive of a coagulation defect. In postsurgical patients, persistent bleeding from a single site is more likely to indicate surgical bleeding than a bleeding disorder.
• Family history. While a positive family history may be present in patients with inherited disorders, the absence of affected relatives does not exclude a hereditary bleeding diathesis; about one-third of cases of haemophilia arise in individuals without a family history, and deficiencies of factor VII, X and XIII are recessively inherited.
• Drugs. Use of antithrombotic, anticoagulant and
fibrinolytic drugs must be elicited. Drug interactions with warfarin and drug-induced thrombocytopenia should be considered. Some ‘herbal’ remedies may result in a bleeding diathesis.Clinical examination may reveal different patterns of
skin bleeding. Petechial purpura is minor bleeding into
the dermis that is flat and non-blanching . Petechiae are typically found in patients with thrombocytopenia
or platelet dysfunction. Palpable purpura occurs in vasculitis. Ecchymosis, or bruising, is more extensive bleeding into deeper layers of the skin.
The lesions are initially dark red or purple but become
yellow as haemoglobin is degraded.
Retroperitoneal bleeding presents with a flank haematoma.
Telangiectasia of lips and tongue points to hereditary haemorrhagic telangiectasia .Joints should be examined for evidence of haemarthroses. A full examination is important, as it may give clues to an underlying associated systemic illness such as a haematological or other malignancy, liver disease, renal failure, connective tissue disease and possible causes of splenomegaly.Investigations
If the patient has a history that is strongly suggestive of a bleeding disorder and all the preliminary screening tests give normal results, further investigations, such as measurement of von Willebrand factor and assessment of platelet function, should be performed .
Causes of non-thrombocytopenic purpura
• Senile purpura• Factitious purpura
• Henoch– Schِnlein purpura
• Vasculitis
• Paraproteinaemias
• Purpura fulminans, e.g. in DIC secondary to sepsis
Thrombocytopenia (low platelet count)
May arise by one of two mechanisms:
• Decreased or abnormal production (bone marrow
failure and hereditary thrombocytopathies)
• Increased consumption following release into the
circulation (immune-mediated, DIC or sequestration).
Spontaneous bleeding does not usually occur until
the platelet count falls < 20 × 109/L, unless their
function is also compromised. Purpura and spontaneous
bruising are characteristic but there may also be oral,
nasal, gastrointestinal or genitourinary bleeding.
Severe thrombocytopenia (< 10 × 109/L) may result in retinal haemorrhage and potentially fatal intracranial bleeding, but this is rare.
Investigations are directed at the possible causes
listed in Box. A blood film is the single most usefulinitial investigation. Examination of the bone marrow
may reveal increased megakaryocytes in consumptive
causes of thrombocytopenia, or the underlying cause of
bone marrow failure in leukaemia, hypoplastic anaemia
or myelodysplasia.
Treatment (if required) depends on the underlying
cause. Platelet transfusion is rarely required and is
usually confined to patients with bone marrow failure
and platelet counts below 10 × 109/L, or to clinical situations with actual or predicted serious haemorrhage.

Causes of thrombocytopenia
Increased consumptionImmune mechanisms
• Idiopathic thrombocytopenic purpura*
• Neonatal alloimmune thrombocytopenia
• Post-transfusion purpura
• Drug-associated, especially quinine and vancomycin
Coagulation activation
• Disseminated intravascular coagulation
Mechanical pooling
• Hypersplenism
Thrombotic microangiopathies
• Haemolytic uraemic syndrome
• Liver disease
• Thrombotic thrombocytopenic purpura
• Pre-eclampsia
Others
• Gestational thrombocytopenia
• Type 2B von Willebrand disease
*Associated conditions include collagen vascular diseases (particularly SLE),
B cell malignancy, HIV infection and antiphospholipid syndrome.
Thrombocytosis (high platelet count)
The most common reason for a raised platelet count is
that it is reactive to another process such as infection,
connective tissue disease, malignancy, iron deficiency,
acute haemolysis or gastrointestinal bleeding .
The presenting clinical features are usually those of the
underlying disorder and haemostasis is rarely affected.
Reactive thrombocytosis is distinguished from the myeloproliferative disorders by the presence of uniform small platelets, lack of splenomegaly, and the presence of an associated disorder. The key to diagnosis is the clinical history and examination, combined with observation of the platelet count over time (reactive thrombocytosis gets better with resolution of the underlying cause).
The platelets are a product of an abnormally expanding
clone of cells in the myeloproliferative disorders,chronic myeloid leukaemia and some forms of myelodysplasia.
Patients with PRV, essential thrombocythaemia and occasionally myelofibrosis may present with thrombosis or, rarely, bleeding. Stroke and transient ischaemic attacks, amaurosis fugax, and digital ischaemia or gangrene are also features. In addition, patients with myeloproliferative disorders present with features such as itching after exposure to water (aquagenic pruritus), splenomegaly and systemic upset.
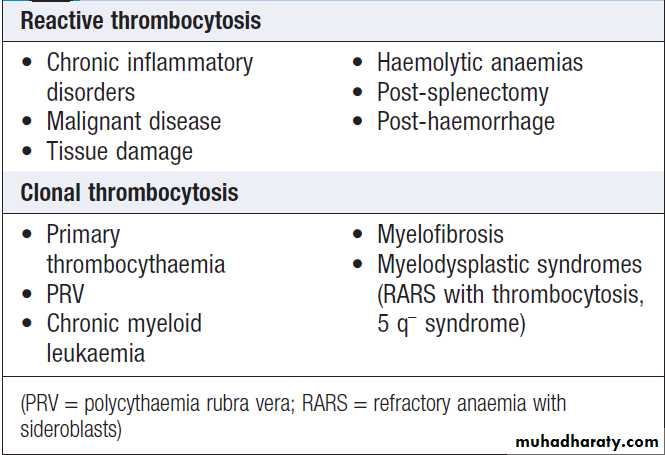
Causes of a raised platelet count
PancytopeniaPancytopenia refers to the combination of anaemia,
leucopenia and thrombocytopenia. It may be due to
reduced production of blood cells as a consequence of
bone marrow suppression or infiltration, or there may
be peripheral destruction or splenic pooling of mature
cells. A bone marrow aspirate and trephine are usually required to establish the diagnosis.
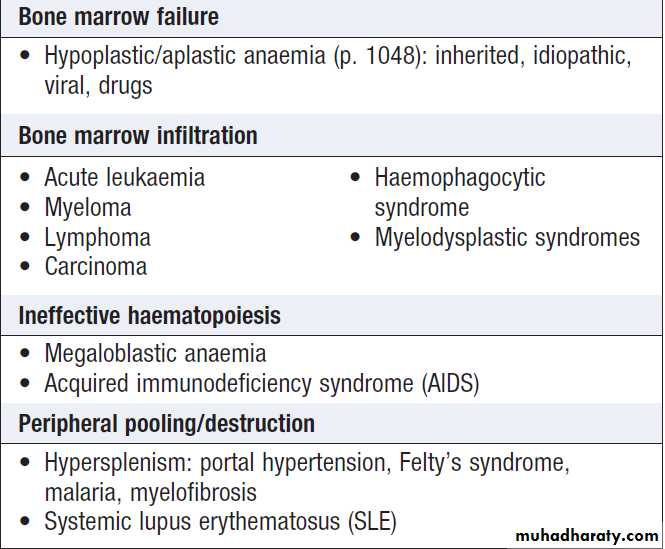
Causes of pancytopenia
Venous thrombosisWhile the most common presentation of venous thromboembolic disease (VTE) is with deep vein thrombosis
(DVT) of the leg and/or pulmonary embolism, similar principles apply to rarer manifestations such as jugular vein thrombosis, upper limb DVT, cerebral sinus thrombosis and intra-abdominal venous thrombosis (e.g. Budd– Chiari syndrome).
DVT has an annual incidence of approximately 1 : 1000 in Western populations and the case mortality is 1–3%. It is increasingly common with ageing, and many of the deaths are related to coexisting medical conditions,
such as active cancer.
Clinical assessment
Lower limb DVT characteristically starts in the distalveins, causing pain, swelling, an increase in temperature and dilatation of the superficial veins. Often, however,
symptoms and signs are minimal. It is typically unilateral
but may be bilateral, and clot may extend proximally
into the inferior vena cava. Bilateral DVT is more commonly
seen with underlying malignancy or anomalies of
the inferior vena cava. The differential diagnosis of unilateral leg swelling includes a spontaneous or traumatic
calf muscle tear or a ruptured Baker’s cyst, both characterised by sudden onset and localised tenderness.
Infective cellulitis is usually distinguished by marked skin
erythema and heat localised within a well-demarcated
area of the leg and may be associated with an obvious
source of entry of infection (e.g. insect bite, leg ulcer). Risk factors for DVT should be considered and examination should include assessment for malignancy. Symptoms and signs of PE should be sought , particularly in those with proximal thrombosis; asymptomatic PE is thought to be present in approximately 30% of patients with lower limb DVT.
Clinical criteria can be used to rank patients according
to their likelihood of DVT or PE: for example, by using
scoring systems such as the Wells score .
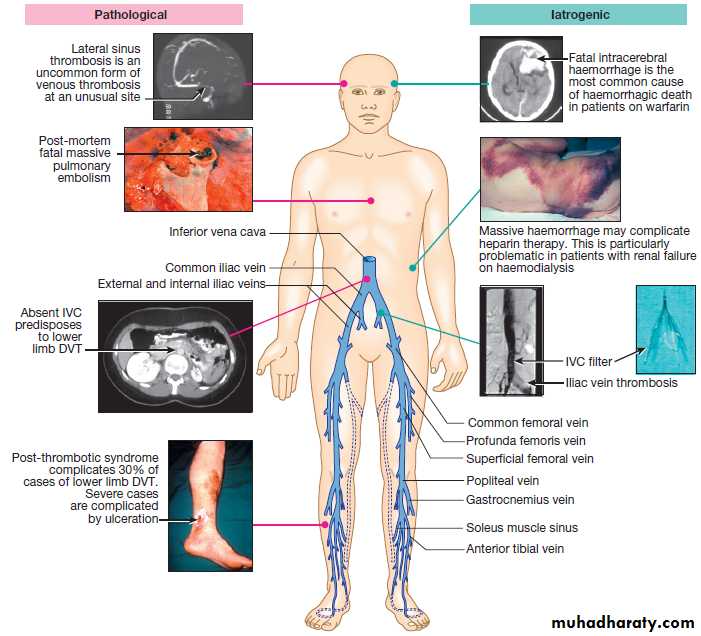
Causes and consequences of venous thromboembolic disease and its treatment.
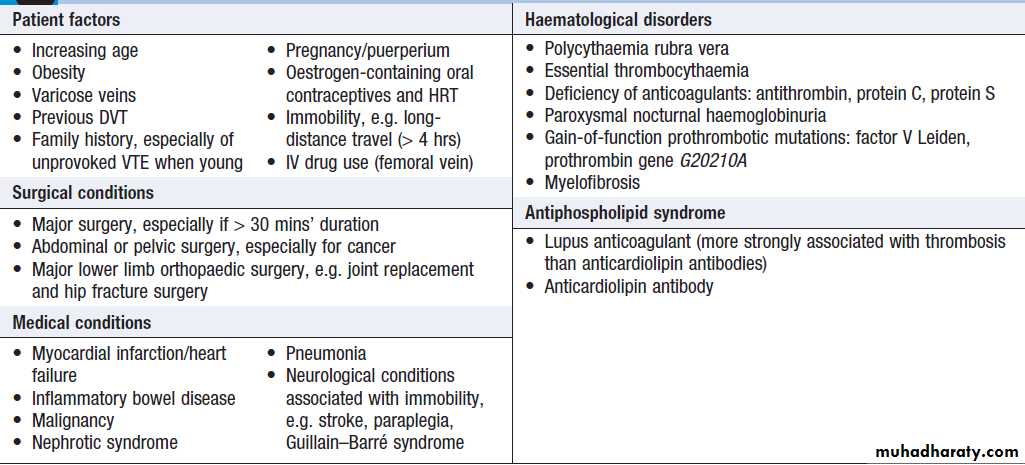
Factors predisposing to venous thrombosis
InvestigationsIn patients with a low (‘unlikely’) pre-test probability of DVT, D-dimer levels can be measured; if these are normal, further investigation for DVT is unnecessary. In those with a
moderate or high (‘likely’) probability of DVT or with
elevated D-dimer levels, objective diagnosis of DVT
should be obtained using appropriate imaging. Compression ultrasound is the imaging modality of choice in most centres. It has a sensitivity for proximal DVT (clot involving the popliteal vein or above) of 99.5%. Sensitivity and specificity are lower for diagnosing calf vein thrombosis. Contrast venography is an alternative that is now rarely used.
In patients with proven DVT, further imaging to diagnose PE is not required unless massive PE is clinically suspected or there is otherwise unexplained breathlessness Predisposing factors, particularly pelvic malignancy and those listed in Box, should be considered and investigation pursued. In occasional patients, further
investigation for an underlying thrombophilic condition may be considered .
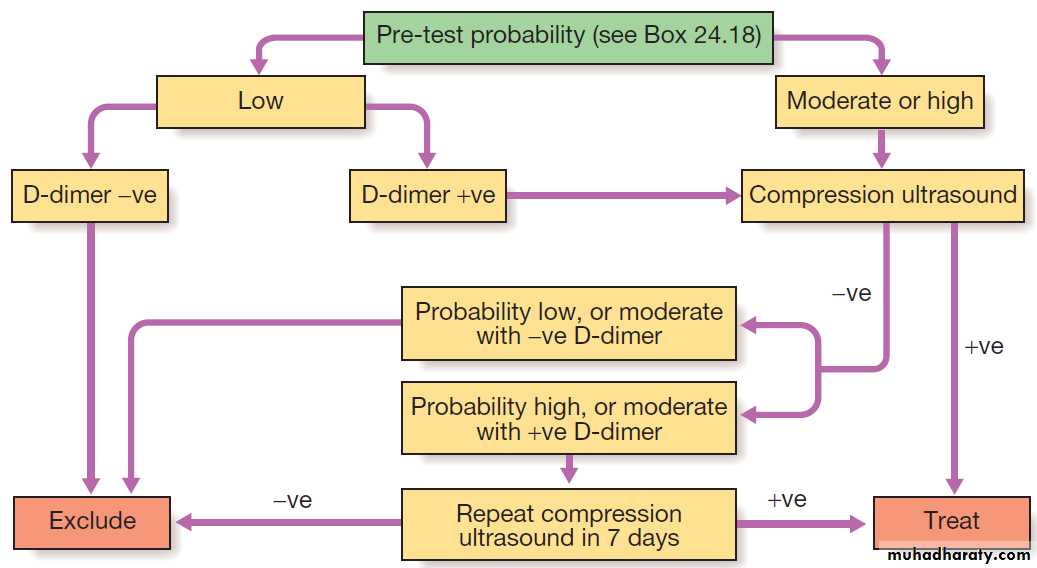
Investigation of suspected deep vein thrombosis.
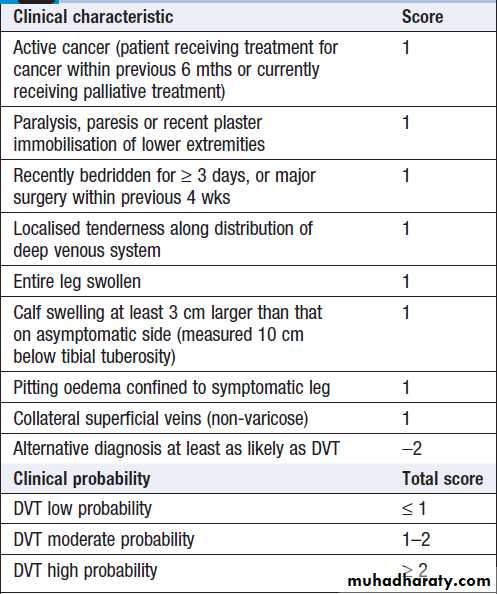
Predicting the pre-test probability of deep vein thrombosis using the Wells score
ManagementThe management of leg DVT includes elevation and
analgesia. Thrombolysis may be considered for limb- threatening DVT, but the mainstay of treatment is anticoagulation with low molecular weight heparin (LMWH), followed by a coumarin anticoagulant, such as warfarin. An alternative is the oral Xa inhibitor, rivaroxaban, which has a rapid onset of action and can be used immediately from diagnosis without the need for LMWH. Treatment of acute VTE with LMWH should continue for at least 5 days. If a coumarin is being introduced, the heparin should continue until the INR has been in the target
range (2–3) for 2 days.
Patients who have had a DVT and have a strong contraindication to anticoagulation, and those who, despite therapeutic anticoagulation, continue to have new pulmonary emboli, should have an inferior vena cava filter inserted to prevent life-threatening PE. The optimal initial duration of anticoagulation is between 6 weeks and 6 months. Patients who have thrombosis in the presence of a temporary risk factor, which is then removed, can usually be treated for shorter periods (e.g. 3 months) than those who sustain unprovoked thrombosis.
In patients with active cancer and VTE, there is evidence that LMWH should be continued for 6 months rather than being replaced by a coumarin.
Evidence indicates that periods of anticoagulation
of more than 6 months do not alter the rate
of recurrence following discontinuation of therapy.
Recurrence of DVT is about 2–3% per annum in
patients who have a medical temporary risk factor at
presentation and about 8% per annum in those with
apparently unprovoked DVT. Recurrence plateaus at
around 30–40% at 5 years.
Post-thrombotic syndrome is due to damage of venous valves by the thrombus. It results in persistent leg swelling, heaviness and discoloration.
The most severe complication of this syndrome
is ulceration around the medial malleolus.

Treatment of venous thromboembolism
BLOOD PRODUCTS AND TRANSFUSIONBlood transfusion from an unrelated donor to a recipient
inevitably carries some risk, including adverse immunological interactions between the host and infused blood and transmission of infectious agents. Although
there are many compelling clinical indications for blood
component transfusion, there are also many clinical circumstances in which transfusion is conventional but the
evidence for its effectiveness is limited. In these settings,
allogeneic transfusion may be avoided by following protocols that recommend use of low haemoglobin thresholds for red cell transfusion , perioperative blood salvage and antifibrinolytic drugs.

Red cell transfusion in critically ill patients
Blood productsBlood components are prepared from whole blood collected from individual donors and include red cells, platelets, plasma and cryoprecipitate .
Plasma derivatives are licensed pharmaceutical products
produced on a factory scale from large volumes of human plasma obtained from many people and treated
to remove transmissible infection. Examples include:
• Coagulation factors. Concentrates of factors VIII and
IX are used for the treatment of conditions such as
haemophilia A, haemophilia B and von Willebrand
disease. Coagulation factors made by recombinant
DNA technology are now preferred due to
perceived lack of infection risk but plasma-derived
products are still used in many countries.
• Immunoglobulins. Intravenous immunoglobulin (IVIgG) is administered as regular replacement therapy to reduce infective complications in patients with immunodeficiency. A short, high-dose course of IVIgG may also be effective in some immunological disorders, including immune thrombocytopenia and Guillain– Barré syndrome. IVIgG can cause acute reactions and must be infused strictly according to the manufacturer’s product information. There is a risk of renal dysfunction in susceptible patients and, in these circumstances, immunoglobulin products containing low or no sucrose are preferred. Anti-zoster immunoglobulin has a role in the prophylaxis of varicella zoster . Anti-Rhesus D immunoglobulin is used in
pregnancy to prevent haemolytic disease of the
newborn .
• Human albumin. This is available in two strengths.
The 5% solution can be used as a colloid
resuscitation fluid, but it is no more effective and
is more expensive than crystalloid solutions . Human albumin 20% solution is used in the
management of hypoproteinaemic oedema in
nephrotic syndrome and ascites in chronic
liver disease . It is hyperoncotic and expands
plasma volume by more than the amount infused.

Fluid resuscitation in critically ill patients
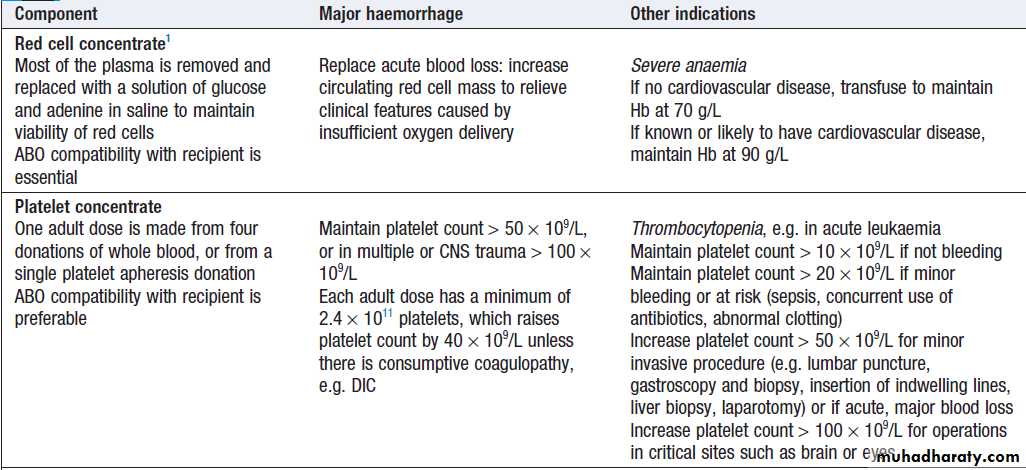
Blood components and their use

Blood components and their use – cont’d

Blood donation
A safe supply of blood components depends on a well organised system with regular donation by healthy individuals who have no excess risk of infections transmissible in blood . Blood donations are obtained by either venesection of a unit of whole blood or collectionof a specific component, such as platelets, by apheresis.
During apheresis, the donor’s blood is drawn via a
closed system into a machine which separates the components by centrifugation and collects the desired fraction into a bag, returning the rest of the blood to the
donor. Each donation must be tested for hepatitis B
(HBV), hepatitis C (HCV), HIV and human T cell lymphotropic (HTLV) virus nucleic acid and/or antibodies.
Platelet concentrates may be tested for bacterial contamination.
The need for other microbiological tests dependson local epidemiology. For example, testing for Trypanosoma cruzi (Chagas’ disease) is necessary in areas
of South America and the USA where infection is prevalent; tests for West Nile virus have been required in the USA since this agent became prevalent; plasma donated in the UK is not used at present for producing pooled plasma derivatives in view of concerns about transmission of variant Creutzfeldt– Jakob disease .
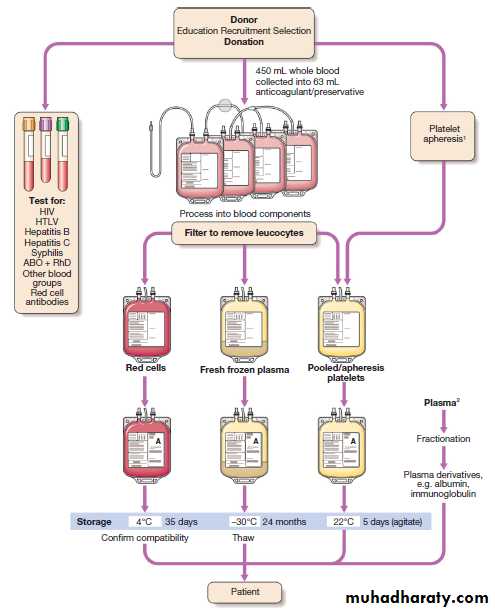
Blood donation, processing and storage. 1Platelet apheresis involves circulating the donor’s blood through a cell separator to remove
platelets before returning other blood components to the donor. 2In the UK, plasma for fractionation is imported as a precautionary measure against variant Creutzfeldt – Jakob disease. (HIV = human immunodeficiency virus;
HTLV = human T cell lymphotropic virus)
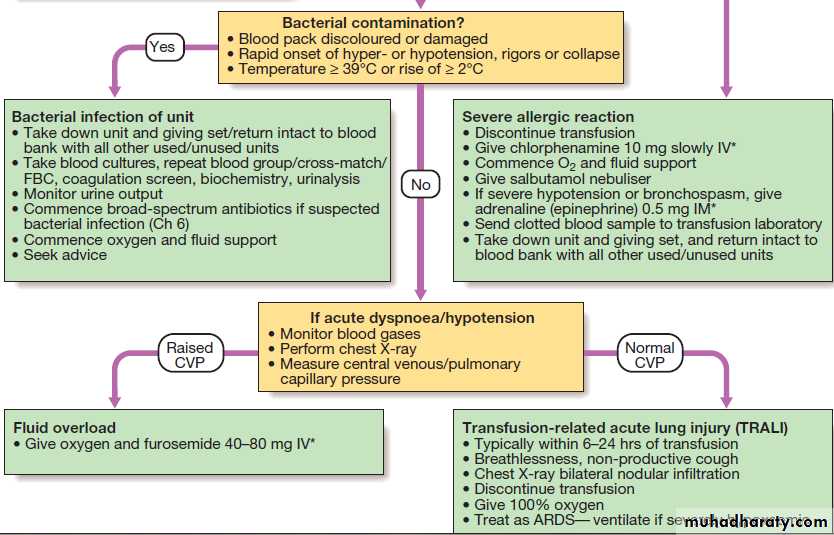
Adverse effects of transfusion
Death directly attributable to transfusion is rare, at less
than 0.3 per 100 000 transfusions. However, relatively
minor symptoms of transfusion reactions (fever, itch or
urticaria) occur in up to 3% of transfusions, usually in
patients who have had repeated transfusions. Any
symptoms or signs that arise during a transfusion must
be taken seriously, as they may be the first warnings of
a serious reaction.
Red cell incompatibility
Red blood cell membranes contain numerous cell surface
molecules which are potentially antigenic. The ABO and Rh(D) antigens are the most important in routine transfusion and antenatal practice.
ABO blood groups
The frequency of the ABO antigens varies among differentpopulations. The ABO blood group antigens are
Oligosaccharide chains that project from the red cell
surface. These chains are attached to proteins and lipids
that lie in the red cell membrane. The ABO gene encodes
a glycosyltransferase that catalyses the final step in the
synthesis of the chain which has three common alleles:
A, B and O. The O allele encodes an inactive enzyme, leaving the ABO antigen precursor (called the H antigen) unmodified. The A and B alleles encode enzymes that differ by four amino acids and hence attach different sugars to the end of the chain.
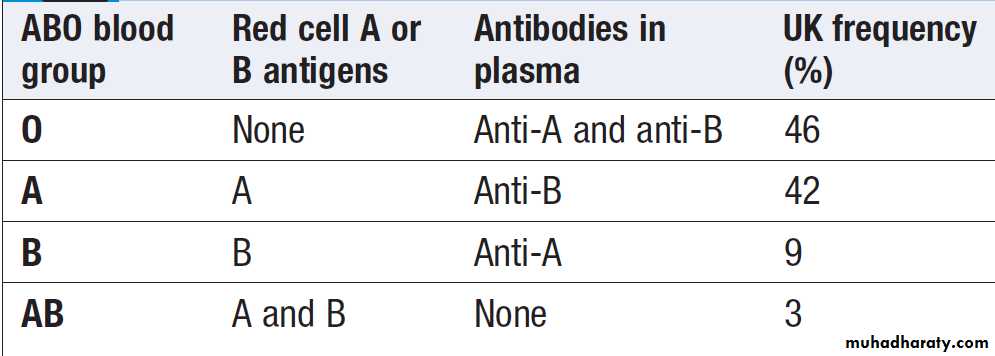
ABO blood group antigens and antibodies
ABO-incompatible red cell transfusionIf red cells of an incompatible ABO group are transfused
(especially if a group O recipient is transfused with
group A, B or AB red cells), the recipient’s IgM anti-A,
anti-B or anti-AB binds to the transfused red cells. This
activates the full complement pathway , creating
pores in the red cell membrane and destroying the transfused red cells in the circulation (intravascular haemolysis).
The anaphylatoxins C3a and C5a, released by
complement activation, liberate cytokines such as tumour
necrosis factor (TNF), interleukin 1 (IL-1) and IL-8, and
stimulate degranulation of mast cells with release of
vasoactive mediators.
All these substances may lead to inflammation, increased vascular permeability and hypotension, which may, in turn, cause shock and renal failure. Inflammatory mediators can also cause platelet aggregation, lung peribronchial oedema and smooth muscle contraction.
About 20–30% of ABO-incompatible transfusions cause some degree of morbidity, and 5–10% cause or contribute to a patient’s death. The main reason for this relatively low morbidity is the lack of potency of
ABO antibodies in group A or B subjects; even if the
recipient is group O, those who are very young or very
old usually have weaker antibodies that do not lead to
the activation of large amounts of complement.
The Rhesus D blood group and haemolytic disease of the newborn
About 15% of Caucasians are Rhesus-negative; that is,
they lack the Rhesus D (RhD) red cell surface antigen. In other populations (e.g. in Chinese and Bengalis), only 1–5% are Rhesus-negative. RhD-negative individuals do not normally produce substantial amounts of anti-RhD antibodies. However, if RhD-positive red cells enter the circulation of an RhD-negative individual, IgG antibodies are produced. This can occur during pregnancy
if the mother is exposed to fetal cells via feto-maternal
haemorrhage, or following transfusion
If a woman is so sensitised, during a subsequent pregnancy anti-RhD antibodies can cross the placenta; if the fetus is RhD-positive, haemolysis with severe fetal anaemia and hyperbilirubinaemia can result.
This can cause severe neurological damage or death due to haemolytic disease of the newborn (HDN). Therefore, an RhD-negative female who may subsequently become pregnant should never be transfused with RhD-positive blood.
In RhD-negative women, administration of anti-RhD
immunoglobulin (anti-D) perinatally can block the
immune response to RhD antigen on fetal cells and is
the only effective product for preventing the development of Rhesus antibodies .
HDN can also be caused by other alloantibodies against red cell antigens, usually after previous pregnancies
or transfusions. These antigens include Rhc, RhC, RhE, Rhe, and the Kell, Kidd and Duffy antigen systems.
HDN can also occur if there is fetomaternal
ABO incompatibility, most commonly seen in a group
O mother with a group A fetus. The fetus is generally
less severely affected by ABO incompatibility than by
RhD, Rhc or Kell antigen mismatch.

