Pediatric surgery

Attendance: Obligatory
Type of lecture: theory
Place : Hall no.4 college of medicine – Al-mustansiriyah University .
Date : Tuesday 7th of March 2017.
Time : 9:00 – 10:00 AM.
Students: 4th year / college of medicine / Mustansiriyah University
By : Dr. Ali E. Joda M.B.Ch.B. - F.I.C.M.S. - pediatric surgeon
Mobile no. : 07725465090
E. mail : alieagle677@yahoo.com
Topics :-
Hirschsprung's disease.Anorectal malformation.
Inguinal hernia.
Infantile hydrocele.
Undescended testes.
Objectives:-
What congenital surgical problem the neonate may developed? How can be treated?
The significance of inguinoscrotal swelling in pediatric age group. One should not be neglected or treated lately.
To correct some false ideas that repair of inguinal hernia can be delayed at older age.
There should be good attention to the presence of two testes in the scrotum.
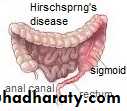
Hirschsprung's disease
(congenital megacolon , colonic aganglionosis)
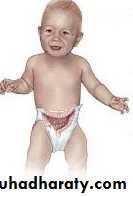
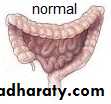
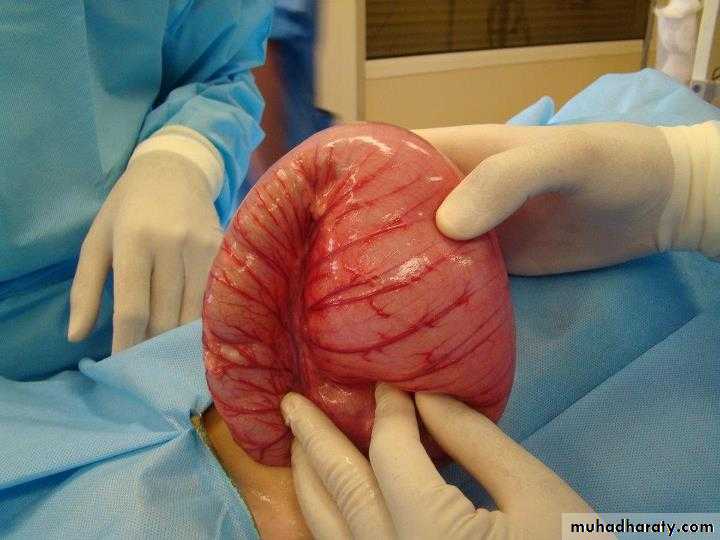
Is a functional intestinal obstruction due to congenital absence of ganglion cells in the recto-sigmoid region & extend proximally for varying length result in failure of propagation of peristalsis & absence of relaxation of internal anal sphincter.so there will be dilated proximal colon (mega) , narrow distal colon , the aganglionosis may be in a short segment , long segment or even total colon.
Incidence 1 in 5000 newborn.
70-80 % in males.25% with family history of the same disease & having other associated anomalies.
Etiology & genetics :-
The ganglion cells are derived from neural crest of embryo, which undergo migration through GIT from proximal to distal after differentiation into mature ganglion cells. In infant with Hirschsprung's disease (HD,) this process is disturbed & the ganglion cells are absent in distal bowel.
The genesis of Hirschsprung's disease is multifactorial (genetic + environmental).
The most common identified gene is RET proto-oncogen. Other genes involved may be GDNF (glial cell line derived neurotrophic factor), END3, ENDB (endotheline) , S10X10 gene , S1P1 gene.
Diagnosis:-
Clinical presentation:-
Neonatal intestinal obstruction: (failure or delayed passage of meconium, abdominal distension, & vomiting).
Chronic constipation: some patients present in later childhood with chronic constipation especially those with short segment HD, they usually had history of delayed passage of meconium for more than 48 hr., failure to thrive, malnutrition, hypoproteinemia, anemia, gross abdominal distension, dependence on laxatives & enemas, but without encopresis or soiling.
Enterocolitis : 10% of children with HD present with fever , abdominal distension , offensive diarrhea due to Hirschsprung's disease associated enterocolitis (HAEC).stasis of fecal materials in the colon lead to bacterial overgrowth & invasion of Clostridium difficilli & Rota virus to the bowel wall causing secondary infection ( enterocolitis )which may cause life threatening toxic megacolon.
Associated anomalies :
Down syndrome (trisomy 21 )
Neurocristopathy syndromes:-
Waardenberg –shah syndrome.
Goldberg – shprintzen syndrome.
Yemenite deaf blind hypopigmentation syndrome.
Piebaldism.
SLOS Smith-Lemli Optiz syndrome.
MEN2 (multiple endocrine neoplasia).
Haddad syndrome (ondine curse).
Rectal examination :-
Hypertonic anal sphincter (long gripping of index finger).
Empty rectum.
Or explosive discharge of fluid stool & gases suggestive of enterocolitis.
Investigation :-
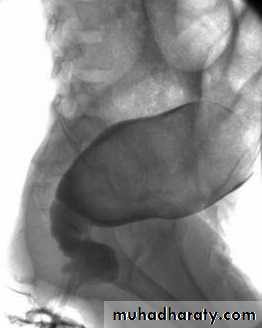
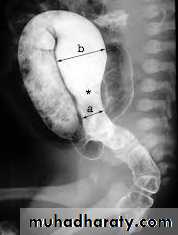
Barium enema :
(Narrow segment distally, transitional zone, dilated segment proximally).
Recto-sigmoid ratio less than 1 (transvers diameter of rectum / transverse diameter of sigmoid).
Retention of contrast material in the colon more than 24hr post filming.
Neonate with HD may have false –ve result.
Anorectal manometry: recto-anal inhibitory reflex is defined as reflex relaxation of internal anal sphincter in response to rectal distension. This is normally present in normal children & absent in HD. But it is not reliable in neonate because of immaturity of the reflex & artifact created by movement & crying of the baby.
Rectal biopsy: give the definitive diagnosis of HD by histological confirmation of (absence of ganglions, hypertrophy of nerve fibers, increase staining of acetyl choline esterase) in both submucosal plexus (meisner) & meyenteric plexus (aurbach). It can be done either by suction rectal biopsy (mucosa & submucosa) or by full thickness rectal biopsy.
Pre- operative management :-
Resuscitation for neonate with intestinal obstruction or children with enterocolitis by I.V.F , electrolytes , broad spectrum antibiotics , nasogastric tube decompression , digital rectal stimulation or colonic irrigation with normal saline enema. Emergency stoma is indicated if the above measures failed.Surgical treatment for Hirschsprung's disease:-
three stages (Usually):-
Stoma formation: as an emergency procedure or electively performed to protect future colonic anastomosis.
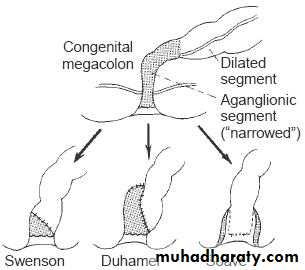
Pull-through procedure: resection of aganglionic segment & bringing the normally innervated bowel down to be anastomosed with the low rectum or anus. The Pull-through procedures may be :-
Swenson pull-through.
Soave ( endorectal pull-through ).
Duhamel (retrorectal pull-through).
Rehbein pull-through.
Trans-anal pull-through.
Laparoscopic pull-through.
Closure of stoma.
Two stages (standard