Rhesus D blood groups in pregnancy
Other immunological complications of transfusionRare but serious complications include transfusion-associated lung injury (TRALI) and transfusion-associated
graft-versus-host disease (TA GVHD). The latter occurs
when there is sharing of a human leucocyte antigen (HLA) haplotype between donor and recipient, which allows transfused lymphocytes to engraft, proliferate and recognise the recipient as foreign, resulting in
acute GVHD .
Prevention is by gamma- or X-ray irradiation of
blood components before their administration to prevent
lymphocyte proliferation.
Those at risk of TA GVHD who must receive irradiated blood components include:
patients with congenital T cell immunodeficienciesOr Hodgkin lymphoma; patients with aplastic anaemia
receiving immunosuppressive therapy with antithymocyte
globulin; recipients of haematopoietic stem
cell transplants or of blood from a family member;
neonates who have received an intrauterine transfusion;
and patients taking T lymphocyte-suppressing drugs,
such as fludarabine and other purine analogues.
Transfusion-transmitted infection
Over the past 30 years, HBV, HIV-1 and HCV have been
identified and effective tests introduced to detect and
exclude infected donations. Where blood is from ‘safe’
donors and correctly tested, the current risk of a donated
unit being infectious is very small. By 2010 in the UK, the
estimated chance that a unit of blood from a ‘safe’ donor
might transmit one of the viruses for which blood is tested
was 1 in 6.4 million units for HIV-1, 1 in 100 million for
HCV and 1 in 1.4 million for HBV. However, some patients
who received transfusions before these tests were available suffered serious consequences from infection; this serves as a reminder to avoid non-essential transfusion, since it is impossible to exclude the emergence of new or currently unrecognised transfusion-transmissible infection.
Licensed plasma derivatives that have been virus- inactivated do not transmit HIV, HTLV, HBV, HCV,
cytomegalovirus or other lipid-enveloped viruses.
vCJD is a human prion disease linked to bovine
spongiform encephalitis (BSE). The risk of a
recipient acquiring the agent of vCJD from a transfusion
is uncertain, but of 16 recipients of blood from donors
who later developed the disease, 3 have died with clinical vCJD and 1 other had postmortem pathological features of infection.
Bacterial contamination of a blood component –
usually platelets – is extremely rare (e.g. no reports in
the UK in either 2010 or 2011) but can result in severe
bacteraemia/ septicaemia in the recipient.
Safe transfusion procedures
The proposed transfusion should be discussed with the patient or, if that is not possible, with a relative.
Pre-transfusion testing
To ensure that red cells supplied for transfusion are
compatible with the intended recipient, the transfusion
laboratory will perform either a ‘group and screen’ procedure or a ‘cross-match’. In the procedure, the red cells from the patient’s blood sample are tested to determine the ABO and RhD type, and the patient’s serum is also tested against an array of red cells expressing the most important antigens to detect any red cell antibodies. Any antibody detected can be identified by further testing, so that red cell units that lack the corresponding antigen can be selected.
The patient’s sample can be held in the laboratory for up to a week, so that the hospital blood bank can quickly prepare compatible blood without the need for a further patient sample. Conventional cross-matching consists of the group and antibody screen, followed by direct confirmation of the compatibility of individual units of red cells with
the patient’s serum. Full cross-matching takes about 45 minutes if no red cell antibodies are present, but may require hours if a patient has multiple antibodies.
Blood can be supplied by ‘electronic issue’, without the need for compatibility cross-matching, if the laboratory’s computer system shows that the patient’s ABO and RhD groups have been identified and confirmed on two separate occasions and their antibody screen is negative.
Bedside procedures for safe transfusion
Errors leading to patients receiving the wrong blood arean important avoidable cause of mortality and morbidity.
Most incompatible transfusions result from failure
to adhere to standard procedures for taking correctly
labelled blood samples from the patient and ensuring
that the correct pack of blood component is transfused
into the intended patient. In the UK in 2011, there were
247 reports of transfusion of an incorrect blood component (8 per 100 000 units transfused).
Every hospital where blood is transfused should have a written transfusion policy used by all staff who order, check or administer blood products .
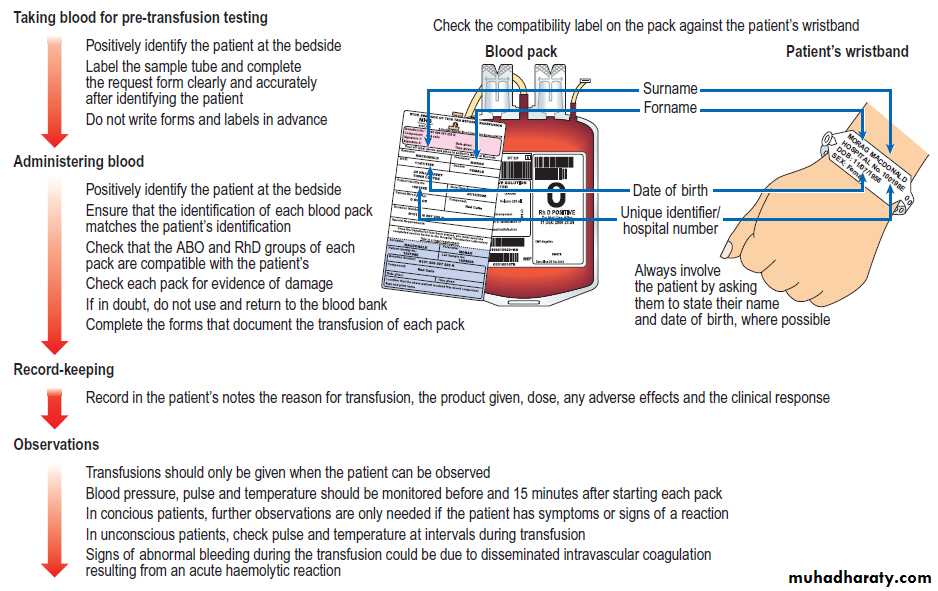
Bedside procedures for safe blood transfusion
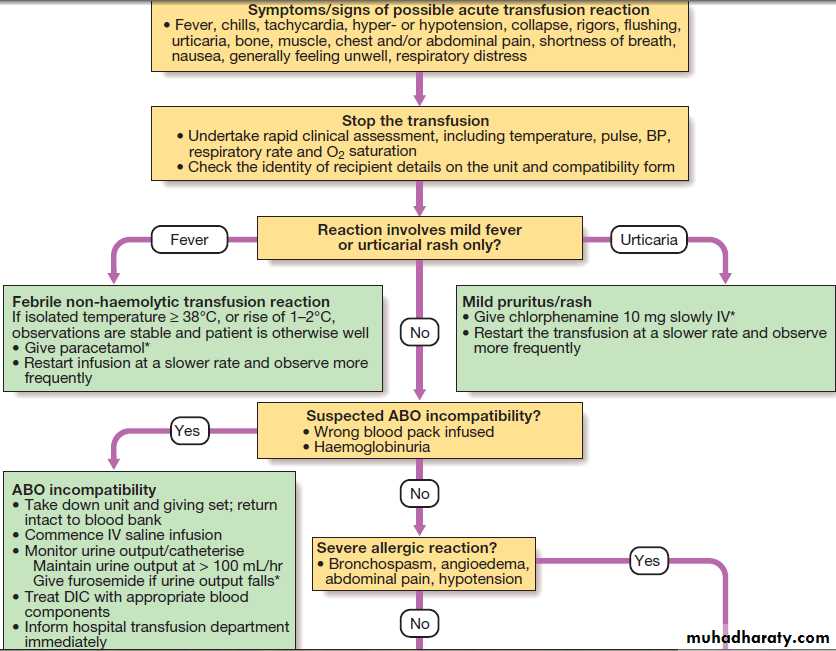
Investigation and management of acute transfusion reactions.
HAEMATOPOIETIC STEM CELL TRANSPLANTATION (HSCT)
Transplantation of HSC has offered the only hope of ‘cure’ in a variety of haematological and non-haematological disorders .The type of HSCT is defined according to the donor and source of stem cells:• In allogeneic HSCT, the stem cells come from a donor
– either related (usually an HLA-identical sibling) or a closely HLA-matched volunteer unrelated donor (VUD).
• In an autologous transplant, the stem cells are
harvested from the patient and stored in the vapour
phase of liquid nitrogen until required. Stem cells can be harvested from the bone marrow or from the blood.
Allogeneic HSCT
Healthy bone marrow or blood stem cells from a donor
are infused intravenously into the recipient, who has
been suitably ‘conditioned’. The conditioning treatment
(chemotherapy with or without radiotherapy) destroys
malignant cells and immunosuppresses the recipient, as well as ablating the recipient’s haematopoietic tissues
(myeloablation). The infused donor cells ‘home’ to the
marrow, engraft and produce enough erythrocytes,
granulocytes and platelets for the patient’s needs after
about 3–4 weeks. During this period of aplasia, patients are at risk of infection and bleeding, and require intensive supportive care.
It may take several years to regain normal immunological function and remain at risk from opportunistic infections, in particular in the first year.
An advantage of receiving allogeneic donor stem
cells is that the donor’s immune system can recognise
residual recipient malignant cells and destroy them. This immunological ‘graft versus disease’ effect is a powerful tool against many haematological tumours and can be boosted post transplantation by the infusion of T cells taken from the donor, so-called donor lymphocyte infusion (DLI).
Considerable morbidity and mortality are associated
with HSCT. The best results are obtained with minimal
residual disease, and in those under 20 years of age who have an HLA-identical sibling donor.
Reduced-intensity HSCT has enabled treatment of older or less fit patients. In this form, rather than using very
intensive conditioning which causes morbidity from organ damage, relatively low doses of drugs, such as fludarabine and cyclophosphamide, are used to immunosuppress the recipient and allow donor stem cells to engraft. The emerging donor immune system then eliminates malignant cells via the ‘graft versus disease’ effect, which may be boosted by the elective use of donor T cell infusions post transplant.
Complications
The risks and outcomes of transplantation depend upon several patient- and disease-related factors. In general, 25% die from procedure-related complications, such as infection and GVHD, and there remains a significant risk of relapse of the haematological malignancy. The long term survival in acute leukaemia is around 50%.
Graft-versus-host disease (GVHD)
Caused by the cytotoxic activity of donor T lymphocytes which become sensitised to their new host, regarding it as foreign. This may cause acute or a chronic form of GVHD.
Acute GVHD occurs in the first 100 days after transplant in one-third of patients. It can affect the skin, causing rashes, the liver, causing jaundice, and the gut, causing diarrhoea, and may vary from mild to lethal.
Prevention includes HLA-matching of the donor,
immunosuppressant drugs, including methotrexate,ciclosporin, alemtuzumab or antithymocyte globulin.
Severe presentations are very difficult to control and,
despite high-dose corticosteroids, may result in death.
Chronic GVHD may follow acute GVHD or arise
independently. It often resembles a connective tissue disorder, and carries an increased risk of infection, although in mild cases a rash may be the only manifestation. Chronic GVHD is usually treated with corticosteroids and prolonged immunosuppression with, for example, ciclosporin. However, associated with chronic GVHD are the graft versus- leukaemia effect and a lower relapse rate of the underlying malignancy.

Indications for allogeneic HSCT
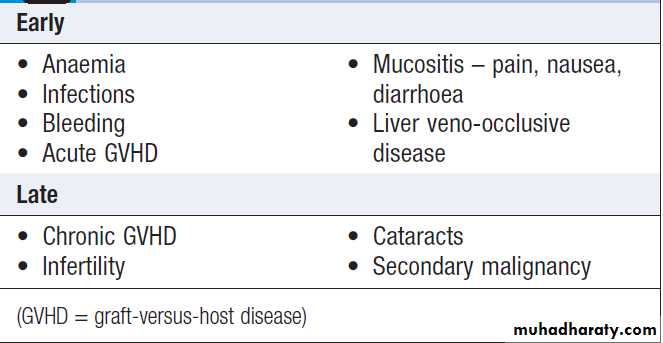
Complications of allogeneic HSCT
Autologous HSCT
The patient’s own stem cells from blood or marrow are first harvested and frozen. After conditioning myeloablative therapy, the autologous stem cells are reinfused into the blood stream in order to rescue the patient from the marrow damage and aplasia caused by chemotherapy. May be used for disorders which do not primarily involve the haematopoietic tissues, or in whom very good remissions have been achieved. The preferred source of stem cells is peripheral blood. These stem cells engraft more quickly, marrow recovery occurring within 2–3 weeks. There is no risk of GVHD and no immunosuppression is required. Carries a lower related mortality rate than allogeneic HSCT at around 5%, but there is a higher rate of recurrence of malignancy.
Infection Time after HSCT Management
Herpes simplex 0–4 wks (aplastic phase) Aciclovir prophylaxisand therapy
Bacterial, fungal 0–4 wks (aplastic phase) As for acute leukaemia– antibiotic
and antifungal prophylaxisand therapy
• Cytomegalovirus 5–21 wks
(cell-mediated immunedeficiency) Antigen screening in
blood (PCR) and
pre-emptive therapy
(e.g. ganciclovir)
Varicella zoster After 13 wks Aciclovir prophylaxis
and therapy
Pneumocystis 8–26 wks Co-trimoxazole
jirovecii
Encapsulated bacteria 8 wks to years Prophylaxis and revaccination
(immunoglobulin deficiency,
prolonged with GVHD)
Infections during recovery from HSCT
ANTICOAGULANT AND ANTITHROMBOTIC THERAPY
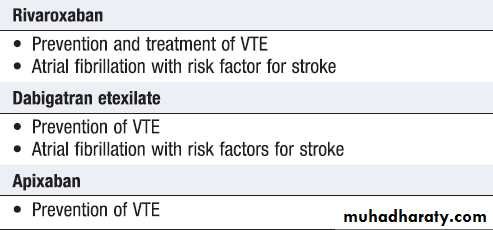
Antiplatelet are of greater efficacy in the prevention of arterial thrombosis and of less value in VTE. Thus, antiplatelet agents, such as aspirin and clopidogrel, are the drugs of choice in acute coronary events and in ischaemic cerebrovascular disease, while warfarin and other anticoagulants are favoured in VTE. In some extremely prothrombotic situations, such as coronary artery stenting, a combination of anticoagulant and antiplatelet drugs is used. Newer agents (dabigatran, rivaroxaban and apixaban), which can be given at fixed doses with predictable effects and no need for monitoring, now approved for the prevention of perioperative VTE, the treatment of VTE and the prevention of cardioembolic stroke in fibrillation.
Unfractionated heparin (UFH) and low molecular weight heparins (LMWH) both act by binding via a specific pentasaccharide to antithrombin which potentiates
its natural anticoagulant activity . Increased cleavage of activated proteases, particularly factor Xa and thrombin (IIa), accounts for the anticoagulant effect. LMWHs are nearly 100% bioavailable and therefore produce reliable dose-dependent anticoagulation,do not require monitoring (except possibly in patients with very low body weight and with a GFR below 30 mL/min).
Half-life of around 4 hours when given subcutaneously, compared with 1 hour for UFH. This permits once-daily dosing by the subcutaneous route, rather than intravenous infusion or prophylactic twice-daily subcutaneous for UFH.
While bleeding rates are similar,the risk of osteoporosis and heparin-induced thrombocytopenia is much lower for LMWH. However, UFH is more completely reversed by protamine sulphate in the event of bleeding and at the end of cardiopulmonary bypass, for which UFH remains of choice. In some situations, UFH is still favoured when rapid reversibility is required, in patients with a high risk of bleeding: for example, those with peptic ulceration or may require surgery.
Also favoured in the treatment of life-threatening thromboembolism: for example, major PE with significant hypoxaemia, hypotension and right sided heart strain, UFH is started with a loading IV dose of 80 U/kg., followed by a continuous infusion of 18 U/kg/ hr.
The level of anticoagulation should be assessed by APTT after 6 hours and, if satisfactory, twice daily thereafter. It is usual to aim for a patient APTT which is 1.5–2.5 times the control time of the test.
Heparin-induced thrombocytopenia (HIT)
Is a rare complication of heparin therapy, caused by induction of anti-heparin/PF4 antibodies which bind to and activate platelets via an Fc receptor. This results in platelet activation and a prothrombotic state, with a paradoxical thrombocytopenia. HIT is more common in surgical than medical patients (especially cardiac and orthopaedic patients), with use of UFH rather than LMWH, and with higher doses of heparin.Clinical features
Patients present, typically 5–14 days after startingheparin treatment, with a fall in platelet count of more
than 30% from baseline. The count may still be in the
reference range. They may be asymptomatic, or develop
venous or arterial thrombosis and skin lesions, including
overt skin necrosis. Affected patients may complain of
pain or itch at injection sites and of systemic symptoms,
such as shivering, following heparin injections. Patients
who have received heparin in the preceding 100 days
and who have preformed antibodies may develop acute
systemic symptoms and an abrupt fall in platelet count
in the first 24 hours after re-exposure.
Investigations
The pre-test probability of the diagnosis is assessed
using the 4Ts scoring system. This assigns a score
based on:
• the thrombocytopenia
• the timing of the fall in platelet count
• the presence of new thrombosis
• the likelihood of another cause for the
thrombocytopenia.
Individuals at low risk need no further test; those
with intermediate and high likelihood scores should
have the diagnosis confirmed or refuted using an anti-
PF4 enzyme-linked immunosorbent assay (ELISA).
Management
Heparin should be discontinued as soon as HIT is diagnosed and an alternative anticoagulant which does not cross-react with the antibody substituted.Argatroban (adirect thrombin inhibitor) and danaparoid (a heparin analogue) are licensed for use in the UK. In asymptomatic patients with HIT who do not receive an alternative anticoagulant, around 50% will sustain a thrombosis in the subsequent 30 days. Patients with established thrombosis have a poor prognosis.
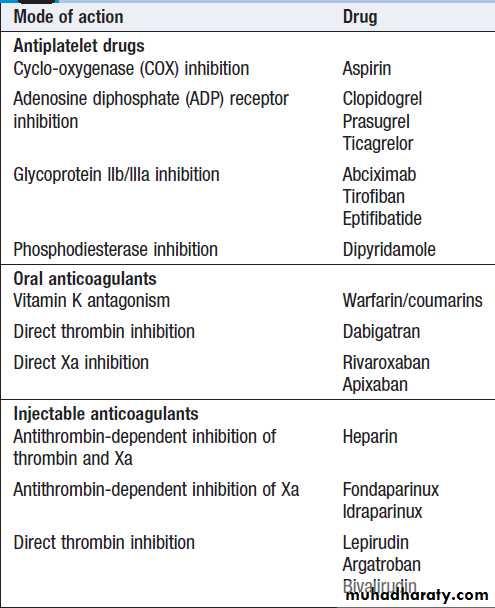
Modes of action of anticoagulant and
antithrombotic drugs
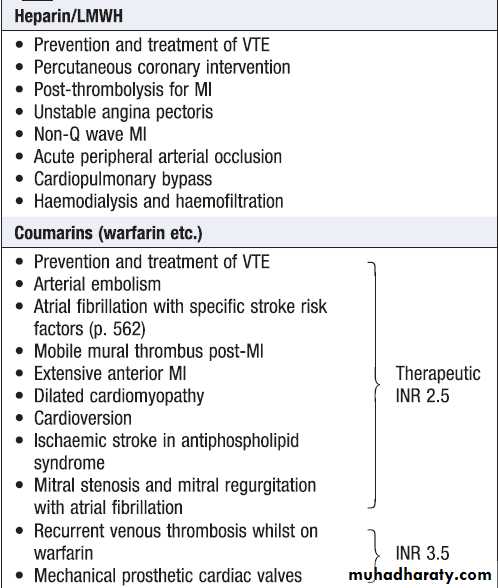
Indications for anticoagulation
CoumarinsAlthough several coumarin anticoagulants are used around the world, warfarin is the most common. Coumarins inhibit the vitamin K-dependent posttranslational carboxylation of factors II, VII, IX and X in the liver. This is monitored by the INR, a standardised test based on measurement of the prothrombin time . Warfarin anticoagulation typically takes 3–5 days to become established, even using initial loading doses.
Patients who require rapid initiation of therapy may receive higher initiation doses of warfarin. A typical regime in this situation is to give 10 mg warfarin on the first and second days, with 5 mg on the third day; subsequent doses are titrated against the INR.
Patients without an urgent need for anticoagulation (e.g. atrial fibrillation) can be introduced slowly using lower doses, associated with a lower risk of bleeding.
The major problems with warfarin are:
• a narrow therapeutic window
• metabolism that is affected by many factors
• numerous drug interactions.
Drug interactions are common through protein binding and metabolism by the cytochrome P450 system.
Major bleeding occurs in 1–2% each year.
Fatal haemorrhage, most commonly intracranial, occurs in about 0.25% per annum. There are scoring systems which predict the annual bleeding risk and can be used to help compare the risks and benefits.

How to assess risks of anticoagulation
Management of warfarin includes strategies for over-anticoagulation and for bleeding:• If the INR is above the therapeutic level, warfarin
should be withheld or the dose reduced. If the patient is not bleeding, it may be appropriate to give a small dose of vitamin K either orally or IV (1–2.5 mg), especially if the INR is greater than 8.
• In the event of bleeding, withhold further warfarin. Minor bleeding can be treated with 1–2.5 mg of vitamin K IV. Major haemorrhage should be treated as an emergency with vitamin K 5–10 mg slowly IV, combined with coagulation factor replacement. This should optimally be a prothrombin complex concentrate (30–50 U/kg) which contains factors II, VII, IX and X; if that is not available, fresh frozen plasma (15–30 mL/kg) should be given.
Prophylaxis of venous thrombosis
All patients admitted to hospital should be assessed fortheir risk of developing VTE and appropriate prophylactic
measures put in place. Early mobilisation is important to prevent DVT. Patients at medium or high risk require additional antithrombotic measures; these may be pharmacological or mechanical. There is increasing evidence in high-risk groups, such as patients who have had major lower limb orthopaedic surgery and abdominal or pelvic cancer surgery, for protracted thromboprophylaxis for as long as 30 days after the procedure.
Particular care should be taken in patients with a high risk of bleeding or risks related to the site of surgery or the use of spinal or epidural anaesthesia
Antithrombotic prophylaxis
IndicationsPatients in the following categories should be considered for specific antithrombotic prophylaxis:
Moderate risk of DVT
Major surgery
• In patients > 40 yrs or with other risk factor for VTE
Major medical illness, e.g.
• Heart failure • MI with complications • Sepsis • Inflammatory conditions, including inflammatory bowel disease • Active malignancy • Nephrotic syndrome
• Stroke and other conditions leading to lower limb paralysis
High risk of DVT
• Major abdominal or pelvic surgery for malignancy or with history of DVT or known thrombophilia • Major hip or knee surgery
• Neurosurgery
Methods of VTE prophylaxis
Mechanical
• Intermittent pneumatic compression
• Mechanical foot pumps
• Graduated compression stockings
Pharmacological
• LMWHs
• Unfractionated heparin
• Fondaparinux
• Dabigatran
• Rivaroxaban
• Apixaban
• Warfarin
ANAEMIAS
Around 30% of the total world population is anaemic and half of these, have iron deficiency. The classification of anaemia by the size of the red cells (MCV) indicates the likely cause. Whilst in the marrow compartment, red cell precursors undergo cell division, driven by erythropoietin.If red cells cannot acquire haemoglobin at a normal rate, they will undergo more divisions than normal and will have a low MCV when finally released into the blood. The MCV is low because component parts of the haemoglobin molecule are not fully available: that is, iron in iron deficiency, globin chains in thalassaemia, haem ring in congenital sideroblastic anaemia and, occasionally, poor iron utilisation in the anaemia of chronic disease.
In megaloblastic anaemia, the biochemical consequence
of vitamin B12 or folate deficiency is an inabilityto synthesise new bases to make DNA. A similar defect
of cell division is seen in the presence of cytotoxic drugs
or myelodysplasia. In these states, cells haemoglobinise normally but undergo fewer cell divisions, resulting in circulating red cells with a raised MCV. The red cell membrane is composed of a lipid bilayer which will freely exchange with the plasma pool of lipid. Conditions such as liver disease, hypothyroidism, hyperlipidaemia and pregnancy are associated with raised lipids and may also
cause a raised MCV. Reticulocytes are larger than mature red cells, so when raised – for example, in haemolysis – this may also increase the MCV.
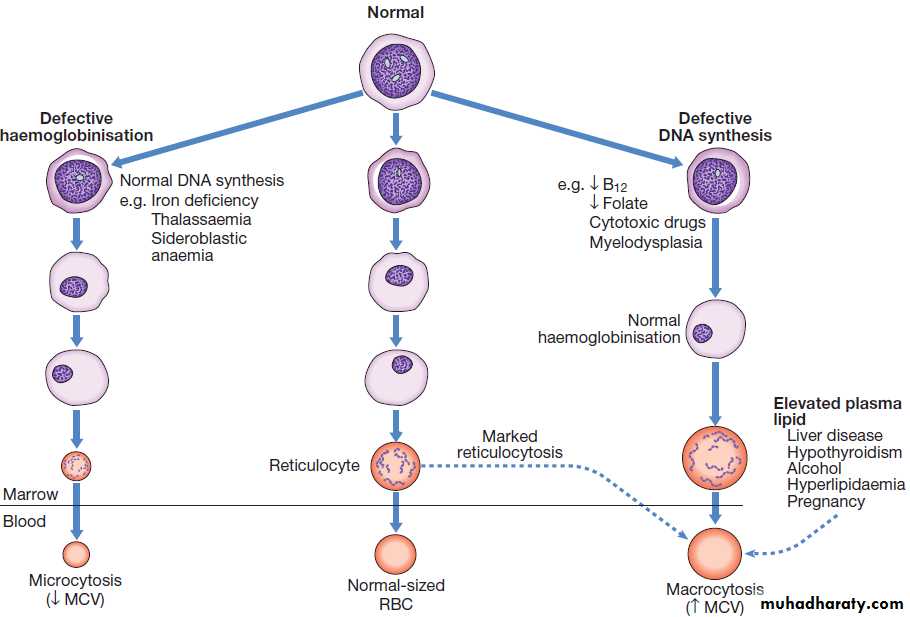
Factors which influence the size of red cells in anaemia. In microcytosis, the
MCV is < 76 fL. In macrocytosis, the MCV is > 100 fL. (MCV = mean cell volume.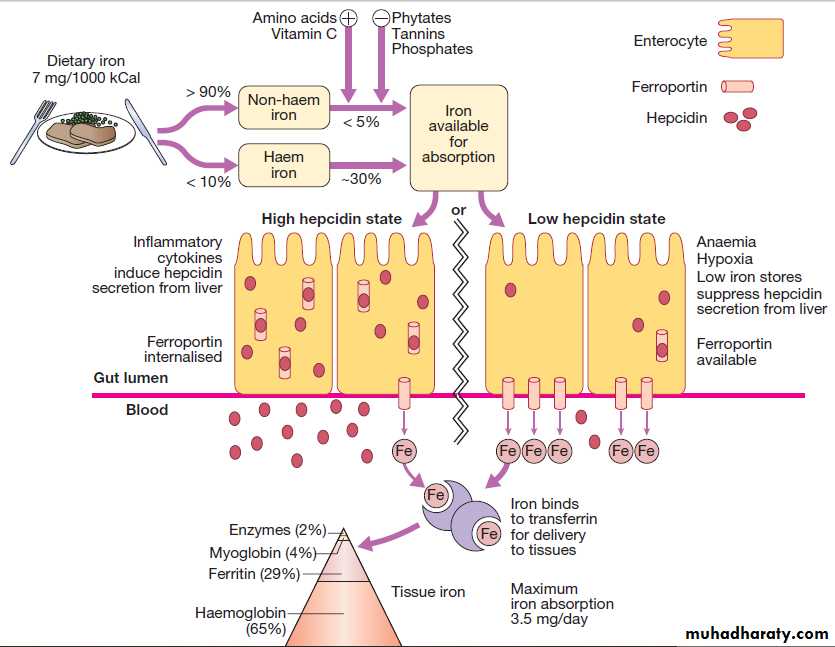
The regulation of iron absorption, uptake and distribution in the body. The transport of iron is regulated in a similar fashion to enterocytes in other iron-transporting cells such as macrophages.
Iron deficiency anaemia
This occurs when iron losses or physiological requirementsexceed absorption.
Blood loss
The most common explanation in men and postmenopausal
women is gastrointestinal blood loss .This may result from occult gastric or colorectal malignancy, gastritis, peptic ulceration, inflammatory bowel disease, diverticulitis, polyps and angiodysplastic lesions. Worldwide, hookworm and schistosomiasis are the most common causes of gut blood loss. Gastrointestinal blood loss may be exacerbated
by the chronic use of aspirin or non-steroidal anti-inflammatory drugs (NSAIDs), which cause intestinal erosions and impair platelet function.
In women of child-bearing age, menstrual blood loss, pregnancy and breastfeeding contribute to iron deficiency by depleting iron stores. Very rarely, chronic haemoptysis or haematuria may cause iron deficiency.
Malabsorption
A dietary assessment should be made in all patients to
ascertain their iron intake .Gastric acid is required to release iron from food and helps to keep iron in the soluble ferrous state . Achlorhydria in the elderly or due to drugs such as proton pump inhibitors may contribute to the lack of iron availability from the diet, as may previous gastric surgery. Iron is absorbed actively in the upper small intestine and hence can be affected by coeliac disease .
Physiological demands
At times of rapid growth, such as infancy and puberty,
iron requirements increase and may outstrip absorption.
In pregnancy, iron is diverted to the fetus, the placenta
and the increased maternal red cell mass, and is lost with
bleeding at parturition .
Investigations
Confirmation of iron deficiency
Plasma ferritin is a measure of iron stores in tissues and is the best single test to confirm iron deficiency . It is a very specific test; a subnormal level is due to iron deficiency or, very rarely, hypothyroidism or vitamin C deficiency.
Ferritin levels can be raised in liver disease and in the acute phase response.
Plasma iron and total iron binding capacity (TIBC)
are measures of iron availability; hence they are affected by many factors besides iron stores. Plasma iron has a marked diurnal and day-to-day variation and becomes very low during an acute phase response but is raised in liver disease and haemolysis. Levels of transferrin, the binding protein for iron, are lowered by malnutrition, liver disease, the acute phase response and nephrotic syndrome, but raised by pregnancy and the oral contraceptive pill. A transferrin saturation (i.e. iron/TIBC × 100) of less than 16% is consistent with iron deficiencybut is less specific than a ferritin measurement.
In difficult cases, it may still be necessary to examine a bone marrow aspirate for iron stores.
Investigation of the cause
This will depend upon the age and sex of the patient, aswell as the history and clinical findings. In men and in
post-menopausal women with a normal diet, the upper
and lower gastrointestinal tract should be investigated
by endoscopy or radiological studies. Serum antiendomysial or anti- transglutaminase antibodies and
possibly a duodenal biopsy are indicated to
detect coeliac disease. In the tropics, stool and urine
should be examined for parasites .
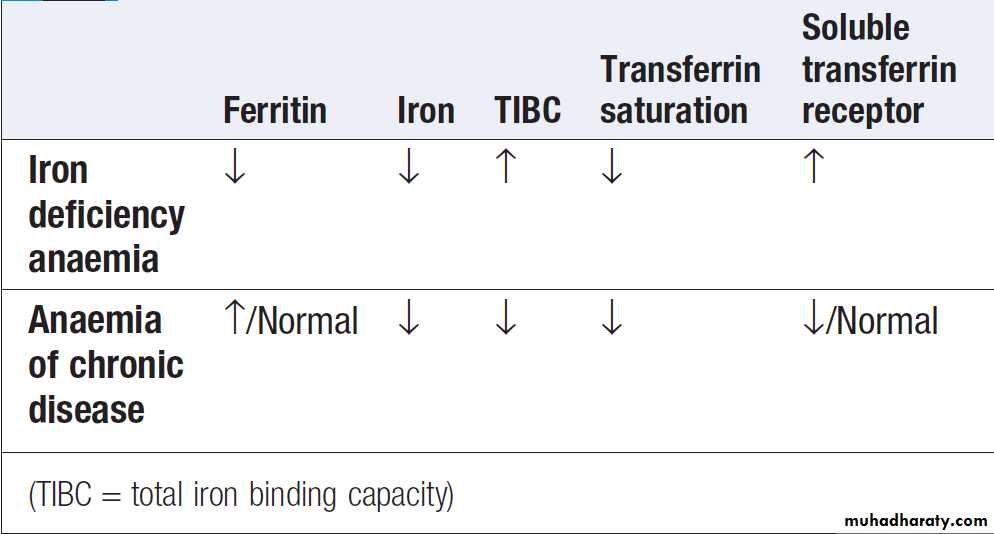
Investigations to differentiate anaemia of
chronic disease from iron deficiency anaemiaManagement
Unless the patient has angina, heart failure or evidenceof cerebral hypoxia, transfusion is not necessary and oral iron replacement is appropriate. Ferrous sulphate
200 mg 3 times daily (195 mg of elemental iron per day) is adequate and should be continued for 3–6 months to replete iron stores. Many patients suffer gastrointestinal
side-effects with ferrous sulphate, including dyspepsia
and altered bowel habit. When this occurs, reduction in
dose to 200 mg twice daily or a switch to ferrous gluconate 300 mg twice daily (70 mg of elemental iron per day) should be tried. Delayed-release preparations are not useful, since they release iron beyond the upper
small intestine, where it cannot be absorbed.
The haemoglobin should rise by around 10 g/L every
7–10 days and a reticulocyte response will be evidentwithin a week. A failure to respond adequately may be
due to non-compliance, continued blood loss, malabsorption or an incorrect diagnosis. Patients with malabsorption or chronic gut disease may need parenteral iron therapy. Previously, iron dextran or iron sucrose was
used, but new preparations of iron isomaltose and iron
carboxymaltose have fewer allergic effects and are preferred.
Doses required can be calculated based on the patient’s starting haemoglobin and body weight. Observation for anaphylaxis following an initial test dose is recommended.
• Full blood count: increased plasma volume (40%) lowers normal Hb (to > 105 g/L at 28 wks). The MCV may increase by 5 fL. A progressive neutrophilia occurs. Gestational thrombocytopenia (rarely < 60 × 109/L) is a benign phenomenon.
• Depletion of iron stores: iron deficiency is a common cause of anaemia in pregnancy and, should be treated with oral iron .
• Vitamin B12: physiologically low , deficiency is uncommon.
• Folate: tissue stores may become depleted, and folate supplementation is recommended in all pregnancies .
• Coagulation factors: from the second trimester, procoagulant factors increase approximately threefold, particularly fibrinogen, VWF and factor VIII. This causes activated protein C resistance and a shortened APTT and contributes to a prothrombotic state.
• Anticoagulants: levels of protein C increase from the second trimester, while protein S fall as C4b binding protein increases.
Haematological physiology in pregnancy
Anaemia of chronic disease (ACD)
ACD is a common type of anaemia, particularly in hospital populations. It occurs in the setting of chronic infection, chronic inflammation or neoplasia. The anaemia is not related to bleeding, haemolysis or marrow infiltration, is mild, with haemoglobin in the range of 85–115 g/L, and is usually associated with a normal MCV (normocytic, normochromic), though this may be reduced in long-standing inflammation. The serum iron is low but iron stores are normal or increased , as indicated by the ferritin or stainable marrow iron.Pathogenesis
It has recently become clear that the key regulatoryprotein that accounts for the findings characteristic of
ACD is hepcidin, which is produced by the liver , induced by proinflammatory cytokines, especially IL-6.
Hepcidin binds to ferroportin on the membrane of iron-exporting cells, such as small intestinal enterocytes and macrophages, internalising the ferroportin and thereby inhibiting the export of iron from these cells into the blood. The iron remains trapped inside the cells in the form of ferritin, levels of which are therefore normal or high in the face of significant anaemia.
Inhibition or blockade of hepcidin is a potential target for treatment of this form of anaemia.
Diagnosis and management
It is often difficult to distinguish ACD associated with alow MCV from iron deficiency.
Examination of the marrow may ultimately be required to assess iron stores directly. A trial of oral iron can be given in difficult situations.
A positive response occurs in true iron deficiency
but not in ACD.
Measures which reduce the severity of the underlying disorder generally help to improve the ACD.
Investigations to differentiate anaemia of
chronic disease from iron deficiency anaemiaMegaloblastic anaemia
Results from a deficiency of vitamin B12 or folic acid. Folate is an important substrate of, and vitamin B12 a co-factor for, the generation of the essential amino acid methionine from homocysteine. This reaction produces tetrahydrofolate, which is converted to thymidine monophosphate for incorporation into DNA.Deficiency of vitamin B12 or folate will therefore produce high plasma levels of homocysteine and impaired DNA synthesis. The end result is cells with arrested nuclear maturation but normal cytoplasmic development: so-called nucleocytoplasmic asynchrony.
All proliferating cells will exhibit megaloblastosis; hence changes are evident in the buccal mucosa, tongue, small intestine, cervix, vagina and uterus.
The high proliferation rate of bone marrow results in striking changes in the haematopoietic system in megaloblastic anaemia. Cells become arrested in development and die within the marrow; this ineffective erythropoiesis results in an expanded hypercellular marrow. Haemolysis within the marrow results in a raised bilirubin and LDH, but without the reticulocytosis characteristic of other forms of haemolysis . Iron stores are usually raised. The mature red cells are large and oval, and sometimes contain nuclear remnants. Nuclear changes are seen in the immature granulocyte precursors and a characteristic appearance is that of ‘giant’ metamyelocytes with a large ‘sausage-shaped’ nucleus.
The mature neutrophils show hypersegmentation of their nuclei, with cells having six or more nuclear lobes.
If severe, a pancytopenia may be present in the peripheral blood.
Vitamin B12 deficiency, but not folate deficiency, is
associated with neurological disease in up to 40% of
cases, although advanced neurological disease due to B12 deficiency is now uncommon in the developed world.
The main pathological finding is focal demyelination
affecting the spinal cord, peripheral nerves, optic nerves and cerebrum.
The most common manifestations are sensory, with peripheral paraesthesiae and ataxia of gait.
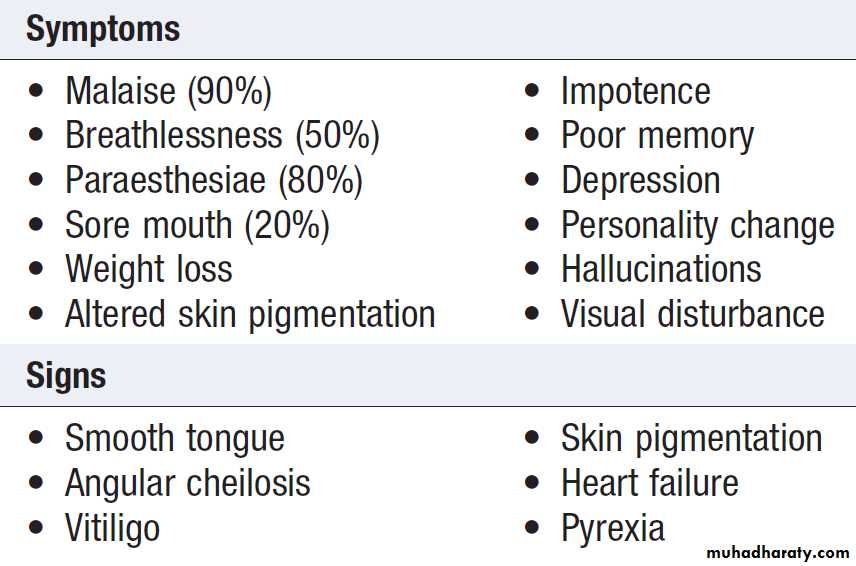
Clinical features of megaloblastic anaemia
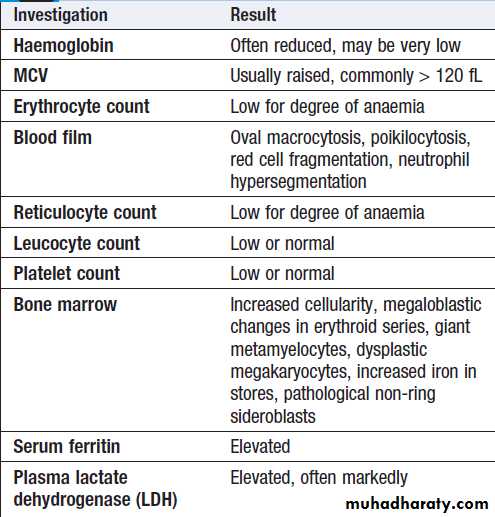
Investigations in megaloblastic anaemia
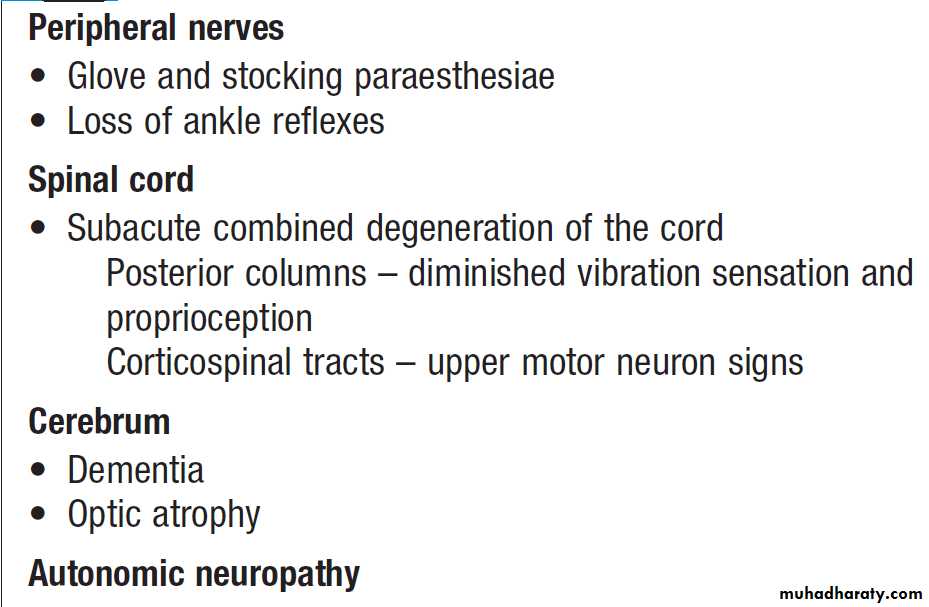
Neurological findings in B12 deficiency
Vitamin B12Vitamin B12 absorption
The average daily diet contains 5–30 μg of vitamin B12,
mainly in meat, fish, eggs and milk – well in excess of
the 1 μg daily requirement.
In the stomach, gastric enzymes release vitamin B12 from food and at gastric pH it binds to a carrier protein termed R protein. The parietal cells produce intrinsic factor, a vitamin B12-binding protein.
As gastric emptying occurs, pancreatic secretion raises the pH and vitamin B12 released from the diet switches from the R protein to intrinsic factor which optimally binds vitamin B12 at pH 8. Bile also contains vitamin B12 which is available for reabsorption in the intestine.
The vitamin B12–intrinsic factor complex binds
to specific receptors in the terminal ileum, and vitamin
B12 is actively transported by the enterocytes to plasma
where it binds to transcobalamin II, a transport protein
produced by the liver, which carries it to the tissues for
utilisation.
The liver stores enough vitamin B12 for 3 years and this, together with the enterohepatic circulation, means that vitamin B12 deficiency takes years to become manifest, even if all dietary intake is stopped or severe B12 malabsorption supervenes.
Blood levels of vitamin B12 provide a reasonable indication of tissue stores and are usually diagnostic of deficiency.
Levels of cobalamins fall in normal pregnancy.
Reference ranges vary between laboratories but levels below 150 ng/L are common and, in the last trimester, 5–10% of women have levels below 100 ng/L. Spuriously low B12 values occur in women using the oral contraceptive pill and in patients with myeloma, in whom paraproteins can interfere with B12 assays.
Causes of vitamin B12 deficiency
1. Dietary deficiencyThis only occurs in strict vegans but the onset of clinical
features can occur at any age between 10 and 80 years.
Less strict vegetarians often have slightly low vitamin
B12 levels but are not tissue vitamin B12-deficient.
2. Gastric pathology
Release of vitamin B12 from the food requires normal gastric acid and enzyme secretion, and this is impaired by hypochlorhydria in elderly patients or following gastric surgery. Total gastrectomy invariably results in vitamin B12 deficiency within 5 years, often combined with iron deficiency; these patients need life-long 3-monthly vitamin B12 injections.
After partial gastrectomy, B12 deficiency only develops in 10–20% of patients by 5 years; an annual injection of vitamin B12 should prevent deficiency in this group.
3. Pernicious anaemia
This is an organ-specific autoimmune disorder in which
the gastric mucosa is atrophic, with loss of parietal cells
causing intrinsic factor deficiency. In the absence of
intrinsic factor, less than 1% of dietary vitamin B12 is
absorbed. Average age of onset of 60 years .It is more common in individuals with other autoimmune disease (Hashimoto’s thyroiditis, Graves’ disease, vitiligo, hypoparathyroidism or Addison’s disease) or a family history of these or pernicious anaemia. The finding of anti-intrinsic factor antibodies in the context of B12 deficiency is diagnostic of pernicious anaemia without further investigation. Antiparietal cell antibodies negative result makes pernicious less likely.
Antiparietal cell antibodies are present in over 90% of cases but are also present in 20% of normal females over the age of 60 years; a negative result makes pernicious anaemia less likely but a positive result is not diagnostic.
The Schilling test, involving measurement of absorption of radio- labelled B12 after oral administration before and after replacement of intrinsic factor, has fallen out of favour with the availability of autoantibody tests, greater caution in the use of radioactive tracers, and limited availability of intrinsic factor.
4. Small bowel pathology
One-third of patients with pancreatic exocrine insufficiency fail to transfer dietary vitamin B12 from R protein to intrinsic factor. This usually results in slightly low vitamin B12 values but no tissue evidence of B12 deficiency. Motility disorders or hypogammaglobulinaemia can result in bacterial overgrowth, and the ensuing competition for free vitamin B12 can lead to deficiency.This is corrected to some extent by appropriate
antibiotics. A small number of people heavily infected with the fish tapeworm develop vitamin B12 deficiency.
Inflammatory disease of the terminal ileum, such as
Crohn’s disease, may impair the absorption of B12–intrinsic factor complex, as may surgery on that part of the bowel.
Folate
Folate absorptionFolates are produced by plants and bacteria; hence
dietary leafy vegetables (spinach, broccoli, lettuce),
fruits (bananas, melons) and animal protein (liver,
kidney) are a rich source. An average Western diet
contains more than the minimum daily intake of 50 μg
but excess cooking destroys folates. Most dietary folate
is present as polyglutamates; these are converted to
monoglutamate in the upper small bowel and actively
transported into plasma. Plasma folate is loosely bound
to plasma proteins such as albumin and there is an
Enterohepatic circulation. Total body stores of folate are small and deficiency can occur in a matter of weeks.
Folate deficiency
The causes and diagnostic features of folate deficiency.
The edentulous elderly or psychiatric patient is particularly susceptible to dietary deficiency and this is exacerbated in the presence of gut disease or malignancy. Pregnancy-induced folate deficiency is the most common cause of megaloblastosis worldwide and is more likely in the context of twin pregnancies, multiparity and hyperemesis gravidarum. Serum folate is very sensitive to dietary intake; a single folate-rich meal can normalise it in a patient with true folate deficiency, whereas anorexia,
alcohol and anticonvulsant therapy can reduce it.
For this reason, red cell folate levels are a more accurate indicator of folate stores and tissue folate deficiency.
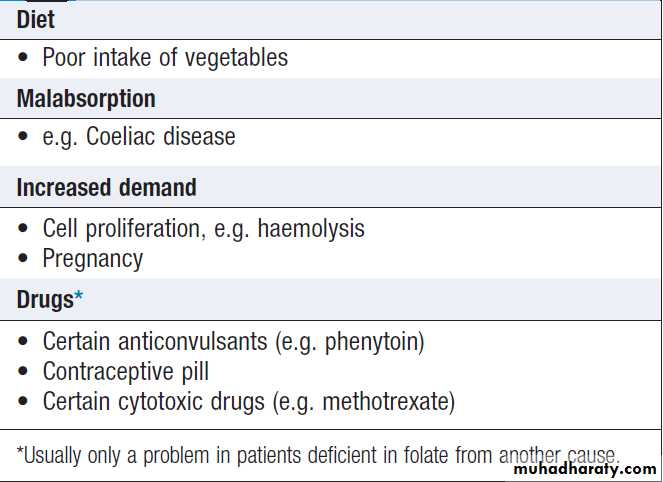
Causes of folate deficiency

Investigation of folic acid deficiency
Management of megaloblastic anaemiaIf a patient with a severe megaloblastic anaemia is very
ill and treatment must be started before vitamin B12 and
red cell folate results are available, that treatment should always include both folic acid and vitamin B12. The use of folic acid alone in the presence of vitamin B12 deficiency may result in worsening of neurological deficits. Rarely, if severe angina or heart failure is present, transfusion can be used in megaloblastic anaemia. The cardiovascular system is adapted to the chronic anaemia present in megaloblastosis, and the volume load imposed by transfusion may result in decompensation and severe cardiac failure. In such circumstances, exchange transfusion or slow administration of 1 U of red cells with diuretic
cover may be given cautiously.
Vitamin B12 deficiency
Treated with hydroxycobalamin 1000 μg IM for 6 doses 2 or 3 days apart, followed by maintenance therapy of 1000 μg every 3 months for life.
The reticulocyte count will peak by the 5th–10th day
after starting replacement therapy. The haemoglobin
will rise by 10 g/L every week until normalised. The
response of the marrow is associated with a fall in
plasma potassium levels and rapid depletion of iron
stores. If an initial response is not maintained and the
blood film is dimorphic (shows a mixture of microcytic
and macrocytic cells), the patient need additional iron therapy. A sensory neuropathy may take 6–12 months to correct; long-standing neurological damage may not improve.
Folate deficiency
Oral folic acid 5 mg daily for 3 weeks will treat acutedeficiency and maintenance 5 mg once weekly is adequate. Prophylactic folic acid in pregnancy prevents megaloblastosis in women at risk, and reduces the risk of fetal neural tube defects . Prophylactic supplementation is also given in chronic haematological disease associated with reduced red cell lifespan (e.g. haemolytic anaemias). There is some evidence that supraphysiological supplementation (400 μg/day) can reduce the risk of coronary and cerebrovascular disease by lowering plasma homocysteine levels. This has led the US Food and Drug Administration to introduce fortification of bread, flour and rice with folic acid.
Hemolytic anemia
• Hereditary disorders include erythrocyte membrane , enzymatic defects and hemoglobin abnormalities.• G6PD deficiency, Pyruvate kinase deficiency enzy
• Hereditary spherocytosis , Hereditary elliptocytosis memb
• Sickle cell anemia , thalassemia hemo
• Acquired hemolytic conditions
• Autoimmune HA result from warm , cold, rarely mixed autoantibody.PCH.
• Alloimmune HA caused by antibodies against non-self red cells, occurs after unmatched
transfusion or after maternal sensitisation to paternal antigens on fetal cells (haemolytic
disease of the newborn).
c . Non-immune haemolytic anaemia
Physical trauma
• Mechanical heart valves. • March haemoglobinuria. • Thermal injury
• Microangiopathic haemolytic anaemia.
Infection
Plasmodium falciparum malaria. Clostridium perfringens septicaemia
Chemicals or drugs
Dapsone , sulfasalazine. Arsenic gas, copper, chlorates, nitrites and nitrobenzene derivatives may all cause haemolysis.
Rare,PNH
Haemolytic anaemia
Haemolysis indicates that there is shortening of the normal red cell lifespan of 120 days. To compensate, the bone marrow may increase its output of red cells six- to eightfold by increasing the proportion of red cells produced, expanding the volume of active marrow, and releasing reticulocytes prematurely. Anaemia only occurs if the rate of destruction exceeds this increased production rate. Red cell destruction overloads pathways for haemoglobin breakdown in the liver, causing a modest rise in unconjugated bilirubin in the blood and mild jaundice. Increased reabsorption of urobilinogen from the gut results in an increase in urinary urobilinogen .Red cell destruction releases LDH into the serum.
The bone marrow compensation results in a reticulocytosis, and sometimes nucleated red cell precursors appear in the blood. Increased proliferation of the bone marrow can
result in a thrombocytosis, neutrophilia and, if marked,
immature granulocytes in the blood, producing a leuco- erythroblastic blood film. The appearances of the red
cells may give an indication of the likely cause of the
haemolysis:
• Spherocytes are small, dark red cells which suggest
autoimmune haemolysis or hereditary spherocytosis.
• Sickle cells suggest sickle-cell disease.
• Red cell fragments indicate microangiopathic
haemolysis.
The compensatory erythroid hyperplasia may give rise to folate deficiency, with megaloblastic blood features.
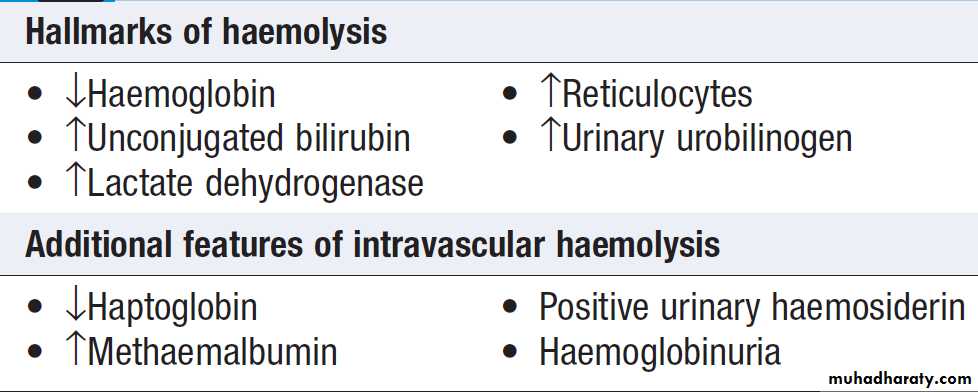
Investigation results indicating active haemolysis
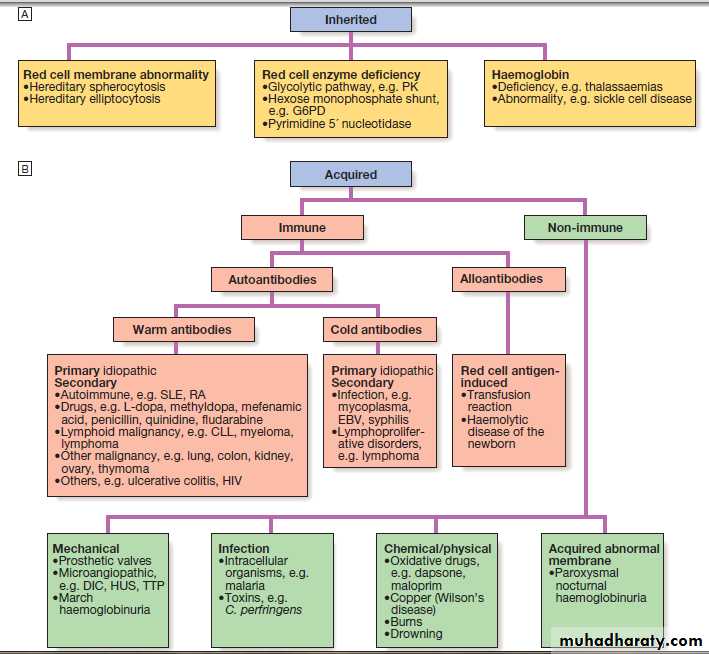
Causes of haemolysis. A Inherited causes.
B Acquired causes. (CLL = chronic lymphatic leukaemia; DIC = disseminated intravascularcoagulation; EBV = Epstein–Barr virus; G6PD = glucose-6-phosphate dehydrogenase; HUS = haemolytic uraemic syndrome; PK = pyruvate kinase;
RA = rheumatoid arthritis; SLE = systemic lupus erythematosus; TTP = thrombotic thrombocytopenic purpura)
Extravascular haemolysis
Physiological red cell destruction occurs in the RE cells in the liver or spleen, so avoiding free haemoglobin in the plasma. In most haemolytic states, haemolysis is predominantly extravascular. To confirm the haemolysis, patients’ red cells can be labelled with 51chromium. When re-injected, they can be used to determine red cell survival; when combined with body surface radioactivity counting, this test may indicate whether the liver or the spleen is the main source of red cell destruction. However, it is seldom performed in clinical practice.Intravascular haemolysis
Less commonly, red cell lysis occurs within the bloodstream due to membrane damage by complement(ABO transfusion reactions, PNH ), infections (malaria, Clostridium perfringens), mechanical trauma (heart valves, DIC) or oxidative damage (e.g. drugs such as dapsone and maloprim). Free haemoglobin is released into the plasma. Haptoglobin is an α2-globulin produced by the liver, which binds free haemoglobin, resulting in a fall in its levels during active haemolysis.
Once haptoglobins are saturated, free haemoglobin is oxidised to form methaemoglobin, which binds to albumin, in turn forming methaemalbumin, which can be detected spectrophotometrically in the Schumm’s test.
Methaemoglobin is degraded and any free haem is
bound to a second binding protein called haemopexin.If all the protective mechanisms are saturated, free haemoglobin may appear in the urine (haemoglobinuria).
When fulminant, this gives rise to black urine as in
severe falciparum malaria infection.
In smaller amounts, renal tubular cells absorb the haemoglobin, degrade it and store the iron as haemosiderin.
When the tubular cells are subsequently sloughed into the urine, they give rise to haemosiderinuria, which is always indicative of intravascular haemolysis.
Red cell membrane defects
The basic structure is a cytoskeleton ‘stapled’ on to the lipid bilayer by special protein complexes.This structure ensures great deformability and elasticity; the red cell diameter is 8 μm but the narrowest capillaries in the circulation are in the spleen, measuring just 2 μm in diameter. When the normal red cell structure is disturbed, usually by a quantitative or functional deficiency of one or more proteins in the cytoskeleton, cells lose their elasticity. Each time such cells pass through the spleen, they lose membrane relative to their cell volume. This results in an increase in mean cell haemoglobin concentration (MCHC), abnormal cell shape and reduced red cell survival
due to extravascular haemolysis.
Hereditary spherocytosis
Autosomal dominant condition, although 25% of cases have no family history and represent new mutations. The most common abnormalities are deficiencies of beta spectrin or ankyrin .The severity of spontaneous haemolysis varies. Most cases are associated with an asymptomatic compensated chronic haemolytic state withspherocytes present on the blood film, a reticulocytosis
and mild hyperbilirubinaemia.
Pigment gallstones are present in up to 50% of patients and may cause symptomatic cholecystitis. Occasional cases are associated with more severe haemolysis.
The clinical course may be complicated by crises:
• A haemolytic crisis occurs when the severity ofhaemolysis increases; this is rare, and usually
associated with infection.
• A megaloblastic crisis follows the development of
folate deficiency; this may occur as a first
presentation of the disease in pregnancy.
• An aplastic crisis occurs in association with
parvovirus B19 infection .Parvovirus causes
a common exanthem in children, but if individuals
with chronic haemolysis become infected, the virus
directly invades red cell precursors and temporarily
switches off red cell production. Patients present
with severe anaemia and a low reticulocyte count.
Investigations
The patient and other family members should be
screened for features of compensated haemolysis .This may be all that is required to confirm the diagnosis. Haemoglobin levels are variable, depending on the degree of compensation. The blood film will show spherocytes but the direct Coombs test is negative, excluding immune haemolysis. An osmotic fragility test may show increased sensitivity to lysis in hypotonic saline solutions but is limited by lack of sensitivity and specificity. More specific flow cytometric tests, detecting binding of eosin-5-maleimide to red cells, are recommended in borderline cases.
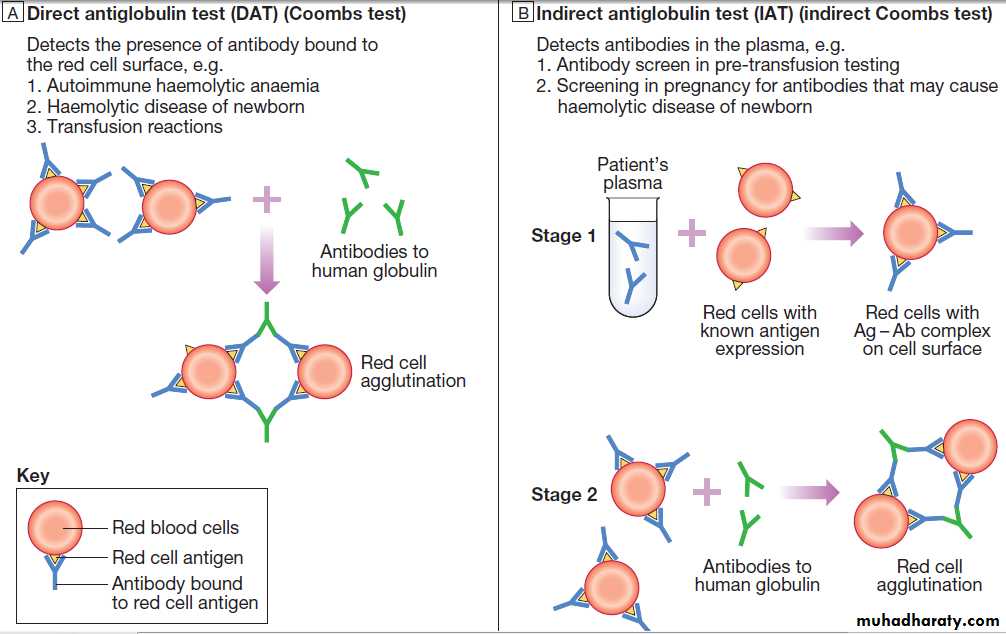
Direct and indirect antiglobulin tests.
ManagementFolic acid prophylaxis, 5 mg daily, should be given for
life. Consideration may be given to splenectomy, which
improves but does not normalise red cell survival.
Potential indications include moderate to severe haemolysis with complications (anaemia and gallstones),
although splenectomy should be delayed until after
6 years of age in view of the risk of sepsis. Acute, severe haemolytic crises require transfusion support, but blood must be cross-matched carefully and transfused slowly as haemolytic transfusion reactions may occur .

Management of the splenectomised patient
Hereditary elliptocytosis
This term refers to a heterogeneous group of disorders
that produce an increase in elliptocytic red cells on the
blood film and a variable degree of haemolysis. This is
due to a functional abnormality of one or more anchor
proteins in the red cell membrane, e.g. alpha spectrin or protein 4.1. Inheritance may be autosomal dominant or recessive. Hereditary elliptocytosis is less common than hereditary spherocytosis in Western countries. The clinical course is variable ,most cases present as an asymptomatic blood film abnormality, but occasional cases result in neonatal haemolysis or a chronic compensated haemolytic state. Management of the latter is the same as for hereditary spherocytosis.
Red cell enzymopathies
The mature red cell must produce energy via ATP to maintain a normal internal environment and cell volume whilst protecting itself from the oxidative stress presented by oxygen carriage. Anaerobic glycolysis via the Embden–Meyerhof pathway generates ATP, and the hexose monophosphate shunt produces NADPH and glutathione to protect against oxidative stress. The impact of functional or quantitative defects in the enzymes in these pathways depends upon the importance of the steps affected and the presence of alternative pathways. In general, defects in the hexose monophosphate shunt pathway result in periodic haemolysis precipitated by episodic oxidative stress, whilst those in the Embden–Meyerhof pathway resultin shortened red cell survival and chronic haemolysis.
Glucose-6-phosphate dehydrogenase deficiency
The enzyme glucose-6-phosphate dehydrogenase (G6PD) is pivotal in the hexose monophosphate shunt pathway. Deficiencies result in the most common human enzymopathy, affecting 10% of the world’s population, with a geographical distribution which parallelsthe malaria belt because heterozygotes are protected
from malarial parasitisation. The enzyme is a heteromeric structure made of catalytic subunits which are encoded by a gene on the X chromosome. The deficiency therefore affects males and rare homozygous females ,but it is carried by females. Carrier heterozygous females are usually only affected in the neonatal period or in the presence of extreme lyonisation, producing selective inactivation of the non-affected X chromosome.
Over 400 subtypes of G6PD are described. The most
common types associated with normal activity are theB+ enzyme present in most Caucasians and 70% of Afro- Caribbeans, and the A+ variant present in 20% of Afro- Caribbeans. The two common variants associated with reduced activity are the A− variety in approximately 10% of Afro- Caribbeans, and the Mediterranean or B− variety in Caucasians.
Management aims to stop any precipitant drugs
and treat any underlying infection. Acute transfusion
support may be life-saving.

Glucose-6-phosphate dehydrogenase
deficiencyPyruvate kinase deficiency
This is the second most common red cell enzyme defect.It results in deficiency of ATP production and a chronic
haemolytic anaemia. It is inherited as an autosomal
recessive trait. The extent of anaemia is variable; the
blood film shows characteristic ‘prickle cells’ which
resemble holly leaves. Enzyme activity is only 5–20% of
normal. Transfusion support may be necessary.
Autoimmune haemolytic anaemia
This results from increased red cell destruction due to red cell autoantibodies. The antibodies may be IgG or M, or more rarely IgE or A. If an antibody avidly fixes complement, it will cause intravascular haemolysis, but if complement activation is weak, the haemolysis will be extravascular.Antibody-coated red cells lose membrane to macrophages in the spleen and hence spherocytes are present in the blood.
The optimum temperature at which the antibody is active (thermal specificity) is used to classify immune haemolysis:
• Warm antibodies bind best at 37°C and account for 80% of cases. The majority are IgG ,often react against Rhesus antigens.
• Cold antibodies bind best at 4°C but can bind up to 37°C in some cases. They are usually IgM and bind complement. To be clinically relevant, they must act within the range of normal body temperatures.
They account for the other 20% of cases.
Warm autoimmune haemolysis
It occurs at all ages but is more common in middle age and in females. No underlying cause in up to 50% of cases. The remainder are secondary
Investigations
There is evidence of haemolysis and spherocytes on the blood film. The diagnosis is confirmed by the direct Coombs or antiglobulin test .The patient’s red cells are mixed with Coombs reagent, which contains antibodies against human IgG/M/complement. If the red cells have been coated by antibody in vivo, the reagent will induce agglutination ,detected visually. The relevant antibody can be eluted from the red cell surface and tested against a panel of typed red cells to determine against which red cell antigen it is directed.
The most common specificity is Rhesus and most often anti-e; this is helpful when choosing blood to cross-match. The direct Coombs test can be negative in the presence of brisk haemolysis.
A positive test requires about 200 antibody molecules to attach to each red cell; with a very avid complement-fixing antibody, haemolysis may occur at lower levels of antibody binding.
The standard Coombs reagent will miss IgA or IgE antibodies. Around 10% of all warm autoimmune
haemolytic anaemias are Coombs test-negative.
Management
If the haemolysis is secondary to an underlying cause,this must be treated and any implicated drugs stopped.
It is usual to treat patients initially with prednisolone
1 mg/kg orally. A response is seen in 70–80% of cases
but may take up to 3 weeks; a rise in haemoglobin will
be matched by a fall in bilirubin, LDH and reticulocyte
levels. Once the haemoglobin has normalised and the
reticulocytosis resolved, the corticosteroid dose can be
reduced slowly over about 10 weeks. Corticosteroids
work by decreasing macrophage destruction of antibody coated red cells and reducing antibody production.
Transfusion support may be required for life threatening
problems, such as the development of heart failure or rapid unabated falls in haemoglobin.
The least incompatible blood should be used but this may still give rise to transfusion reactions or the development of alloantibodies.
If the haemolysis fails to respond to corticosteroids or
can only be stabilised by large doses, then splenectomy
should be considered. This removes a main site of red
cell destruction and antibody production, with a good
response in 50–60% of cases. The operation can be performed laparoscopically with reduced morbidity. If
splenectomy is not appropriate, alternative immunosuppressive therapy with azathioprine or cyclophosphamide may be considered. This is least suitable for young patients, in whom long-term immunosuppression carries a risk of secondary neoplasms. The anti-CD20 (B cell) monoclonal antibody, rituximab, has shown some success in difficult cases.
Cold agglutinin disease
This is due to antibodies, usually IgM, which bind to thered cells at low temperatures and cause them to agglutinate.
It may cause intravascular haemolysis if complement
fixation occurs. This can be chronic when the
antibody is monoclonal, or acute or transient when the
antibody is polyclonal.
Chronic cold agglutinin disease
This affects elderly patients and may be associated with an underlying low-grade B cell lymphoma. It causes a low-grade intravascular haemolysis with cold, painful and often blue fingers, toes, ears or nose (so-called acrocyanosis).The latter is due to red cell agglutination in
the small vessels in these colder exposed areas. The
blood film shows red cell agglutination and the
MCV may be spuriously high because the automated analysers detect aggregates as single cells.
Monoclonal IgM usually has anti-I or, less often, anti-i specificity.
Treatment is directed at any underlying lymphoma but
if the disease is idiopathic, then patients must keep
extremities warm, especially in winter. Some patients
respond to corticosteroid therapy and blood transfusion may be considered, but the cross-match sample must be placed in a transport flask at a temperature of 37°C and blood administered via a blood-warmer. All patients should receive folic acid supplementation.
Other causes of cold agglutination
Can occur in association with Mycoplasma pneumoniae or with infectious mononucleosis.Paroxysmal cold haemoglobinuria is a very rare cause seen in children, in association with viral or bacterial infection. An IgG antibody binds to red cells in the peripheral circulation but lysis occurs in the central
circulation when complement fixation takes place.
This antibody is termed the Donath–Landsteiner antibody and has specificity against the P antigen on the red cells.
Alloimmune haemolytic anaemia
Alloimmune haemolytic anaemia is caused by antibodiesagainst non-self red cells, and occurs after unmatched
transfusion , or after maternal sensitisation to
paternal antigens on fetal cells (haemolytic disease of the newborn).
Non-immune haemolytic anaemia
Physical traumaPhysical disruption of red cells , presence of red cell fragments on the blood film and markers of IV haemolysis:
• Mechanical heart valves. High flow through incompetent valves or periprosthetic leaks through the suture ring holding a valve in place result in shear stress damage.
• March haemoglobinuria. Vigorous exercise, such as prolonged marching or marathon running, can cause red cell damage in the capillaries in the feet.
• Thermal injury.Severe burns cause thermal damage to RBC. • Microangiopathic haemolytic anaemia. Fibrin deposition in capillaries can cause red cell disruption. It may occur in disseminated carcinomatosis, malignant or pregnancy-induced hypertension, HUS ,TTP and DIC.
Infection
Plasmodium falciparum malaria may be associated
with intravascular haemolysis; when severe, this is
termed blackwater fever because of the associated
haemoglobinuria. Clostridium perfringens septicaemia
usually in the context of ascending cholangitis,
may cause severe intravascular haemolysis due to bacterial production of a lecithinase which destroys the membrane.
Chemicals or drugs
Dapsone and sulfasalazine cause haemolysis by oxidative denaturation of haemoglobin. Denatured haemoglobin forms Heinz bodies in the red cells, visible on supravital staining with brilliant cresyl blue.
Arsenic gas, copper, chlorates, nitrites and nitrobenzene derivatives may all cause haemolysis.
Paroxysmal nocturnal haemoglobinuria (PNH)
Is a rare acquired, non-malignant clonal expansion of haematopoietic stem cells deficient in GPI-anchor protein; it results in intravascular haemolysis and anaemia because of increased sensitivity of red cells to lysis by complement.Episodes of haemolysis result in haemoglobinuria, most noticeable in early morning urine, which has a characteristic red–brown colour. The disease is associated with an increased risk of venous thrombosis in unusual sites, such as the liver or abdomen. PNH is also associated with hypoplastic bone marrow failure, aplastic anaemia and myelodysplastic syndrome.
Management is supportive with transfusion and treatment of thrombosis. Recently, the anti-complement C5 monoclonal antibody eculizumab was shown to be effective in reducing haemolysis.
Haemoglobinopathies
Caused by mutations affecting the genes encoding the globin chains of the haemoglobin molecule.Normal haemoglobin is comprised of two αα and two non-αα globin chains. Alpha globin chains are produced throughout life, including in the fetus, so severe mutations may cause intrauterine death.
Production of non-alpha chains varies with age; fetal
haemoglobin (HbF-αα/γγ) has two gamma chains, while
the predominant adult haemoglobin (HbA-αα/ββ) has
two beta chains. Thus, disorders affecting the beta chains do not present until after 6 months of age.
A constant small amount of haemoglobin A2 (HbA2-αα/ δδ, usually less than 2%) is made from birth. The haemoglobinopathies can be classified into qualitative or quantitative abnormalities.
The α chain is present in HbA, HbA2, and HbF; α-chain mutations thus cause abnormalities in all three.
The α-globin hemoglobinopathies are symptomatic
in utero and after birth because normal function
of the α-globin gene is required throughout gestation
and adult life.
In contrast, infants with β-globin hemoglobinopathies
tend to be asymptomatic until 3–9 months
of age when HbA has largely replaced HbF.

SICKLE CELL SYNDROMES
The sickle cell syndromes are caused by a mutation inthe β-globin gene that changes the sixth amino acid
from glutamic acid to valine.
HbS polymerizes
reversibly when deoxygenated to form a gelatinous
network of fibrous polymers that stiffen the RBC
membrane, increase viscosity, and cause dehydration due
to potassium leakage and calcium influx . These changes also produce the sickle shape. Sickled cells lose the pliability needed to traverse small capillaries.
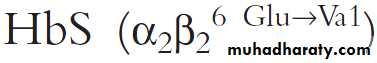
They possess altered sticky membranes (especially reticulocytes) that adhere abnormally to the endothelium of small venules.
These abnormalities provoke unpredictable episodes of microvascular vasoocclusion and premature RBC destruction (hemolytic anemia). Hemolysis occurs
because the spleen destroys the abnormal RBC. The
rigid adherent cells also clog small capillaries and
venules, causing tissue ischemia, acute pain, and gradual
end-organ damage. This venoocclusive component usually
dominates the clinical course. Prominent manifestations
include episodes of ischemic pain (i.e., painful crises) and ischemic malfunction or frank infarction in the spleen, central nervous system, bones, liver, kidneys, and lungs.
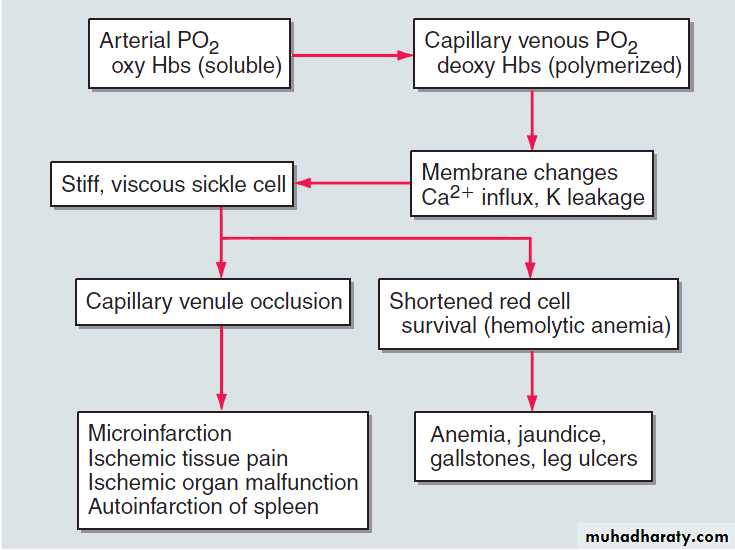
Qualitative abnormalities – abnormal haemoglobins
There is a functionally important alteration in the amino acid structure of the polypeptide chains of the globin chains. Several hundred such variants are known; they were originally designated by letters of the alphabet, e.g. S, C, D or E, but are now described by names usually taken from the town or district in which they were first described. The best-known example is haemoglobin S, found in sickle-cell anaemia. Mutations around the haem-binding pocket cause the haem ring to fall out of the structure and produce an unstable haemoglobin.These substitutions often change the charge of the globin chains, producing different electrophoretic mobility, and this forms the basis for the diagnostic use of haemoglobin electrophoresis to identify
haemoglobinopathies.
Quantitative abnormalities – thalassaemias
In quantitative abnormalities (the thalassaemias), there
are mutations causing a reduced rate of production of
one or other of the globin chains, altering the ratio of
alpha to non-alpha chains.
In alpha- thalassaemia excess beta chains are present, whilst in
beta- thalassaemia excess alpha chains are present.
The excess chains precipitate, causing red cell membrane damage and reduced red cell survival.
Qualitative abnormalities : Sickle-cell anaemia
Results from a single glutamic acid to valine substitution at position 6 of the beta globin polypeptide chain. It is inherited as an autosomal recessive trait .Homozygotes only produce abnormal beta chains that make haemoglobin S (HbS, termed SS), and this results in the clinical syndrome of sickle-cell disease.
Heterozygotes produce a mixture of normal and abnormal beta chains that make normal HbA and HbS (termed AS), and this results in the clinically asymptomatic sickle-cell trait.
Epidemiology
The heterozygote frequency is over 20% in tropicalAfrica . In black American populations,
sickle-cell trait has a frequency of 8%. Individuals with
sickle-cell trait are relatively resistant to the lethal effects of falciparum malaria in early childhood; the high prevalence in equatorial Africa can be explained by the survival advantage it confers in areas where falciparum malaria is endemic.
However, homozygous patients with sickle-cell anaemia do not have correspondingly greater resistance to falciparum malaria.
Pathogenesis
When haemoglobin S is deoxygenated, the molecules
of haemoglobin polymerise to form pseudocrystalline
structures known as ‘tactoids’. These distort the red cell
membrane and produce characteristic sickle-shaped
cells .The polymerisation is reversible when
re-oxygenation occurs. The distortion of the red cell membrane, however, may become permanent and the red cell ‘irreversibly sickled’. The greater the
concentration of sickle-cell haemoglobin in the individual
cell, the more easily tactoids are formed, but this
process may be enhanced or retarded by the presence of
other haemoglobins. Thus, the abnormal haemoglobin C variant participates in the polymerisation more readily than haemoglobin A, whereas haemoglobin F strongly inhibits polymerisation.
Clinical features
Sickling is precipitated by hypoxia,acidosis,dehydrationand infection. Irreversibly sickled cells have a
shortened survival and plug vessels in the microcirculation.
This results in a number of acute syndromes, termed ‘crises’, and chronic organ damage :
• Painful vaso-occlusive crisis. Plugging of small vessels in the bone produces acute severe bone pain. This affects areas of active marrow:
the hands and feet in children (so-called dactylitis) or the femora, humeri, ribs, pelvis and vertebrae in adults.
Patients usually have a systemic response with tachycardia, sweating and a fever. This is the most common crisis.
• Sickle chest syndrome. This may follow a vasoocclusive
crisis and is the most common cause of death in adult sickle disease. Bone marrow infarction results in fat emboli to the lungs, which cause further sickling and infarction, leading to ventilatory failure if not treated.
• Sequestration crisis. Thrombosis of the venous outflow from an organ causes loss of function and acute painful enlargement. In children, the spleen is the most common site. Massive splenic enlargement may result in severe anaemia, circulatory collapse and death. Recurrent sickling in the spleen in childhood results in infarction and adults may have no functional spleen. In adults, the liver may undergo sequestration with severe pain due to capsular stretching. Priapism is a complication seen in affected men.
• Aplastic crisis. Infection with human parvovirus B19
results in a severe but self-limiting red cell aplasia.This produces a very low haemoglobin, which may
cause heart failure.
Unlike in all other sickle crises,
the reticulocyte count is low.
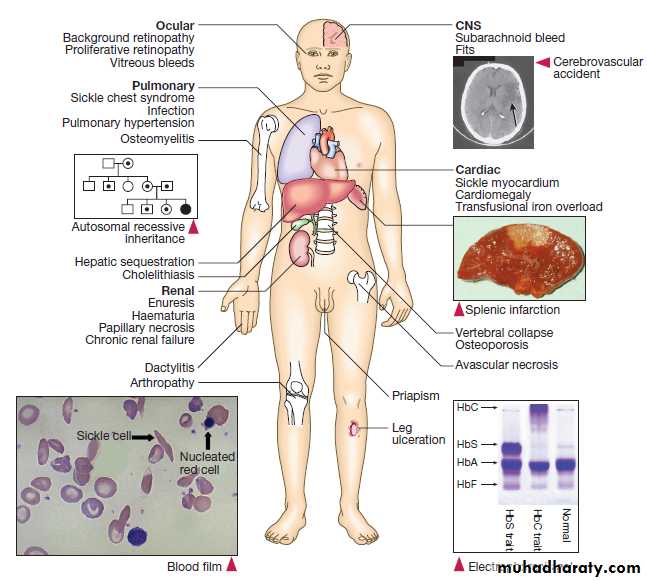
Clinical and laboratory features of sickle-cell disease
InvestigationsPatients with sickle-cell disease have a compensated
anaemia, usually around 60–80 g/L. The blood film
shows sickle cells, target cells and features of hyposplenism. A reticulocytosis is present. The presence of HbS can be demonstrated by exposing red cells to a reducing agent such as sodium dithionite; HbA gives a clear solution, whereas HbS polymerises to produce a turbid solution. This forms the basis of emergency screening tests before surgery in appropriate ethnic groups but cannot distinguish between sickle-cell trait and disease. The definitive diagnosis requires haemoglobin electrophoresis to demonstrate the absence of HbA,
2–20% HbF and the predominance of HbS. Both parents of the affected individual will have sickle-cell trait.
Management
All patients with sickle-cell disease should receive
prophylaxis with daily folic acid, and penicillin V to
protect against pneumococcal infection, which may be
lethal in the presence of hyposplenism. These patients
should be vaccinated against pneumococcus, meningococcus, Haemophilus influenzae B, hepatitis B and seasonal influenza.
Vaso-occlusive crises are managed by aggressive
rehydration, oxygen therapy, adequate analgesia (which often requires opiates) and antibiotics.
Transfusion should be with fully genotyped blood wherever possible.
Simple top-up transfusion may be used in asequestration or aplastic crisis. A regular transfusion
programme to suppress HbS production and maintain
the HbS level below 30% may be indicated in patients
with recurrent severe complications, such as cerebrovascular accidents in children or chest syndromes in
adults.
Exchange transfusion, in which a patient is simultaneously venesected and transfused to replace
HbS with HbA, may be used in life-threatening crises or
to prepare patients for surgery.
A high HbF level inhibits polymerisation of HbS and
reduces sickling. Patients with sickle-cell disease andhigh HbF levels have a mild clinical course with few crises. Some agents are able to increase synthesis of HbF and this has been used to reduce the frequency of severe crises. The oral cytotoxic agent hydroxycarbamide (hydroxyurea) ,shown to have clinical benefit with acceptable side effects in children and adults who have recurrent severe crises. Relatively few allogeneic stem cell transplants from HLA-matched siblings have been performed but this procedure appears to be potentially curative .
Prognosis In Africa, few children with anaemia survive to adult life without medical attention. Even with standard medical care, approximately 15% die by the age 20 years and 50% by 40.
Other abnormal haemoglobins
Another beta chain haemoglobinopathy, haemoglobin C (HbC) disease, is clinically silent but associated with microcytosis and target cells on the blood film. Compound heterozygotes inheriting one HbS gene and one HbC gene from their parents have haemoglobin SC disease, which behaves like a mild form of sickle-cell disease. SC disease is associated with a reduced frequency of crises but is not uncommonly linked with complications in pregnancy and retinopathy.
Quantitative abnormalities
The thalassaemiasThalassaemia is an inherited impairment of haemoglobin
production, in which there is partial or complete failure
to synthesise a specific type of globin chain. In alpha thalassaemia, disruption of one or both alleles on chromosome 16 may occur, with production of some or no
alpha globin chains. In beta- thalassaemia, defective production usually results from disabling point mutations
causing no (β0) or reduced (β–) beta chain production.
Beta- thalassaemia
Failure to synthesise beta chains (beta-thalassaemia) isthe most common type of thalassaemia, most prevalent
in the Mediterranean area. Heterozygotes have thalassaemia minor, a condition in which there is usually mild anaemia and little or no clinical disability, which may be detected only when iron therapy for a mild microcytic
anaemia fails. Homozygotes (thalassaemia major) either are unable to synthesise haemoglobin A or, at best,
produce very little; after the first 4–6 months of life, they
develop profound hypochromic anaemia.
Intermediate grades of severity occur.

Diagnostic features of beta-thalassaemia
Management and preventionCure is now a possibility for selected children, with allogeneic haematopoietic stem cell transplantation.
It is possible to identify a fetus with homozygous
beta- thalassaemia by obtaining chorionic villous material for DNA analysis sufficiently early in pregnancy to allow termination. This examination is only appropriate if both parents are known to be carriers (beta-thalassaemia minor) and will accept a termination.
Problem Management
Erythropoietic failure Allogeneic HSCT fromHLA-compatible sibling
Transfusion to maintain
Hb > 100 g/L
Folic acid 5 mg daily
Iron overload Iron therapy contraindicated
Iron chelation therapy
Splenomegaly causing
mechanical problems, Splenectomy
excessive transfusion;
(Hb = haemoglobin; HLA = human leucocyte antigen;
HSCT = haematopoietic stem cell transplantation)
Treatment of beta-thalassaemia major
Alpha- thalassaemia
Reduced or absent alpha chain synthesis is common in Southeast Asia. There are two alpha gene loci on chromosome 16 and therefore each individual carries four alpha gene alleles.
If one is deleted, there is no clinical effect.
If two are deleted, a mild hypochromic anaemia.
Three are deleted, the patient has haemoglobin H disease.
Four are deleted, the baby is stillborn (hydrops fetalis).
Haemoglobin H is a beta-chain tetramer, formed from the excess of beta chains, functionally useless, patients rely on their low levels of HbA for oxygen transport. Treatment of haemoglobin H disease is similar to that of beta-Th- of intermediate severity, involving folic acid supplementation,
transfusion if required and avoidance of iron therapy.

Anaemia in old age
HAEMATOLOGICAL MALIGNANCIESArise when the processes controlling proliferation or apoptosis are corrupted in blood cells. If mature differentiated cells are involved, produce indolent neoplasms, such as the low-grade lymphomas or chronic leukaemias. If more primitive stem cells are involved, the cells can have the highest growth fractions, producing rapidly progressive, life-threatening illnesses such as the acute leukaemias or high-grade lymphomas. Involvement of pluripotent stem cells produces the most aggressive acute leukaemias.
In general, haematological neoplasms are diseases of elderly, the exceptions being acute LL, which affects children, and Hodgkin, affects people aged 20–40 years.
Leukaemias
Are malignant disorders of the haematopoieticstem cell compartment, characteristically associated
with increased numbers of white cells in the bone
marrow and/or peripheral blood. Males are affected more frequently than females, the ratio being about 3 : 2 in acute leukaemia, 2 : 1 in CLL and 1.3 : 1 in CML . ALL shows a peak of incidence in children aged 1–5 years. AML have their lowest incidence in young adult life and there is a striking rise over the age of 50. Chronic leukaemias occur mainly in middle and old age.
The cause of the leukaemia is unknown in the majority.
Several risk factors, however, have been identified .

WHO classification of acute leukaemia
Terminology and classificationLeukaemias are traditionally classified into four main groups:
• acute lymphoblastic leukaemia (ALL)
• acute myeloid leukaemia (AML)
• chronic lymphocytic leukaemia (CLL)
• chronic myeloid leukaemia (CML).

Risk factors for leukaemia
In acute leukaemia, there is proliferation of primitivestem cells, leading to an accumulation of blasts, predominantly in the bone marrow, which causes bone
marrow failure. In chronic leukaemia, the malignant
clone is able to differentiate, resulting in an accumulation of more mature cells.
Lymphocytic and lymphoblastic cells are those derived from the lymphoid stem cell (B cells and T cells).
Myeloid refers to the other lineages:
that is, precursors of red cells, granulocytes,
monocytes and platelets .
The diagnosis of leukaemia is usually suspected from
an abnormal blood count, often a raised white count,
and is confirmed by examination of the bone marrow.
This includes the morphology of the abnormal cells,
analysis of cell surface markers (immunophenotyping),
clone-specific chromosome abnormalities and molecular
changes. These results are incorporated in the World
Health Organization (WHO) classification of tumours of
haematopoietic and lymphoid tissues. The features in the bone marrow not only provide an accurate diagnosis but also give valuable prognostic information, allowing therapy to be tailored to the patient’s disease.
Acute leukaemia
There is a failure of cell maturation in acute leukaemia.Proliferation of cells which do not mature leads to an
accumulation of primitive cells which take up more and
more marrow space at the expense of the normal haematopoietic elements. Eventually, this proliferation
spills into the blood. Acute myeloid leukaemia (AML) is
about four times more common than acute lymphoblastic
leukaemia (ALL) in adults. In children, the proportions
are reversed, the lymphoblastic variety being
more common. The clinical features are usually those of
bone marrow failure (anaemia, bleeding or infection ).
Investigations
Blood examination usually shows anaemia with a normal or raised MCV. The leucocyte count may vary from as low as 1 × 109/L to as high as 500 × 109/L or more. In the majority of patients, the count is below 100 × 109/L. Severe thrombocytopenia is usual but not invariable. Frequently, blast cells are seen in the blood film but sometimes blast cells may be infrequent or absent. A bone marrow examination will confirm the diagnosis. The bone marrow is usually hypercellular, with replacement of normal elements by leukaemic blast cells in varying degrees (but more than 20% of the cells) .The presence of Auer rods in the cytoplasm of blast cells indicates a myeloblastic type of leukaemia. Classification and prognosis are determined by immunophenotyping, chromosome and molecular analysis.
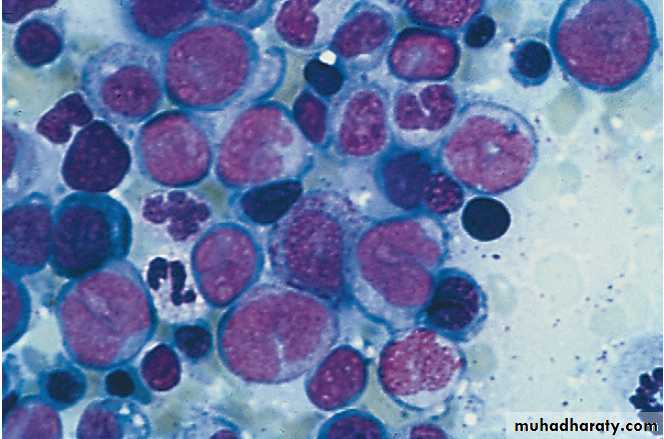
Acute myeloid leukaemia. Bone marrow aspirate showing
infiltration with large blast cells which display nuclear folding andprominent nucleoli.
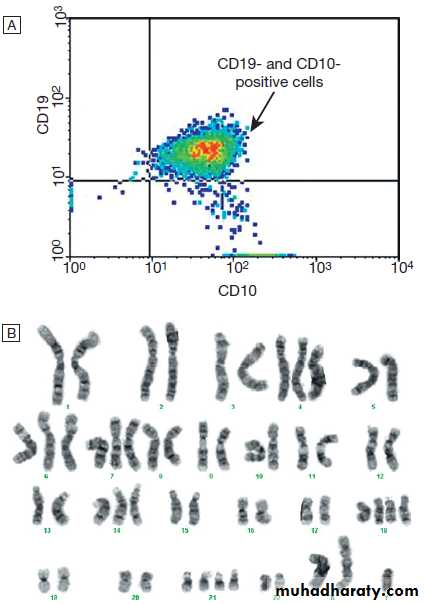
Investigation of acute lymphoblastic leukaemia (ALL).
A Flow cytometric analysis of blasts labelled with the fluorescent antibodies anti-CD19 (y axis) and anti-CD10 (x axis). ALL blasts arepositive for both CD19 and CD10 (arrow).
B Chromosome analysis
(karyotype) of blasts showing additional chromosomes X, 4, 6, 7, 14, 18 and 21.
Management
The first decision must be whether or not to give specific treatment. This is generally aggressive, has numerous side-effects, and may not be appropriate for the very elderly or patients with serious comorbidities . In these patients, supportive treatment can effect considerable improvement in well-being.Specific therapy
If a decision to embark on specific therapy has been
taken, the patient should be prepared as recommended. The aim of treatment is to destroy the leukaemic
clone of cells without destroying the residual normal
stem cell compartment from which repopulation of the
haematopoietic tissues will occur.
There are three phases:
• Remission induction. In this phase, the bulk of the
tumour is destroyed by combination chemotherapy.
The patient goes through a period of severe bone
marrow hypoplasia, requiring intensive support
and inpatient care from a specially trained
multidisciplinary team.
• Remission consolidation. If remission has been
achieved, residual disease is attacked by therapy
during the consolidation phase. This consists of a
number of courses of chemotherapy, again resulting
in periods of marrow hypoplasia. In poor-prognosis
leukaemia, this may include haematopoietic stem
cell transplantation.
• Remission maintenance. If the patient is still in
remission after the consolidation phase for ALL,a period of maintenance therapy is given, with
the individual as an outpatient and treatment
consisting of a repeating cycle of drug
administration. This may extend for up to 3 years
if relapse does not occur.
In patients with ALL, it is necessary to give prophylactic treatment to the central nervous system, as this is a sanctuary site where standard therapy does not penetrate. This usually consists of a combination of cranial irradiation, intrathecal chemotherapy and high dose methotrexate, which crosses the blood–brain barrier.
Thereafter, specific therapy is discontinued and the
patient observed. Generally, if a patient fails to go into remission with induction treatment, alternative drug combinations may be tried, but the outlook is poor unless remission can be achieved. Disease which relapses during treatment or soon after the end of treatment carries a poor prognosis and is difficult to treat. The longer after the end of treatment that relapse occurs, the more likely it is that further treatment will be effective. In some patients, alternative palliative chemotherapy, not designed to achieve remission, may be used to curb excessive leucocyte proliferation. Drugs used for this purpose include hydroxycarbamide and mercaptopurine. The aim is to reduce the blast count without inducing BM failure.
Supportive therapy
Aggressive and potentially curative therapy, whichinvolves periods of severe bone marrow failure, would
not be possible without appropriate supportive care.
The following problems commonly arise.
Anaemia. Anaemia is treated with red cell concentrate
transfusions.
Bleeding. Thrombocytopenic bleeding requires platelet
transfusions, unless the bleeding is trivial. Prophylactic
platelet transfusion should be given to maintain the
platelet count above 10 × 109/L.
Coagulation abnormalities occur and need accurate diagnosis and treatment.
Infection. Fever (> 38°C) lasting over 1 hour in a neutropenic patient indicates possible septicaemia . Parenteral broad-spectrum antibiotic therapy
is essential. Empirical therapy is given according to local bacteriological resistance patterns: for example, with a combination of an aminoglycoside (e.g. gentamicin) and a broad-spectrum penicillin (e.g. piperacillin/ tazobactam) or a single-agent beta-lactam (e.g. meropenem).
The organisms most commonly associated with severe neutropenic sepsis are Gram-positive bacteria, such as Staph aureus and Staph. epidermidis, which are present on the skin and gain entry via cannulae and central lines.
Gram-negative infections often originate from the gastrointestinal tract, which is affected by chemotherapy-induced mucositis; organisms such as Escherichia coli, Pseudomonas and Klebsiella spp. are likely to cause rapid clinical deterioration and must be covered with the initial empirical antibiotic therapy. Gram-positive infection may require vancomycin therapy.
If fever has not resolved after 3–5 days, empirical antifungal therapy (e.g. a liposomal amphotericin B preparation, voriconazole or caspofungin) is added.
Patients with ALL are susceptible to infection with
Pneumocystis jirovecii ,which causes a severepneumonia. Prophylaxis with co- trimoxazole is given
during chemotherapy. Diagnosis may require either
bronchoalveolar lavage or open lung biopsy. Treatment
is with high-dose co- trimoxazole, initially intravenously,
changing to oral treatment as soon as possible.
Oral and pharyngeal candida infection is common.
Fluconazole is effective for the treatment of established local infection and for prophylaxis against systemic candidaemia.Prophylaxis against other systemic fungal infections, including Aspergillus, using itraconazole or posaconazole, for example, is usual practice during
high-risk intensive chemotherapy. This is often used
along with sensitive markers of early fungal infection to guide treatment initiation (a ‘pre-emptive approach’).
For systemic fungal infection with Candida or aspergillosis, intravenous liposomal amphotericin or voriconazole is required.
Reactivation of herpes simplex infection occurs frequently around the lips and nose during ablative therapy for acute leukaemia, and is treated with aciclovir. This may also be prescribed prophylactically to patients with a history of cold sores or elevated antibody titres to herpes simplex. Herpes zoster manifesting as chickenpox or, after reactivation, as shingles should be treated in the early stage with high-dose aciclovir, as it can be fatal in immunocompromised patients.
The value of isolation facilities, such as laminar flow
rooms, is debatable but may contribute to staff awareness
of careful reverse barrier nursing practice. The isolation
can be psychologically stressful for the patient.
Metabolic problems.
Frequent monitoring of fluid balance and renal, hepatic and haemostatic function is necessary. Patients are often severely anorexic and diarrhoea is common as a consequence of the side-effects of therapy; they may find drinking difficult and hence
require intravenous fluids and electrolytes. Renal toxicity
occurs with some antibiotics (e.g. aminoglycosides)
and antifungal agents (amphotericin). Cellular breakdown
during induction therapy (tumour lysis syndrome) releases intracellular ions and nucleic acid breakdown products, causing hyperkalaemia, hyperuricaemia, hyperphosphataemia and hypocalcaemia. This may cause renal failure. Allopurinol and intravenous hydration are given to try to prevent this. In patients at high risk of tumour lysis syndrome, prophylactic rasburicase (a recombinant urate oxidase enzyme) can be used. Occasionally, dialysis may be required.
Psychological problems. Psychological support is a key
aspect of care. Patients should be kept informed, andtheir questions answered and fears allayed as far as possible.
A multidisciplinary approach to patient care
involves input from many services, including psychology.
Key members of the team include haematology
specialist nurses, who are often the central point of
contact for patients and families throughout the illness.
Haematopoietic stem cell transplantation
High-risk acute leukaemia, allogeneic HSCT can improve 5-year survival from 20% to around 50%.
Prognosis
Without treatment, the median survival of patients withacute leukaemia is about 5 weeks. This may be extended to a number of months with supportive treatment. Patients who achieve remission with specific therapy have a better outlook. Around 80% of adult patients under 60 years of age with ALL or AML achieve remission, although remission rates are lower for older
patients. However, the relapse rate continues to be high.
Box : shows the survival in ALL and AML, and the
influence of prognostic features.
Advances in treatment have led to steady improvement
in survival from leukaemia. Advances include the
introduction of drugs such as ATRA (all transretinoic
acid) in acute promyelocytic leukaemia, which has
greatly reduced induction deaths from bleeding in this
good-risk leukaemia. Current trials aim to improve survival, especially in standard and poor-risk disease, with strategies that include allogeneic HSCT and targeted therapies such as anti-CD33 monoclonal antibodies and FLT3 inhibitors.
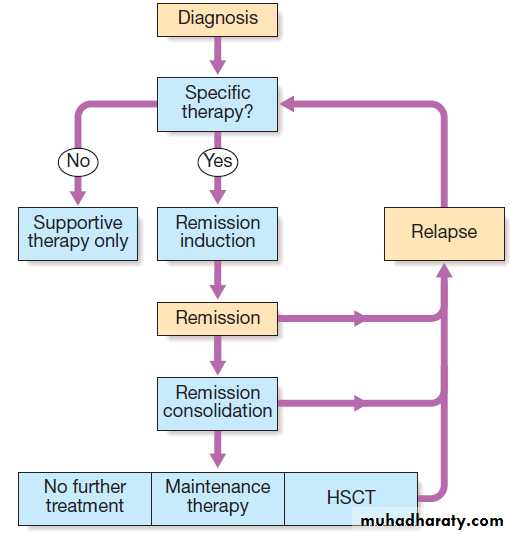
Treatment strategy in acute leukaemia.
(HSCT =haematopoietic stem cell transplantation)
Preparation for specific therapy in
acute leukaemia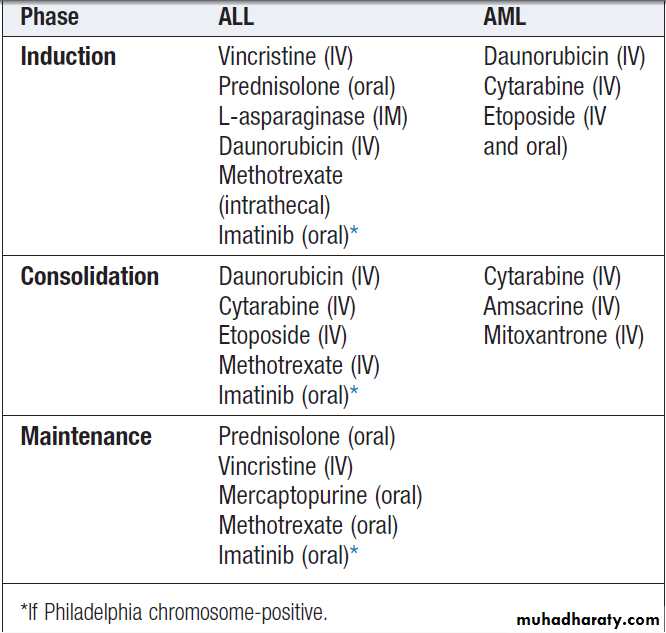
Drugs commonly used in the treatment of acute leukaemia
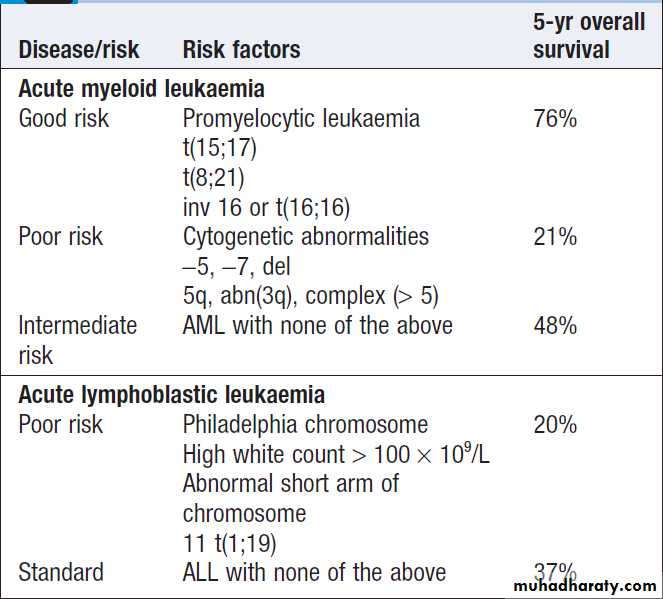
Outcome in adult acute leukaemia

Consequences of haematological malignancy in adolescence
Chronic myeloid leukaemia (CML)Chronic myeloid leukaemia is a myeloproliferative
stem cell disorder resulting in proliferation of all
haematopoietic lineages but manifesting predominantly in the granulocytic series. Maturation of cells proceeds fairly normally. The disease occurs chiefly between the ages of 30 and 80 years, with a peak incidence at 55 years.
It is rare, with an annual incidence in the UK of 1.8/100 000, and accounts for 20% of all leukaemias. The defining characteristic of CML is the chromosome
abnormality known as the Philadelphia (Ph) chromosome. This is a shortened chromosome 22 resulting from a reciprocal translocation of material with chromosome 9.
The break on chromosome 22 occurs in the breakpoint cluster region (BCR). The fragment from chromosome 9 that joins the BCR carries the abl oncogene, which forms a fusion gene with the remains of the BCR. This BCR ABL fusion gene codes for a 210 kDa protein with tyrosine kinase activity, which plays a causative role in the disease as an oncogene , influencing cellular proliferation, differentiation and survival.
In some patients in whom conventional chromosomal analysis does not detect a Ph chromosome, the BCR ABL gene product is detectable by molecular techniques.
Natural history :The disease has three phases:
• A chronic phase, in which the disease is responsive to
treatment and is easily controlled, which used to
last 3–5 years. With the introduction of imatinib
therapy, this phase has been prolonged to longer
than 8 years in many patients.
• An accelerated phase (not always seen), in which
disease control becomes more difficult.
• Blast crisis, in which the disease transforms into an
acute leukaemia, either myeloid (70%) or lymphoblastic (30%), which is relatively refractory to treatment. This is the cause of death in the majority of patients. Prior to imatinib approximately 10% of patients per year would transform. In those treated with imatinib for up to 5 years, only between 0.5 and 2.5% have transformed each year.
Clinical features
Symptoms at presentation may include lethargy, weightloss, abdominal discomfort and sweating, but about 25%
of patients are asymptomatic at diagnosis.
Splenomegaly is present in 90%; in about 10%, the enlargement is massive, extending to over 15 cm below the costal margin.
A friction rub may be heard in cases of splenic
infarction.
Hepatomegaly occurs in about 50%. Lymphadenopathy is unusual.
Investigations
FBC results are variable between patients. There isusually a normocytic, normochromic anaemia. The leucocyte count can vary from 10 to 600 × 109/L.
In about one-third of patients, there is a very high platelet count, sometimes as high as 2000 × 109/L.
In the blood film, the full range of granulocyte precursors, from myeloblasts to mature neutrophils, is seen but the predominant cells are neutrophils and myelocytes .
Myeloblasts usually constitute < 10% of all white cells.
There is often an absolute increase in eosinophils and basophils, and nucleated red cells are common.
If the disease progresses through an accelerated
phase, the percentage of more primitive cells
increases.
Blast transformation is characterised by a dramatic increase in the number of circulating blasts. In patients with thrombocytosis, very high platelet counts may persist during treatment, in both chronic and accelerated phases, but usually drop dramatically at blast transformation.
Basophilia tends to increase as the disease progresses.
Bone marrow should be obtained to confirm the diagnosis and phase of disease by morphology, chromosome analysis to demonstrate the presence of the Ph chromosome, and RNA analysis to demonstrate the presence of the BCR ABL gene product.
Blood LDH levels are elevated and the uric acid level may be high due to increased cell breakdown.
Management :Chronic phase
Imatinib, dasatinib and nilotinib specifically inhibit BCRABL tyrosine kinase activity and reduce the uncontrolled
proliferation of white cells. They are recommended as first-line therapy in chronic-phase CML, producing complete cytogenetic response (disappearance of the Ph chromosome) in 76% at 18 months of therapy .Patients are monitored by repeated BM examination until there is a complete cytogenetic response, and then by 3-monthly real-time quantitative PCR for BCR ABL mRNA transcripts in blood.
For those failing to respond or progress on imatinib, options include second-generation tyrosine kinase inhibitors, such as dasatinib or nilotinib, allogeneic HSCT ,or classical cytotoxic such as hydroxycarbamide (hydroxyurea) or interferon.
Hydroxycarbamide (hydroxyurea) was previously used widely for initial control of disease, and
is still useful in this context or in palliative situations. It
does not diminish the frequency of the Ph chromosome
or affect the onset of blast cell transformation.
Interferon alfa was considered first-line treatment before imatinib was developed. It was given alone or with the chemotherapy agent Ara-C, and controlled CML chronic phase in about 70% of patients.
Accelerated phase and blast crisis
Management is more difficult. For patients presenting in
accelerated phase, imatinib is indicated if the patient has
not already received it. Hydroxycarbamide can be an
effective single agent and low-dose cytarabine can also
be tried. When blast transformation occurs, the type of
blast cell should be determined. Response to appropriate
acute leukaemia treatment is better if disease is lymphoblastic than if it is myeloblastic. Given
the very poor response in myeloblastic transformation,
there is a strong case for supportive therapy only, particularly in older patients. Patients progressing to advanced-phase disease on imatinib may respond to a second-generation tyrosine kinase inhibitor and may be considered for allogeneic HSCT .
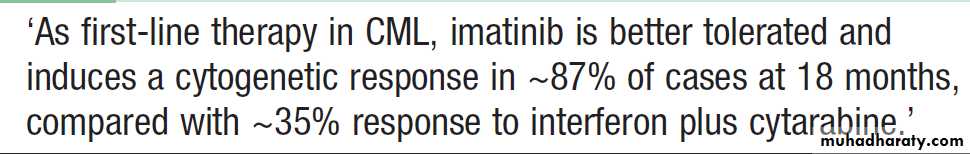
Tyrosine kinase inhibition in chronic myeloid leukaemia
Chronic lymphocytic leukaemia (CLL)Chronic lymphocytic leukaemia is the most common variety of leukaemia, accounting for 30% of cases. The male to female ratio is 2 : 1 and the median age at presentation is 65–70 years.
In this disease, B lymphocytes, which would normally respond to antigens by transformation and antibody formation, fail to do so. An ever-increasing mass of immuno-incompetent cells accumulates, to the detriment of immune function and normal bone marrow haematopoiesis.
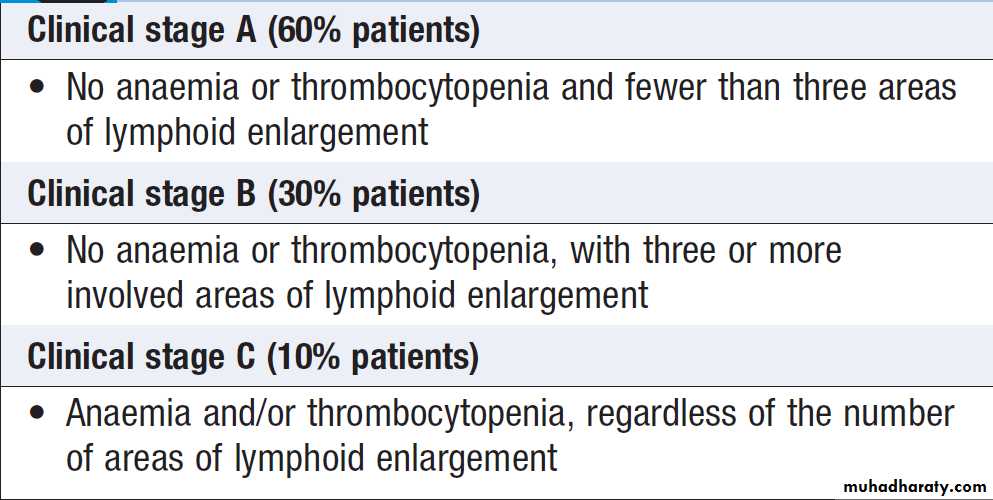
Staging of chronic lymphocytic leukaemia
Clinical featuresThe onset is usually insidious. Indeed, in around 70% of
patients, the diagnosis is made incidentally on a routine
FBC. Presenting problems may be anaemia, infections,
painless lymphadenopathy, and systemic symptoms
such as night sweats or weight loss.
Investigations
The diagnosis is based on the peripheral blood findings
of a mature lymphocytosis (> 5 × 109/L) with characteristic morphology and cell surface markers. Immunophenotyping reveals the lymphocytes to be monoclonal B cells expressing the B cell antigens
CD19 and CD23, with either kappa or lambda immunoglobulin light chains and, characteristically, an aberrant T cell antigen, CD5.
Other useful investigations in CLL include a reticulocyte
count and a direct Coombs test, as autoimmunehaemolytic anaemia may occur .Serum immunoglobulin
levels should be estimated to establish the degree of immunosuppression, which is common and progressive.
Bone marrow examination by aspirate and trephine is not essential for the diagnosis of CLL, but may be helpful in difficult cases, for prognosis (patients with diffuse marrow involvement have apoorer prognosis) and to monitor response to therapy. The main prognostic factor is stage of disease , however, malignant cell characteristics, such as CD38 expression, abnormalities of chromosome 11 or 17, and absence of mutations of IgVH genes, also indicate a poorer prognosis.
Management
No specific treatment is required for most clinical stage
A patients, unless progression occurs. Life expectancy is
usually normal in older patients. The patient should be
offered clear information about CLL, and be reassured
about the indolent nature of the disease, as the diagnosis
of leukaemia inevitably causes anxiety.
Treatment is required if there is evidence of marrow failure, massive or progressive lymphadenopathy or splenomegaly, systemic symptoms such as weight loss or night sweats, a rapidly increasing lymphocyte count or autoimmune haemolytic anaemia or thrombocytopenia. Initial therapy for stages B and C may consist of oral chemotherapy with the alkylating agent chlorambucil.
This will reduce the abnormal lymphocyte mass and produce symptomatic improvement in most patients. More recently, the purine analogue fludarabine, in combination with the alkylating agent cyclophosphamide and the anti-CD20 monoclonal antibody rituximab, has increased remission rates and disease-free survival, although there are increased risks of infection and secondary malignancies.
Bone marrow failure or autoimmune cytopenias may respond to corticosteroids.
Supportive care is increasingly required in progressive
disease, e.g. transfusions for symptomatic anaemiaor thrombocytopenia, prompt treatment of infections
and, for some patients with hypogammaglobulinaemia,
immunoglobulin replacement. Radiotherapy may be
used for lymphadenopathy which is causing discomfort
or local obstruction, and for symptomatic splenomegaly.
Splenectomy may be required to improve low blood
counts due to autoimmune destruction or to hypersplenism,
and can relieve massive splenomegaly.
Prognosis
The majority of clinical stage A patients have a normal
life expectancy but patients with advanced CLL are
more likely to die from their disease or infectious complications.
Survival is influenced by prognostic features
of the leukaemia and whether patients can tolerate
intensive treatment. In those treated with chemotherapy
and rituximab, 90% are alive 4 years later .
Rarely, CLL transforms to an aggressive high-grade
lymphoma, called Richter’s transformation.

Chemotherapy plus anti-CD20 monoclonal
antibody therapy in CLLProlymphocytic leukaemia
This is a variant of chronic lymphocytic leukaemiafound mainly in males over the age of 60 years; 25% of cases are of the T cell variety. There is typically massive splenomegaly with little lymphadenopathy and a very high leucocyte count, often in excess of 400 × 109/L; the characteristic cell is a large lymphocyte with a prominent nucleolus.
Treatment is generally unsuccessful and the prognosis very poor. Leukapharesis, splenectomy and chemotherapy may be tried.
Hairy cell leukaemia
This is a rare chronic B-cell lymphoproliferative disorder.
The male to female ratio is 6 :1and the median age at diagnosis is 50 years. Presenting symptoms are those of general ill health and recurrent infections. Splenomegaly occurs in 90% but lymph node enlargement is unusual. Severe neutropenia, monocytopenia and the characteristic
hairy cells in the blood and bone marrow are typical. These cells usually have a B lymphocyte immunotype
but they also characteristically express CD25
and CD103. Recently, all patients with hairy cell leukaemia
have been found to have a mutation in the BRAF gene. Over recent years, a number of treatments, including
cladribine and deoxycoformycin, have been shown to
produce long-lasting remissions.
Myelodysplastic syndrome
Myelodysplastic syndrome (MDS) consists of a group ofclonal haematopoietic disorders which represent steps
in the progression to the development of leukaemia.
MDS presents with consequences of bone marrow
failure (anaemia, recurrent infections or bleeding),
usually in older people (median age at diagnosis is
69 years). The blood film is characterised by cytopenias
and abnormal-looking (dysplastic) blood cells, including
macrocytic red cells and hypogranular neutrophils with
nuclear hyper- or hyposegmentation. The bone marrow is hypercellular, with dysplastic changes in all three cell lines. Blast cells may be increased but do not reach the 20% level that indicates acute leukaemia.
Chromosome analysis frequently reveals abnormalities, particularly of chromosome 5 or 7.
Inevitably, MDS progresses to AML, although the
time to progression varies (from months to years) with the subtype of MDS, being slowest in refractory anaemia
and most rapid in refractory anaemia with excess of
blasts. An international prognostic scoring system (IPSS)
predicts clinical outcome based upon karyotype and
cytopenias in blood, as well as percentage of bone
marrow blasts. In low-risk patients, median survival is
5.7 years and time for 25% of patients to develop AML
is 9.4 years; equivalent figures in high-risk patients are
0.4 and 0.2 years, respectively.
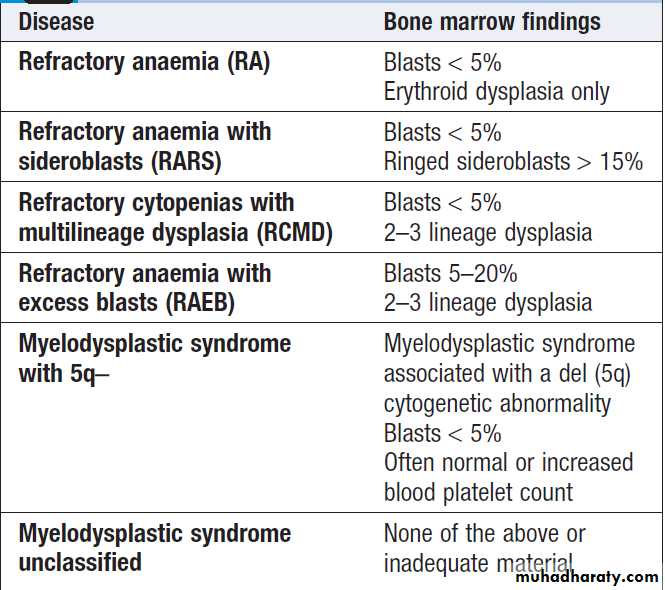
WHO classification of myelodysplastic syndromes
ManagementFor the vast majority of patients who are elderly, the
disease is incurable, and supportive care with red cell
and platelet transfusions is the mainstay of treatment. A
trial of erythropoietin and granulocyte–colony-stimulating factor (G–CSF) is recommended in some patients with early disease to improve haemoglobin and white cell counts. For younger patients with higher-risk disease, allogeneic HSCT preceded by intensive chemotherapy in those with more advanced disease. may afford a cure. More recently, the hypomethylating agent azacytidine has improved survival by a median of 9 months for high-risk patients, and in the UK is recommended for those not eligible for transplantation.
Lymphomas
These neoplasms arise from lymphoid tissues, and are
diagnosed from the pathological findings on biopsy as
Hodgkin or non-Hodgkin lymphoma.
The majority are of B cell origin.
Non-Hodgkin lymphomas are classified
as low- or high-grade tumours on the basis of their proliferation rate.
• High-grade tumours divide rapidly, are typically
present for a matter of weeks before diagnosis, and
may be life-threatening.
• Low-grade tumours divide slowly, may be present for
many months before diagnosis, and typically
behave in an indolent fashion.
Hodgkin lymphoma
The histological hallmark of Hodgkin lymphoma (HL)is the presence of Reed–Sternberg cells, large malignant
lymphoid cells of B cell origin .They are often
only present in small numbers but are surrounded by
large numbers of reactive non-malignant T cells, plasma
cells and eosinophils. Nodular lymphocyte-predominant HL is slow growing, localised and rarely fatal.
Classical HL is divided into four histological subtypes from the appearance of the Reed–Sternberg cells and surrounding reactive cells. The nodular sclerosing more common in young and in women. Mixed cellularity is more common in the elderly. Lymphocyte-rich HL usually presents in men. Lymphocyte-depleted HL is rare and probably represents large-cell or anaplastic non-Hodgkin lymphoma.
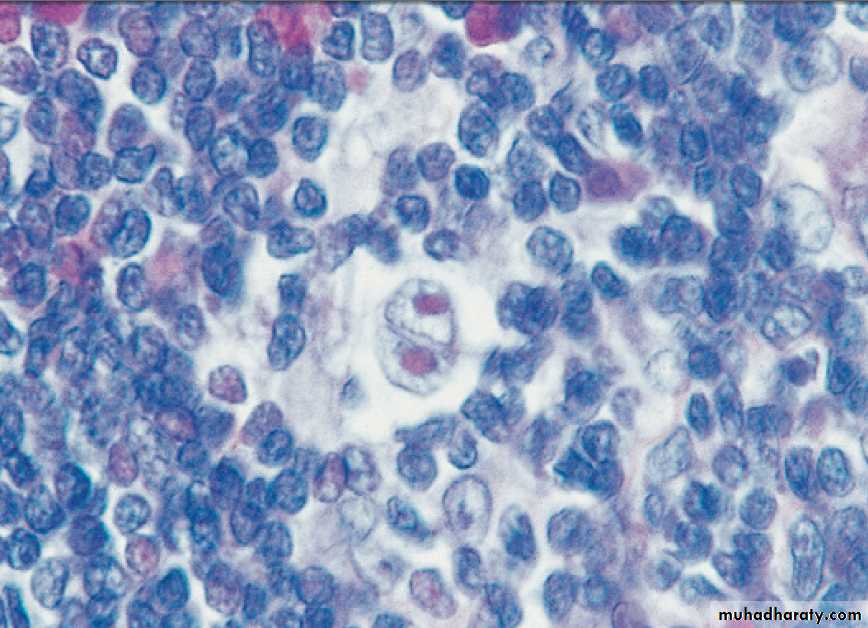
Hodgkin lymphoma. In the centre of this lymph node biopsy
is a large typical Reed–Sternberg cell with two nuclei containing a prominent eosinophilic nucleolus.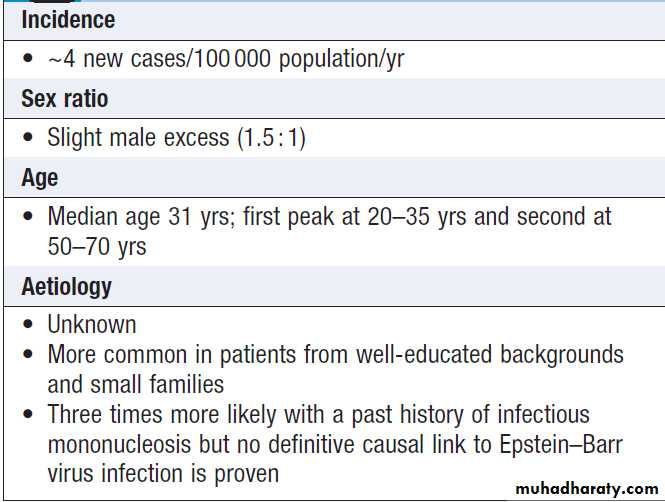
Epidemiology and aetiology of
Hodgkin lymphoma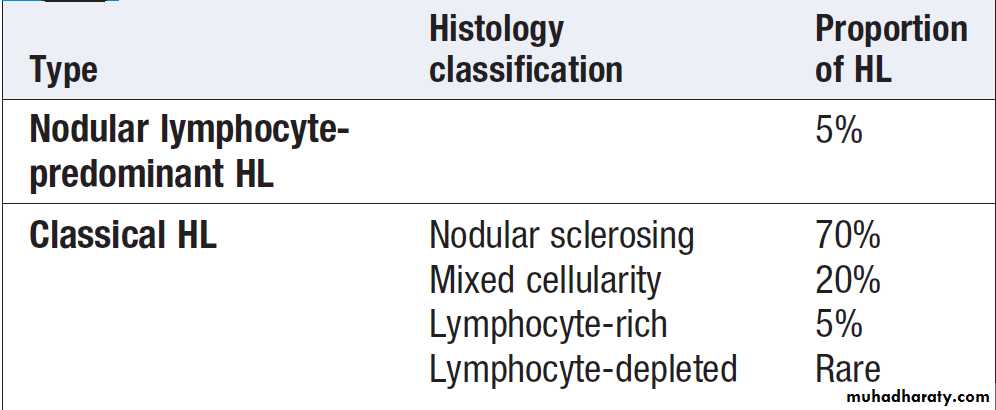
WHO pathological classification of Hodgkin lymphoma
Clinical featuresThere is painless, rubbery lymphadenopathy, usually in
the neck or supraclavicular fossae; the lymph nodes may fluctuate in size. Young patients with nodular sclerosing
disease may have large mediastinal masses which are
surprisingly asymptomatic but may cause dry cough
and some breathlessness. Isolated subdiaphragmatic
nodes occur in fewer than 10% at diagnosis. Hepatosplenomegaly may be present but does not always
indicate disease in those organs. Spread is contiguous
from one node to the next and extranodal disease, such
as bone, brain or skin involvement, is rare.
Investigations
Treatment of HL depends upon the stage at presentation;
therefore investigations aim not only to diagnose
lymphoma but also to determine the extent .
• FBC may be normal. If a normochromic, normocytic
anaemia or lymphopenia is present, this is a poor
prognostic factor. An eosinophilia or a neutrophilia
may be present.
• ESR may be raised.
• Renal function tests are required to ensure function is
normal prior to treatment.
• Liver function may be abnormal in the absence of
disease or may reflect hepatic infiltration. An obstructive pattern may be caused by nodes at the porta hepatis.
• LDH measurements showing raised levels are an
adverse prognostic factor.• Chest X-ray may show a mediastinal mass.
• CT scan of chest, abdomen and pelvis permits
staging. Bulky disease (> 10 cm in a single node
mass) is an adverse prognostic feature.
• Lymph node biopsy may be undertaken surgically or
by percutaneous needle biopsy .
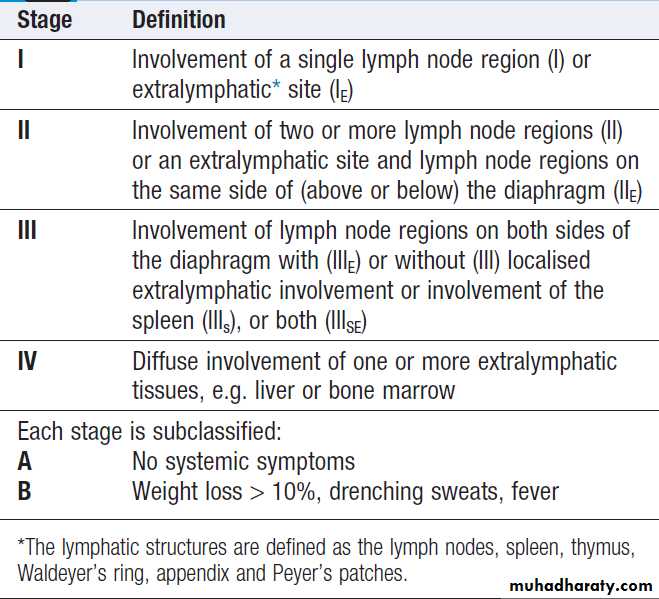
Clinical stages of Hodgkin lymphoma
(Ann Arbor classification)Management
Historically, radiotherapy to lymph nodes alone hasbeen used to treat localised stage IA or stage IIA disease
effectively, with no adverse prognostic features. Careful
planning of radiotherapy is required to limit the doses
delivered to normal tissues. Fertility is usually preserved
after radiotherapy. Young women receiving breast irradiation during the treatment of chest disease have an
increased risk of breast cancer and should participate in
a screening programme. Patients continuing to smoke
after lung irradiation are at particular risk of lung cancer.
Clinical trials have shown that patients with early stage disease have better outcomes if chemotherapy is
included in their treatment. The majority of HL patients
are now treated with chemotherapy and adjunctive
radiotherapy. The ABVD regimen (doxorubicin, vinblastine,
bleomycin and dacarbazine) is widely used in the UK. Standard therapy of early-stage patients usually includes additional treatment with radiotherapy to the involved lymph nodes after four courses of ABVD.Treatment response is assessed clinically and by repeat CT and PET.
ABVD chemotherapy can cause cardiac and pulmonary toxicity, due to doxorubicin and bleomycin, respectively. The incidence of infertility and secondary myelodysplasia/AML is low with this regime.
Patients with advanced-stage disease are most commonly
managed with chemotherapy alone. Standard
treatment in the UK is 6–8 cycles of ABVD, followed by
an assessment of response. As with early disease, achieving
PET-negative remission predicts a better long-term
remission rate. Overall, the long-term disease control/
cure rates are lower with advanced disease.
Patients with disease which is resistant to therapy
may be considered for autologous HSCT .
Prognosis
Over 90% of patients with early-stage achieve completeremission when treated with chemotherapy followed by involved field radiotherapy, and the great majority are cured. Between 50 and 70% of those with advanced-stage HL can be cured. The Hasenclever index can be helpful in assigning approximate chances of cure when discussing treatment plans with patients. Patients who fail to respond to initial chemotherapy or relapse within a year have a poor prognosis but some may
achieve long-term survival after autologous HSCT. Patients relapsing after 1 year may obtain long-term survival with further chemotherapy alone.
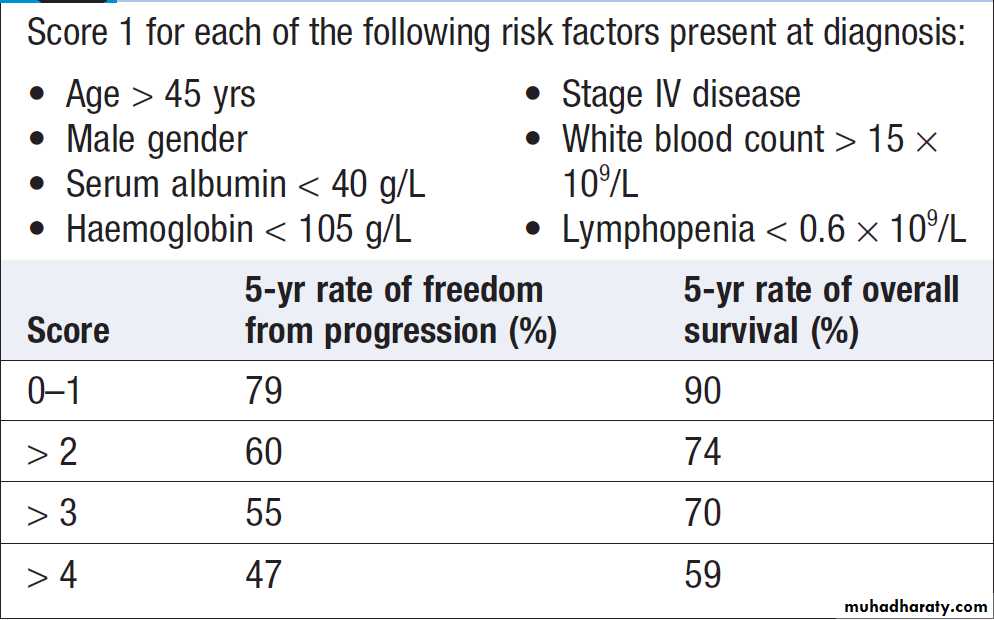
The Hasenclever prognostic index for
advanced Hodgkin lymphomaNon-Hodgkin lymphoma
Non-Hodgkin lymphoma (NHL) represents a monoclonal
proliferation of lymphoid cells of B cell (70%) or
T cell (30%) origin. The incidence of these tumours
increases with age. Previous classifications were based principally on histological appearances.
The current WHO classification stratifies according to cell lineage (T or B cells) and incorporates clinical features, histology, chromosomal abnormalities and cell surface markers of the malignant cells. Clinically, the most important factor is grade, which is a reflection of proliferation rate.
High-grade NHL has high proliferation rates, rapidly produces symptoms, is fatal if untreated,
but is potentially curable.
Low-grade NHL has low proliferation rates, may be asymptomatic for many months before presentation, runs an indolent course, but is not curable by conventional therapy. Of all cases of NHL in the developed world, over two thirds are either diffuse large B-cell NHL (high-grade) or follicular NHL (low-grade).
Other forms of NHL, including Burkitt lymphoma, mantle cell lymphoma, MALT lymphomas and T-cell lymphomas, are less common.
Epidemiology and aetiology of non-Hodgkin lymphoma
Incidence• 12 new cases/100 000 people/year
Sex ratio
• Slight male excess
Age
• Median age 65–70 yrs
Aetiology
• No single causative abnormality described
• Lymphoma is a late manifestation of HIV infection
• Specific lymphoma types are associated with viruses: e.g. Epstein–Barr virus (EBV) with post-transplant NHL, human herpesvirus 8 (HHV8) with a primary effusion lymphoma, and
human T-cell lymphotropic virus (HTLV) with adult T-cell leukaemia lymphoma
• Gastric lymphoma can be associated with Helicobacter pylori infection
• Some lymphomas are associated with specific chromosomal translocations; the t(14;18) in follicular lymphoma results in the dysregulated expression of the BCL-2 gene product,
which inhibits apoptotic cell death. The t(8;14) found in Burkitt lymphoma and the t(11;14) in mantle cell lymphoma alter function of c- myc and cyclin D1, respectively, resulting
in malignant proliferation
• Lymphoma occurs in congenital immunodeficiency states and in immunosuppressed patients after organ transplantation
Clinical features
Unlike Hodgkin, NHL is often widely disseminated
at presentation, including in extranodal sites. Patients present with lymph node enlargement, which may be associated with systemic upset: weight loss, sweats, fever and itching. Hepatosplenomegaly may be present. Sites of extranodal involvement include the bone marrow, gut, thyroid, lung, skin, testis, brain and, more rarely, bone.
Bone marrow involvement is more common in low-grade (50–60%) than high-grade (10%)
disease. Compression syndromes may occur, including
gut obstruction, ascites, superior vena cava obstruction
and spinal cord compression.
The same staging system is used for both HL and NHL, but NHL is more likely to be stage III or IV at presentation.
Investigations
As for HL, but in addition the following should be performed:• Bone marrow aspiration and trephine.
• Immunophenotyping of surface antigens to distinguish T from B cell tumours. This may be done on blood, marrow or nodal material.
• Cytogenetic analysis to detect chromosomal translocations and molecular testing for T cell receptor or immunoglobulin gene rearrangements,if available.
• Immunoglobulin determination. Some lymphomas are associated with IgG or IgM paraproteins, which serve as markers for treatment response.
• Measurement of uric acid levels. Some very aggressive
high-grade NHLs are associated with very high urate levels, which can precipitate renal failure when treatment is started.
• HIV testing. This may be appropriate if risk factors are present .
Management
Low-grade NHLAsymptomatic patients may not require therapy. Indications for treatment include marked systemic symptoms, lymphadenopathy causing discomfort or disfigurement, bone marrow failure or compression syndromes. In follicular lymphoma, the options are:
• Radiotherapy. This can be used for localised stage I
disease, which is rare.
• Chemotherapy. Most patients will respond to oral chlorambucil, which is well tolerated but not curative.
More intensive IV chemotherapy in younger patients produces better quality of life but no survival benefit.
Humanised monoclonal antibodies (‘biological’ therapy;) can be used to target surface antigens on tumour cells, and induce tumour cell apoptosis directly. The anti-CD20 antibody rituximab has been shown to induce durable clinical responses in up to 60% of patients when given alone, and acts synergistically when given with chemotherapy. Rituximab (R) in combination with cyclophosphamide, vincristine and prednisolone
(R-CVP) is commonly used as first-line therapy.
• Transplantation. Particular interest centres on the
role of high-dose chemotherapy and HSCT in patients with relapsed disease. Such high-dose therapy improves disease-free survival but longer follow-up is awaited before conclusions can be drawn about cure.
High-grade NHL
Diffuse large B-cell NHL need treatment at initial presentation:• Chemotherapy. The majority (> 90%) are treated with IV combination chemotherapy, typically with the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisolone).
When combined with CHOP chemotherapy, the biological therapy rituximab (R) increases the complete response rates and improves overall survival.
R-CHOP is currently recommended as first-line therapy for those with stage II or greater diffuse large B-cell lymphoma .
• Radiotherapy. A few stage I patients without bulky disease may be suitable for radiotherapy. Radiotherapy is also indicated for a residual localised site of bulk disease after chemotherapy,
and for spinal cord and other compression syndromes.
• HSCT. Autologous HSCT benefits patients with relapsed chemosensitive disease.

Chemotherapy plus anti-CD20 therapy in
high-grade non-Hodgkin lymphomaPrognosis
Low-grade NHL runs an indolent remitting and relapsing course, with an overall median survival of 10 years. Transformation to a high-grade NHL occurs in 3% per annum and is associated with poor survival.In diffuse large B-cell high-grade NHL treated with R-CHOP, some 75% of patients overall respond initially to therapy and 50% will have disease-free survival at 5 years. The prognosis for patients with NHL is further refined according to the international prognostic index (IPI). For high-grade NHL, 5-year survival ranges from
75% in those with low-risk scores (age < 60 years, stage
I or II, one or fewer extranodal sites, normal LDH and good performance status) to 25% in those with high-risk scores (increasing age, advanced stage, concomitant disease and a raised LDH).
Relapse is associated with a poor response to further
chemotherapy (< 10% 5-year survival), but in patients
under 65 years, HSCT improves survival.
Paraproteinaemias
A gammopathy refers to over-production of one or more
classes of immunoglobulin. It may be polyclonal in association with acute or chronic inflammation, such as
infection, sarcoidosis, autoimmune disorders or some
malignancies. Alternatively, a monoclonal increase in a single IG class may occur in association with normal or reduced levels of the other IGs. Such monoclonal proteins (also called M-proteins, paraproteins or monoclonal gammopathies) occur as a feature of myeloma, lymphoma and amyloidosis, in connective tissue disease such as rheumatoid arthritis or polymyalgia rheumatica, in infection such as HIV, and in solid tumours. They may be present with no underlying disease. Gammopathies are detected by plasma immunoelectrophoresis.
Monoclonal gammopathy of uncertain significance
(MGUS, also known as benign monoclonal gammopathy),a paraprotein is present in the blood but with no
other features of myeloma, Waldenstrِm macroglobulinaemia ,lymphoma or related disease. It is a
common finding associated with increasing age; a paraprotein can be found in 1% of the population aged over 50 years, increasing to 5% over 80 years.
Clinical features and investigations
Patients are usually asymptomatic, and the paraproteinis found on blood testing for other reasons. The routine
blood count and biochemistry are normal, the paraprotein
is usually present in small amounts with no associated
immune paresis, and there are no lytic bone lesions. The bone marrow may have increased plasma cells but these usually constitute <10% of nucleated cells.
Prognosis
After follow-up of 20 years, only one-quarter of cases will progress to myeloma or a related disorder . Patients with low-level IgG paraproteins without reductions in IgM and IgA levels and with normal serum free light chain level are highly unlikely to progress at any time.
Waldenstrِm macroglobulinaemia
This is a low-grade lymphoplasmacytoid lymphoma
associated with an IgM paraprotein, causing clinical features of hyperviscosity syndrome. It is a rare tumour
occurring in the elderly and affects males more
commonly. Patients classically present with features of hyperviscosity, such as nosebleeds, bruising, confusion and visual disturbance. However, presentation may be with anaemia, systemic symptoms, splenomegaly or lymphadenopathy.Patients are found on investigation to
have an IgM paraprotein associated with a raised plasma viscosity. The bone marrow has a characteristic appearance,with infiltration of lymphoid cells and prominent mast cells.
Management
If patients show symptoms of hyperviscosity andanaemia, plasmapheresis is required to remove IgM and make blood transfusion possible.
Chemotherapy with alkylating agents, such as chlorambucil, has been the mainstay of treatment, controlling disease in over 50%.
Fludarabine may be more effective but has more side effects.
Rituximab can also be effective.
The median survival is 5 years.
Multiple myeloma
This is a malignant proliferation of plasma cells. Normalplasma cells are derived from B cells and produce
immunoglobulins which contain heavy and light chains.
Normal immunoglobulins are polyclonal, which means
that a variety of heavy chains are produced and each
may be of kappa or lambda light chain type .
In myeloma, plasma cells produce immunoglobulin of a
single heavy and light chain, a monoclonal protein commonly referred to as a paraprotein.
In some cases, only light chain is produced and this appears in the urine as Bence Jones proteinuria. Although a small number of malignant plasma cells are present in the circulation, the majority are present in the BM .
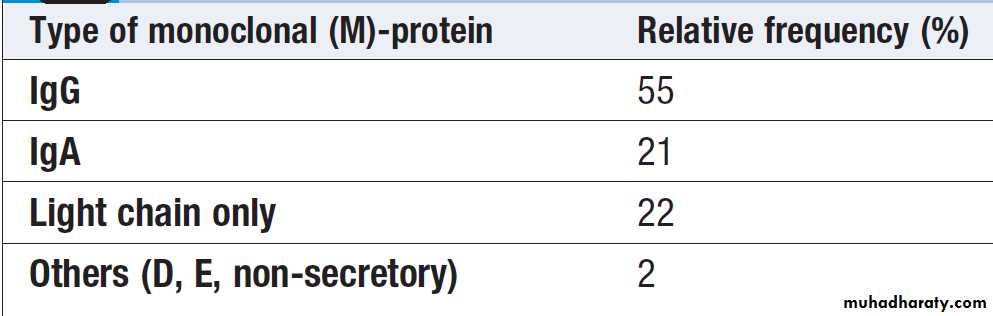
Classification of multiple myeloma
The malignant plasma cells produce cytokines, which stimulate osteoclasts and result in net bone reabsorption. The resulting lytic lesions cause bone pain, fractures and hypercalcaemia. Marrow involvement can result in anaemia or pancytopenia.Clinical features and investigations
The incidence of myeloma is 4/100 000 new cases per annum, with a male to female ratio of 2 : 1. The median age at diagnosis is 60–70 years and the disease is more common in Afro- Caribbeans.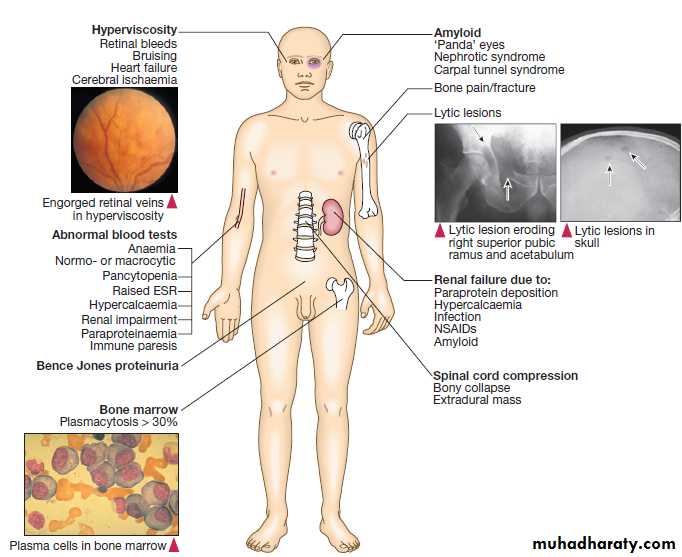
Clinical and laboratory features of multiple myeloma
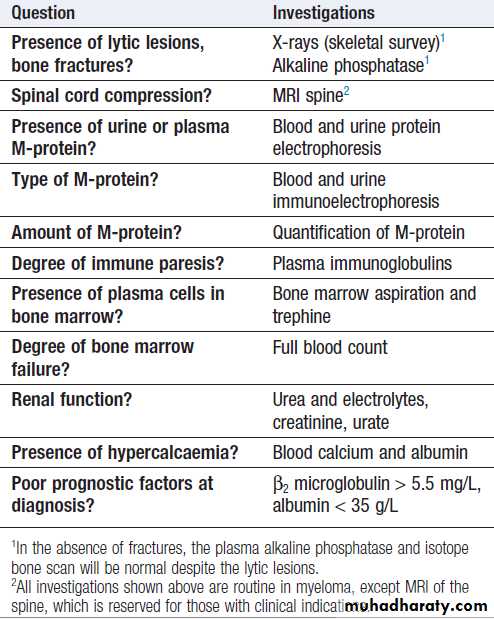
Rationale for investigations in
multiple myelomaDiagnosis of myeloma requires two of the following
criteria:
• increased malignant plasma cells in the BM
• serum and/or urinary M-protein
• skeletal lytic lesions.
BM aspiration, plasma and urinary electrophoresis,
and a skeletal survey are thus required. Normal immunoglobulin levels, i.e. the absence of immunoparesis, should cast doubt on the diagnosis.
Paraproteinaemia can cause an elevated ESR but this is a nonspecific test; only approximately 5% of patients with a persistently elevated ESR above 100 mm/ hr have underlying myeloma.
Management
If patients are asymptomatic with no evidence of endorgan damage (e.g. to kidneys, bone marrow or bone),
treatment may not be required.
Immediate support
• High fluid intake to treat renal impairment and
hypercalcaemia .
• Analgesia for bone pain.
• Bisphosphonates for hypercalcaemia and to delay
other skeletal related events .
• Allopurinol to prevent urate nephropathy.
• Plasmapheresis, if necessary, for hyperviscosity.
Chemotherapy with or without HSCT
Myeloma therapy has improved with the addition of novel agents, initially thalidomide and more recently the proteasome inhibitor bortezomib, to first-line treatments. In older patients, thalidomide combined with the alkylating agent melphalan and prednisolone has increased the median overall survival to more than 4 years. Thalidomide has both anti- angiogenic effects against tumour blood vessels and immunomodulatory effects. It can cause somnolence, constipation, peripheral neuropathy and thrombosis. It is vital that females of child-bearing age use adequate contraception, as thalidomide is teratogenic.
Treatment is administered until paraprotein levels have stopped falling. This is termed ‘plateau phase’ and can last for weeks or years. In younger, fitter patients, standard treatment includes first-line chemotherapy to maximum response and then an autologous HSCT, which improves quality of life and prolongs survival but does not cure myeloma.
The role of allogeneic transplantation
and of reduced-intensity allografting after autologous transplantation in younger patients is under evaluation.When myeloma progresses, treatment is given to
induce a further plateau phase. In the UK at present,
bortezomib is recommended, followed by lenalidomide
if there is subsequent progression.
Radiotherapy
This is effective for localised bone pain not responding
to simple analgesia and for pathological fractures. It is
also useful for the emergency treatment of spinal cord
compression complicating extradural plasmacytomas.
Bisphosphonates
Long-term bisphosphonate therapy reduces bone painand skeletal events. These drugs protect bone .
and may cause apoptosis of malignant plasma cells.
There is evidence that intravenous zoledronate in combination with anti-myeloma therapy confers a survival advantage over oral bisphosphonates. Osteonecrosis of the jaw may be associated with long-term use; therefore regular dental review is advisable.

Autologous haematopoietic stem cell
transplantation in multiple myelomaPrognosis
The international staging system (ISS) identifiesPoor prognostic features, including a high β2-microglobulin and low albumin at diagnosis (ISS stage 3, median survival 29 months). Those with a normal albumin and a low β2-microglobulin (ISS stage 1) have a median survival of 62 months. Use of autologous HSCT and advances in drug therapy have increased survival, with over one-third of patients now surviving for 5 years, compared with only one-quarter 10 years ago. The outlook may improve further with new drugs and combinations of treatments.

Haematological malignancy in old age
APLASTIC ANAEMIAPrimary idiopathic acquired aplastic anaemia
This is a rare disorder in Europe and North America,
with 2–4 new cases per million population per annum.
The disease is much more common in certain other parts
of the world: for example, east Asia. The basic problem
is failure of the pluripotent stem cells, producing hypoplasia of the bone marrow with a pancytopenia in the blood. The diagnosis rests on exclusion of other causes of secondary aplastic anaemia and rare congenital causes, such as Fanconi’s anaemia.
Clinical features and investigations
Patients present with symptoms of bone marrow failure,
usually anaemia or bleeding, and less commonly, infections. An FBC demonstrates pancytopenia, low reticulocytes and often macrocytosis. Bone marrow aspiration and trephine reveal hypocellularity.
Management
All patients will require blood product support and
aggressive management of infection. The prognosis of
severe aplastic anaemia managed with supportive
therapy only is poor and more than 50% of patients die,
usually in the first year. The curative treatment for
patients under 30 years of age with severe idiopathic
aplastic anaemia is allogeneic HSCT .
Those with a compatible sibling donor should proceed to transplantation as soon as possible; they have a 75–90% chance of long-term cure.
In older patients, immunosuppressive therapy
with ciclosporin and antithymocyte globulin gives
5-year survival rates of 75%.
Such patients may relapse or other clonal disorders of haematopoiesis may evolve, such as PNH , myelodysplastic syndrome and AML .They must be followed up long-term.
Secondary aplastic anaemia
It is not practical to list all the drugs which have been suspected of causing aplasia. It is important to check the reported side-effects of all drugs taken over the preceding months.In some instances, the cytopenia is more selective and
affects only one cell line, most often the neutrophils.
Frequently, this is an incidental finding, with no ill
health. It probably has an immune basis but this is difficult to prove.
The clinical features and methods of diagnosis are the
same as for primary idiopathic aplastic anaemia. An underlying cause should be treated or removed but
otherwise management is as for the idiopathic form.
Causes of secondary aplastic anaemia
• Drugs. Cytotoxic drugs. Antibiotics – chloramphenicol, sulphonamides. Antirheumatic agents – penicillamine, gold, phenylbutazone, indomethacin. Antithyroid drugs
Anticonvulsants. Immunosuppressants – azathioprine
• Chemicals
Benzene toluene solvent misuse – glue-sniffing
Insecticides – chlorinated hydrocarbons (DDT),
organophosphates and carbamates
• Radiation
• Viral hepatitis
• Pregnancy
• Paroxysmal nocturnal haemoglobinuria
MYELOPROLIFERATIVE NEOPLASMS
These make up a group of chronic conditions characterisedby clonal proliferation of marrow precursor cells,
and include PRV , essential thrombocythaemia, myelofibrosis, and CML. Although the majority of patients
are classifiable as having one of these disorders, some
have overlapping features and there is often progression from one to another, e.g. PRV to myelofibrosis. The
recent discovery of the molecular basis of these disorders
will lead to changes in classification and treatment;
a mutation in the gene on chromosome 9 encoding the
signal transduction molecule JAK-2 has been found in
more than 90% of PRV cases and 50% of those with
essential thrombocythaemia and myelofibrosis.
Myelofibrosis
In myelofibrosis, the marrow is initially hypercellular,
with an excess of abnormal megakaryocytes which
release growth factors, e.g. platelet-derived growth
factor, to the marrow microenvironment, resulting in a
reactive proliferation of fibroblasts. As the disease
progresses, the marrow becomes fibrosed.
Most patients present over the age of 50 years, with
lassitude, weight loss and night sweats. The spleen can
be massively enlarged due to extramedullary haematopoiesis (blood cell formation outside the bone marrow), and painful splenic infarcts may occur.
The characteristic blood picture is leucoerythroblastic
anaemia, with circulating immature red blood cells(increased reticulocytes and nucleated red blood
cells) and granulocyte precursors (myelocytes). The red
cells are shaped like teardrops (teardrop poikilocytes),
and giant platelets may be seen in the blood. The white
count varies from low to moderately high, and the platelet
count may be high, normal or low. Urate levels may
be high due to increased cell breakdown, and folate
deficiency is common.
The marrow is often difficult to aspirate and a trephine biopsy shows an excess of megakaryocytes, increased reticulin and fibrous tissue replacement. The presence of a JAK-2 mutation supports the diagnosis.
Management and prognosis
Median survival is 4 years from diagnosis, but ranges
from 1 year to over 20 years. Treatment is directed at
control of symptoms, e.g. red cell transfusions for
anaemia. Folic acid should be given to prevent deficiency.
Cytotoxic therapy with hydroxycarbamide may
help control spleen size, the white cell count or systemic
symptoms. Splenectomy may be required for a grossly
enlarged spleen or symptomatic pancytopenia secondary to splenic pooling of cells and hypersplenism. HSCT may be considered for younger patients. Ruxolitinib, an inhibitor of JAK-2, has recently been licensed for use.
Essential thrombocythaemia
Increased proliferation of megakaryocytes results in araised level of circulating platelets that are often dysfunctional.
Prior to making a diagnosis of essential thrombocythaemia
(ET), reactive causes of thrombocytosis must be excluded . The presence of a JAK-2 mutation supports the diagnosis but is not universal. Patients present at a median age of 60 years with vascular occlusion or bleeding, or with an asymptomatic isolated raised platelet count.
A small percentage transform to acute leukaemia and others to myelofibrosis.
It is likely that most patients with ET benefit from
low-dose aspirin to reduce the risk of occlusive vascularevents. Low-risk patients (age < 40 years, platelet count
< 1000 × 109/L and no bleeding or thrombosis) may not require treatment to reduce the platelet count. For those with a platelet count above 1000 × 109/L, with symptoms, or with other risk factors for thrombosis such as
diabetes or hypertension, treatment to control platelet
counts should be given. Agents include oral hydroxycarbamide or anagrelide, an inhibitor of megakaryocyte maturation. Intravenous radioactive phosphorus (32P) may be useful in old age.
Polycythaemia rubra vera (PRV)
occurs mainly in patients over the age of 40 years and presents either as an incidental finding of a high haemoglobin, or with symptoms of hyperviscosity, such as lassitude, loss of concentration, headaches, dizziness, blackouts, pruritus and epistaxis. Some patients present with manifestations of peripheral arterial disease or a cerebrovascular accident.
VTE may also occur. Peptic ulceration is common, sometimes complicated by bleeding.
Patients are often plethoric and many have a palpable
spleen at diagnosis.
The diagnosis of PRV now rests upon the demonstration
of a high haematocrit and the presence of theJAK-2 mutation. In the occasional JAK-2-negative cases,
a raised red cell mass and absence of causes of a secondary erythrocytosis must be established. The spleen is enlarged, neutrophil and platelet counts are frequently raised, an abnormal karyotype may be found in the marrow, and in vitro culture of the marrow can be used to demonstrate autonomous growth in the absence of added growth factors.
Management and prognosis
Aspirin reduces the risk of thrombosis. Venesectiongives prompt relief of hyperviscosity symptoms.
Between 400 and 500 mL of blood (less if the patient is
elderly) are removed and the venesection is repeated
every 5–7 days until the haematocrit is reduced to below 45%. Less frequent but regular venesection will maintain this level until the haemoglobin remains reduced
because of iron deficiency. Suppression of marrow proliferation with hydroxycarbamide or interferon-alfa may reduce the risk of vascular occlusion, control spleen size and reduce transformation to myelofibrosis. Radioactive phosphorus (5 mCi of 32P IV) is reserved for older patients, as it increases the risk of transformation to acute leukaemia by 6- to 10-fold.
Median survival after diagnosis in treated patients
exceeds 10 years. Some patients survive more than20 years; however, cerebrovascular or coronary events occur in up to 60% of patients.
The disease may convert to another myeloproliferative disorder, with about 25% developing acute leukaemia or myelofibrosis.
BLEEDING DISORDERS
Disorders of primary haemostasis
The initial formation of the platelet plug may fail
in thrombocytopenia ,von Willebrand disease
and also in platelet function disorders and
diseases affecting the vessel wall.
Vessel wall abnormalities
Vessel wall abnormalities may be:
• congenital, such as hereditary haemorrhagic
telangiectasia
• acquired, as in a vasculitis or scurvy.
Hereditary haemorrhagic telangiectasia (HHT)
A dominantly inherited condition caused by mutations in the genes encoding endoglin and activin receptor-likekinase, which are endothelial cell receptors for transforming growth factor-beta (TGF-β), a potent angiogenic cytokine. Telangiectasia and small aneurysms are found on the fingertips, face and tongue, and in the nasal passages, lung and gastrointestinal tract. A significant proportion of these patients develop larger pulmonary arteriovenous malformations (PAVMs) that cause arterial hypoxaemia due to a right-to-left shunt. These predispose to paradoxical embolism, resulting in stroke or cerebral abscess. All patients with should be
screened for PAVMs; if these are found, ablation by
percutaneous embolisation should be considered.
Patients present either with recurrent bleeds, particularly
epistaxis, or with iron deficiency due to occult gastrointestinal bleeding. Treatment can be difficult because of the multiple bleeding points but regular iron therapy often allows the marrow to compensate for blood loss.Local cautery or laser therapy may prevent single lesions from bleeding.
A variety of medical therapies have been tried but none has been found to be universally effective.
Ehlers–Dan los disease
Vascular Ehlers– Danlos syndrome (type 4) is a rare autosomal dominant disorder (1/100 000) caused by a defect in type 3 collagen which results in fragile blood vessels and organ membranes, leading to bleeding and organ rupture. Classical joint hypermobility is often
limited in this form of the disease but skin changes and
facial appearance are typical. The diagnosis should be
considered when there is a history of bleeding but
normal laboratory tests.
Scurvy
Vitamin C deficiency affects the normal synthesis of collagen and results in a bleeding disorder characterised by perifollicular and petechial haemorrhage, bruising and subperiosteal bleeding. The key to diagnosis is the dietary history .Platelet function disorders
Bleeding may result from thrombocytopenia or from congenital or acquired abnormalities of platelet function. The most common acquired disorders are iatrogenic, resulting from the use of aspirin, clopidogrel, dipyridamole and the IIb/ IIIa inhibitors to prevent arterial thrombosis. Inherited platelet function abnormalities arerelatively rare. Congenital abnormalities may be due to
deficiency of the membrane glycoproteins, e.g. Glanzmann’s thrombasthenia (IIb/ IIIa) or Bernard–Soulier disease (Ib), or due to the presence of defective platelet granules, e.g. a deficiency of dense (delta) granules giving rise to storage pool disorders.
Glanzmann’s is an autosomal recessive condition
associated with a variable but often severe bleeding disorder.These conditions are usually managed by local
mechanical measures, but antifibrinolytics, such as tranexamic acid, may be useful and, in severe bleeding, platelet transfusion may be required. Recombinant VIIa is licensed for the treatment of resistant bleeding in Glanzmann’s thrombasthenia.
Thrombocytopenia
Idiopathic thrombocytopenic purpura (ITP)Mediated by autoantibodies, most often directed against the platelet membrane glycoprotein IIb/IIIa, which sensitise the platelet, resulting in premature removal from the circulation by cells of the reticulo-endothelial system. It is not a single disorder; some cases occur in isolation while others are associated with underlying immune dysregulation in conditions such as connective tissue diseases, HIV infection, B cell malignancies, pregnancy and certain drug therapies. However, the clinical presentation and pathogenesis are similar, whatever the cause of ITP.
Clinical features and investigations
Spontaneous bleeding typically occurs only when the platelet count is below 20 × 109/L. At higher counts, the patient may complain of easy bruising or sometimes epistaxis or menorrhagia. Many cases with counts of more than 50 × 109/L are discovered by chance.
In adults, ITP more commonly affects females and may have an insidious onset. Unlike ITP in children, it is unusual for there to be a history of a preceding viral infection.
Symptoms or signs of a connective tissue disease may be apparent at presentation or emerge several years later.
Patients aged over 65 years should have a bone marrow examination to look for an accompanying
B cell malignancy and appropriate autoantibody testing performed if a diagnosis of connective tissue disease is likely. HIV testing should be considered. The peripheral blood film is normal, apart from a
greatly reduced platelet number, whilst the bone marrow reveals an obvious increase in megakaryocytes.
Management
Many patients with stable compensated ITP and a platelet count of more than 30 × 109/L do not require treatment to raise the platelet count, except at times of increased bleeding risk, such as surgery and biopsy.First-line therapy for patients with spontaneous bleeding is with prednisolone 1 mg/kg daily to suppress antibody production and inhibit phagocytosis of sensitized platelets by reticuloendothelial cells.
Administration of IVimmunoglobulin (IVIg) can raise the count by blocking antibody receptors on RE cells, and is combined with corticosteroid therapy if there is severe haemostatic failure or a slow response to steroids alone.
Persistent or potentially life-threatening bleeding should be treated with platelet transfusion in addition to the other therapies.
The condition may become chronic, with remissions and relapses. Relapses should be treated by reintroducing corticosteroids.
If a patient has two relapses, or primary refractory disease, splenectomy is considered, Splenectomy produces complete remission in about 70% of patients and improvement in a further 20–25%, so that, following splenectomy, only 5–10% of patients require further medical therapy.
If severe thrombocytopenia with or without significant bleeding persists despite splenectomy, second-line therapy with the thrombopoietin analogue romiplostim or the thrombopoietin receptor agonist eltrombopag should be considered. Low-dose corticosteroid therapy, immunosuppressants such as rituximab, ciclosporin and tacrolimus should be considered in cases where the approaches above are ineffective.
Coagulation disorders
Coagulation factor deficiency may be congenital or acquired, and may affect one or several of the coagulation factors . Inherited disorders are almost uniformly related to decreased synthesis, as a result of mutation in the gene encoding a key protein in coagulation.Von Willebrand disease is the most common inherited bleeding disorder. Haemophilia A and B are the most common single coagulation factor deficiencies, but inherited deficiencies of all the other coagulation factors are seen. Acquired disorders may be due to under-production (e.g. in liver failure), increased consumption (e.g. in disseminated intravascular coagulation) or inhibition of function (such as heparin therapy or immune inhibitors of coagulation, e.g. acquired haemophilia A).
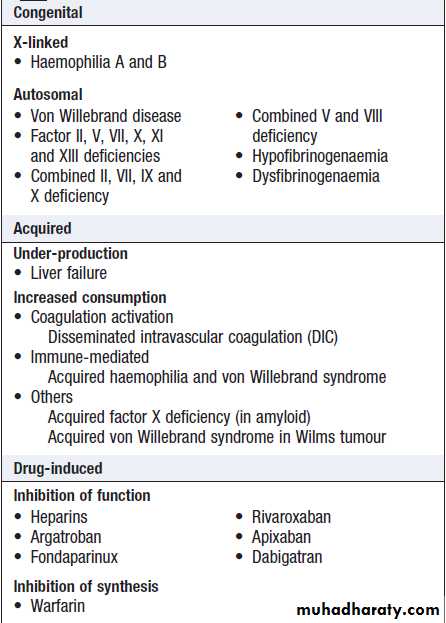
Causes of coagulopathy
Haemophilia AFactor VIII deficiency resulting in haemophilia A affects
1/10 000 individuals. It is the most common congenital coagulation factor deficiency.
Factor VIII is primarily synthesised by the liver and endothelial cells, and has a half-life of about 12 hours. It is protected from proteolysis in the circulation by binding to von Willebrand factor (vWF).
Genetics
The factor VIII gene is located on the X chromosome. Severe haemophilia is associated with large deletions, while single-base changes more often result in moderate or mild disease . As the gene is on the X chromosome, haemophilia A is a sex-linked disorder. Thus all daughters of haemophiliacs are obligate carriers and they, in turn, have a 1 in 4 chance of each pregnancy resulting in the birth of an affected male baby, a normal male baby, a carrier female or a normal female.Antenatal diagnosis by chorionic villous sampling is possible in families with a known mutation.
Haemophilia ‘breeds true’ within a family; all members have the same factor VIII gene mutation and a similarly severe or mild phenotype. Female carriers of haemophilia may have reduced factor VIII levels because of random inactivation of their normal X chromosome in the developing fetus (lyonisation).
This can result in a mild bleeding disorder; thus all known or suspected carriers of haemophilia should have their factor VIII level measured.
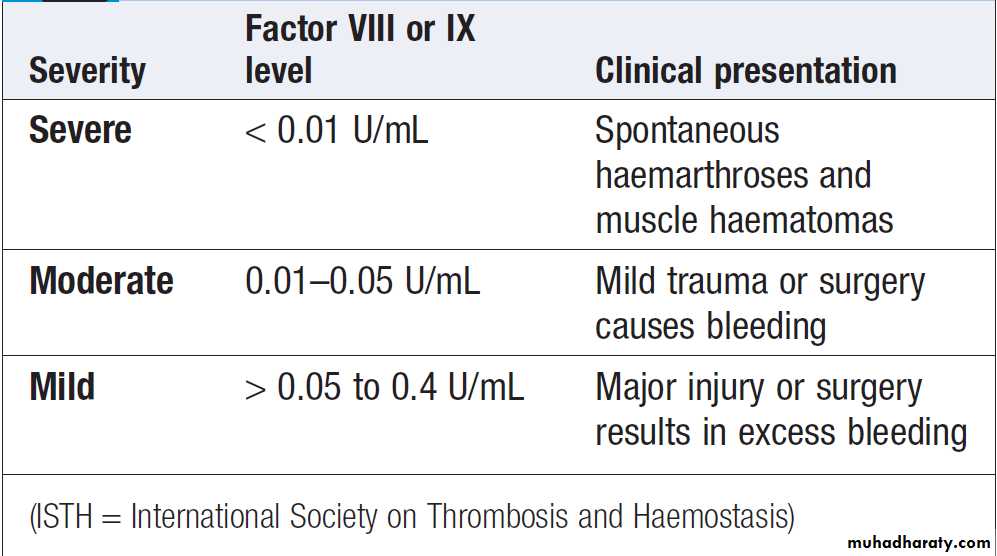
Severity of haemophilia (ISTH criteria)
Clinical features
The extent and patterns of bleeding are closely related
to residual factor VIII levels. Patients with severe haemophilia (< 1% of normal factor VIII levels) present with spontaneous bleeding into skin, muscle and joints. Retroperitoneal and intracranial bleeding is also a feature. Babies with severe haemophilia have an increased risk of intracranial haemorrhage and, although there is insufficient evidence to recommend routine caesarean section for these births, it is appropriate to avoid head trauma and to perform imaging of the newborn within the first 24 hours of life.
Individuals with moderate and mild haemophilia (factor VIII levels 1–40%) present with the same pattern of bleeding, but usually after trauma or surgery, when bleeding is greater than would be expected from the severity of the insult. The major morbidity of recurrent bleeding in severe haemophilia is musculoskeletal. Bleeding is typically into large joints, especially knees, elbows, ankles and hips.
Muscle haematomas are also characteristic, most commonly in the calf and psoas muscles. If early treatment is not given to arrest bleeding, a hot, swollen and very painful joint or muscle haematoma develops. Recurrent bleeding into joints leads to synovial hypertrophy, destruction of the cartilage and secondary osteoarthrosis .
Complications of muscle haematomas depend on their location. A large psoas bleed may extend to compress the femoral nerve; calf haematomas may increase pressure within the inflexible
fascial sheath, causing a compartment syndrome with ischaemia, necrosis, fibrosis, and subsequent contraction and shortening of the Achilles tendon.
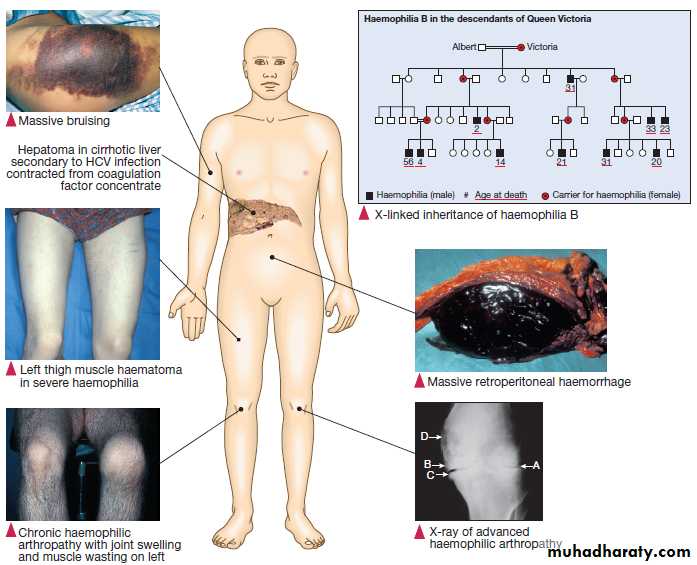
Clinical manifestations of haemophilia.
On the knee X-ray, repeated bleeds have led to broadening of the femoral epicondyles, and there is no cartilage present, as evidenced by the close proximity of the femur and tibia (A); sclerosis (B), osteophyte (C) and bony cysts (D) are present. (HCV =hepatitis C virus)
Management
In severe haemophilia A, bleeding episodes should be treated by raising the factor VIII level, usually by intravenous infusion of factor VIII concentrate. Factor VIII concentrates are freeze-dried and stable at 4°C and can therefore be stored in domestic refrigerators, allowing patients to treat themselves at home at the earliest indication of bleeding. Factor VIII concentrate preparedfrom blood donor plasma is now screened for HBV, HCV and HIV, and undergoes two separate virus inactivation processes during manufacture; these preparations have a good safety record. However, factor VIII concentrates prepared by recombinant technology are now widely available and, although more expensive, are perceived as being safer than those derived from human plasma.
In addition to raising factor VIII concentrations, resting of the bleeding site by either bed rest or a splint reduces continuing haemorrhage. Once bleeding has settled, the patient should be mobilised and physiotherapy used to restore strength to the surrounding muscles. All non-immune potential recipients of pooled blood products should be offered hepatitis A and B immunisation.
The vasopressin receptor agonist DDAVP raises the vWF and factor VIII levels by 3–4-fold, which
is useful in arresting bleeding in patients with mild or
moderate haemophilia A. The dose required for this purpose is higher than that used in diabetes insipidus,
usually 0.3 μg/kg given intravenously or subcutaneously.
Alternatively, the same effect can be achieved by
intranasal administration of 300 μg. Following repeatedadministration of DDAVP, patients need to be monitored
for evidence of water retention, which can result
in significant hyponatraemia. DDAVP is contraindicated
in patients with a history of severe arterial disease
because of a propensity to provoke a thrombotic event.
In addition to treatment ‘on demand’ for bleeding,
factor VIII can be administered 2 or 3 times per week as ‘prophylaxis’ to prevent bleeding in severe haemophilia.
This is most appropriate in children, but its widespread use is limited by the high cost of factor VIII preparations.
New concentrates of factor VIII (and factor IX) will soon add to the treatment options for these conditions.
Complications of coagulation factor therapy
Before 1986, coagulation factor concentrates from human plasma were not virally inactivated with heat or chemicals, and many patients became infected with HIV and hepatitis viruses HBV and HCV. In exposed patients with severe haemophilia, infection with HCV is almost universal, 80–90% have evidence of HBV exposure, and 60% became HIV-positive. Management since 1989, viral inactivation of these blood products has eradicated the risk of viralinfection.
Concern that the infectious agent that causes vCJD
might be transmissible by blood and blood products has been confirmed in recipients of red cell transfusion, and in one recipient of factor VIII.
Pooled plasma products, including factor VIII concentrate,
are now manufactured from plasma collected in
countries with a low incidence of bovine spongiform
encephalopathy.
Another serious complication of factor VIII infusion
is the development of anti-factor VIII antibodies, which
arise in about 20% of severe haemophiliacs. Such antibodies rapidly neutralise therapeutic infusions, making
treatment relatively ineffective.
Infusions of activated clotting factors, e.g. VIIa or factor VIII inhibitor bypass activity (FEIBA), may stop bleeding.
Haemophilia B (Christmas disease)
Aberrations of the factor IX gene, which is also presenton the X chromosome, result in a reduction of the plasma factor IX level, giving rise to haemophilia B. This disorder
is clinically indistinguishable from haemophilia A but is
less common. The frequency of bleeding episodes is
related to the severity of the deficiency of the plasma
factor IX level. Treatment is with a factor IX concentrate,
used in much the same way as factor VIII for haemophilia
A. Although factor IX concentrates shared the problems
of virus transmission seen with factor VIII, they do not
commonly induce inhibitor antibodies (< 1% patients);
when this does occur, however, it may be heralded by
the development of a severe allergic-type reaction.
Von Willebrand disease
A common but usually mild bleeding disorder caused by a quantitative (types 1 and 3) or qualitative (type 2) deficiency ,a protein synthesised by endothelial cells
and megakaryocytes, which is involved in both platelet
function and coagulation. It normally forms a multimeric
structure which is essential for its interaction with subendothelial collagen and platelets . vWF acts as a carrier protein for factor VIII, to which it is non-covalently bound; deficiency of vWF lowers the plasma factor VIII level. vWF also forms bridges between platelets and subendothelial components (e.g. collagen), allowing platelets to adhere to damaged vessel walls; deficiency of vWF therefore leads to impaired platelet plug formation.
Blood group antigens (A and B) are expressed on vWF, reducing its susceptibility to proteolysis; as a result, people with blood group O have lower circulating vWF levels than individuals with non-O groups. This needs to be borne in mind when making a diagnosis of von Willebrand disease. Most patients with von Willebrand disease have a type 1 disorder, characterised by a quantitative decrease in a normal functional protein. Patients with type 2 disorders inherit vWF molecules that are functionally abnormal. The type of abnormality depends on the site of the mutation in the vWD gene.
Patients with mutations in platelet binding have type 2A disease those with mutations in the platelet glycoprotein Ib binding site have type 2B, those with mutations in the factor VIII binding site have type 2N disease, and those
with other abnormalities in platelet binding have type 2M.
The gene for vWF is located on chromosome 12 and the disease is usually inherited as an autosomal dominant, except in cases of type 2N and type 3, when it is recessive.
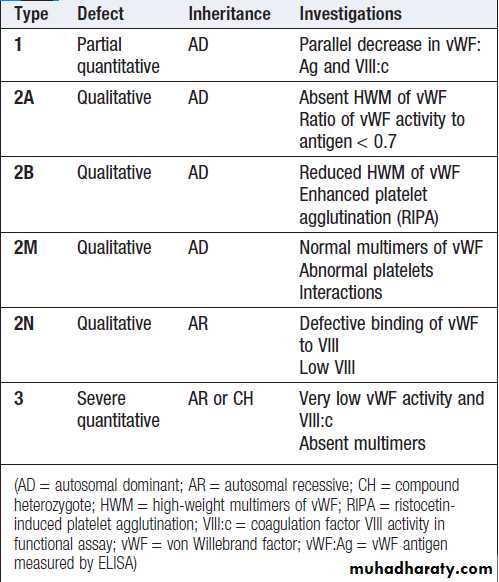
Classification of von Willebrand disease
Clinical featuresPatients present with haemorrhagic manifestations
similar to those in individuals with reduced platelet
function. Superficial bruising, epistaxis, menorrhagia
and gastrointestinal haemorrhage are common. Bleeding episodes are usually much less common than in severe haemophilia and excessive haemorrhage may
only be observed after trauma or surgery. Within a
single family, the disease has variable penetrance, so
that some members may have quite severe and frequent
bleeds, whereas others are relatively asymptomatic.
Investigations
The disorder is characterised by reduced activity of vWF and factor VIII. The disease can be classified using a combination of assays which include functional and
antigenic measures of vWF, multimeric analysis of the
protein, and specific tests of function to determine
binding to platelet glycoprotein Ib (RIPA) and factor VIII.
In addition, analysis for mutations in the vWF gene is informative in most cases.
Management
Many episodes of mild haemorrhage can be successfully
treated by local means or with DDAVP, which raises the
vWF level, resulting in a secondary increase in factor VIII.
Tranexamic acid may be useful in mucosal bleeding. For more serious or persistent bleeds, haemostasis can be achieved with selected factor VIII concentrates which contain considerable quantities of vWF in addition to factor VIII.
Young children and patients with severe arterial
disease should not receive DDAVP, and patients with
type 2B disease develop thrombocytopenia which may
be troublesome following DDAVP.
Bleeding in type 3 patients responds to nothing apart from concentrate.
Rare inherited bleeding disorders
Severe deficiencies of factor VII, X and XIII occur as autosomal recessive disorders. They are rare but are associated with severe bleeding. Typical features include haemorrhage from the umbilical stump and intracranial haemorrhage.Factor XIII deficiency is typically associated with female infertility.
Factor XI deficiency may occur in heterozygous or homozygous individuals. Bleeding is very variable and is not accurately predicted by coagulation factor levels.
In general, severe bleeding is confined to patients with
levels below 15% of normal.
Acquired bleeding disorders
DIC is an important cause of bleeding which begins with exaggerated and inappropriate intravascular coagulation
Liver disease
In severe parenchymal liver disease ,bleeding may arise from many different causes. Pathological sources of potential major bleeding, such as oesophageal varices or peptic ulcer, are more likely. There is reduced hepatic synthesis: for example, of factors V, VII, VIII, IX,
X, XI, prothrombin and fibrinogen.
Clearance of plasminogen activator is reduced. Thrombocytopenia may occur secondary to hypersplenism in portal hypertension.
In cholestatic jaundice, there is reduced vitamin K absorption, leading to deficiency of factors II, VII, IX and X. Treatment with plasma products or platelet transfusion should be reserved for acute bleeds or to cover interventional procedures such as liver biopsy.
Vitamin K deficiency can be readily corrected with parenteral administration of vitamin K.
Renal failure
The severity of the haemorrhagic state in renal failure is proportional to the plasma urea concentration. Bleeding manifestations are those of platelet dysfunction, with GI haemorrhage being particularly common. The causes are multifactorial, including anaemia, mild thrombocytopenia and the accumulation of LMW waste products, which inhibit platelet function.Treatment is by dialysis. Rarely, in severe or persistent bleeding, platelet concentrate infusions and red cell transfusions are indicated.
Increasing the concentration of vWF, either by cryoprecipitate or by DDAVP, may promote haemostasis.
Inherited abnormalities of coagulation
Several inherited conditions predispose to VTE, and have several points in common that are worth noting:• None of them is strongly associated with arterial thrombosis.
• All are associated with increased incidence of adverse outcome of pregnancy, including recurrent early fetal loss, but there are no data to indicate that any specific intervention changes that outcome.
• Apart from in antithrombin deficiency and homozygous factor V Leiden, most carriers of these genes will never have an episode of VTE; if they do, it will be associated with the presence of an
additional temporary risk factor.
• There is little evidence that detection of these abnormalities predicts recurrence of VTE.
• None of these conditions per se requires anticoagulants. Patients who are deemed to be at high risk of thrombosis, e.g. those with antithrombin deficiency in pregnancy, should receive treatment or prophylactic doses of heparin to cover the period of risk only.
Antithrombin deficiency
Antithrombin (AT) is a serine protease inhibitor
(SERPIN) which inactivates the activated coagulation
factors IIa, IXa, Xa and XIa.
Heparins, fondaparinux and idraparinux achieve their therapeutic effect by potentiating the activity of AT. Inherited as an autosomal dominant; homozygosity is not compatible with life. Around 70% of affected individuals will have an episode of VTE before the age of 60 years and the relative risk for thrombosis compared with the background population is 10–20.
Pregnancy is a high-risk period for VTE and this requires fairly aggressive management with doses of LMWH which are greater than the usual prophylactic doses (≥ 100 U/kg/day). AT concentrate (either plasma derived or recombinant) is available; this is required for cardiopulmonary bypass and may be used as an adjunct to heparin in surgical prophylaxis.
Protein C and S deficiencies
Protein C and S are vitamin K-dependent naturalanticoagulants involved in switching off coagulation
factor activation (factors Va and VIIIa) and thrombin
generation .Inherited deficiency of either protein C or S results in a prothrombotic state with a fivefold relative risk of VTE compared with the background population.
Factor V Leiden
Factor V Leiden results from a gain-of-function, single base pair mutation which prevents the cleavage and hence inactivation of activated factor V.
This results in arelative risk of venous thrombosis of 5 in heterozygotes and 50 or more in rare homozygotes.
Prothrombin G20210A
This gain-of-function mutation in the non-coding 3′ endof the prothrombin gene is associated with an increased
plasma level of prothrombin. It is present in about 2%
of Northern Europeans but is rare in native populations
of Korea, China, India and Africa. In the heterozygous
state, it is associated with a 2–3-fold increase in risk of
VTE compared with the background population.
Antiphospholipid syndrome
Antiphospholipid syndrome (APS) is a clinico pathological
entity in which a constellation of clinical conditions,
alone or in combination, is found in association with a
persistently positive test for an antiphospholipid antibody.
The antiphospholipid antibodies are heterogeneous
and typically are directed against proteins which
bind to phospholipids .
Although causal roles for these antibodies have been proposed, the mechanisms underlying the clinical features of APS are not clear.
Antiphospholipid antibody
In clinical practice, two types of test are used, detect:1. Antibodies which bind to negatively charged
phospholipid on an ELISA plate
(anticardiolipin antibody test)
2. Those which interfere with phospholipid-dependent
coagulation tests like the APTT or the dilute Russellviper venom time
(DRVVT; called a lupus anticoagulant test).
The term antiphospholipid antibody encompasses both
a lupus anticoagulant and an anticardiolipin antibody;
individuals may be positive for one or both of these activities.
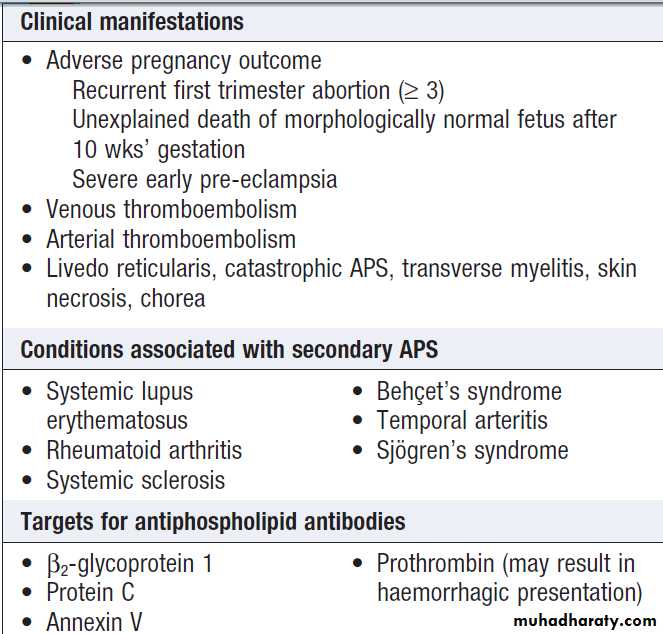
Antiphospholipid syndrome
Clinical features and managementAPS may primary or in association with conditions shown in Box, most typically SLE (secondary APS). Most patients present with a single manifestation and APS is now most frequently diagnosed in women with adverse outcomes of pregnancy. It is extremely important to make the diagnosis in patients with APS, because it affects the prognosis and management of arterial thrombosis, VTE and pregnancy. Arterial thrombosis, typically stroke, associated with APS should be treated with warfarin, as opposed to aspirin. APS-associated VTE is one of the situations in which the predicted recurrence rate is high enough to indicate long-term anticoagulation after a first event. In women with APS, it is likely that intervention with heparin and possibly aspirin increases the chance of a successful pregnancy outcome.
Disseminated intravascular coagulation (DIC)
May complicate a range of illnesses .It is characterised by systemic activation of the pathways involved in coagulation and its regulation. This may result in the generation of intravascular fibrin clots causing multiorgan failure, with simultaneous coagulation factor and platelet consumption causing bleeding. The systemic coagulation activation is induced either through cytokine pathways, which are activated as part of a systemic inflammatory response, or by the release of procoagulant substances such as tissue factor.In addition, suboptimal function of the natural anticoagulant pathways and dysregulated fibrinolysis contribute to DIC.
There is consumption of platelets, coagulation factors (notably factors V and VIII) and fibrinogen.
The lysis of fibrin clot results in production of fibrin degradation products (FDPs), including D-dimers.
Investigations
DIC should be suspected when any of the conditionslisted in Box are met. Measurement of coagulation
times (APTT), along with fibrinogen, platelet count and FDPs, helps in the assessment of prognosis and aids clinical decision-making with regard to both bleeding and thrombotic complications.
Management
Therapy is primarily aimed at the underlying cause.
These patients will often require intensive care to deal
with concomitant issues, such as acidosis, dehydration,
renal failure and hypoxia.
Blood component therapy, such as fresh frozen plasma, cryoprecipitate and platelets, should be given if the patient is bleeding or to cover interventions with high bleeding risk, but should not be prescribed routinely based on coagulation tests and platelet counts alone. Prophylactic doses of heparin should be given, unless there is a clear contraindication.
Established thrombosis should be treated cautiously
with therapeutic doses of unfractionated heparin, unless
clearly contraindicated.
Patients with DIC should not, in general, be treated with antifibrinolytic therapy, e.g. tranexamic acid.

(ISTH = International Society for Thrombosis and Haemostasis)
Disseminated intravascular coagulation
Haemostasis and thrombosis in old age
Thrombotic thrombocytopenic purpura (TTP)Like 1. DIC and 2. heparin-induced thrombocytopenia, TTP is a disorder in which thrombosis is accompanied by paradoxical thrombocytopenia.
TTP is characterised by
a pentad of findings, although few patients have all five components:• thrombocytopenia
• microangiopathic haemolytic anaemia
• neurological sequelae
• fever
• renal impairment.
It is an acute autoimmune disorder mediated by antibodies against ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type-1 motif).
This enzyme normally cleaves vWF multimers to produce normal functional units, and its deficiency results in large vWF multimers which cross-link platelets. The features are of microvascular occlusion by platelet thrombi affecting key organs, principally brain and kidneys.
It is a rare disorder (1 in 750 000 per annum), which may occur alone or in association with drugs (ticlopidine, ciclosporin), HIV, shiga toxins and malignancy.
It should be treated by emergency plasma exchange.
Corticosteroids, aspirin and rituximab also have a role in management. Untreated mortality rates are 90% in the first 10 days, and even with appropriate therapy, the mortality rate is 20–30% at 6 months.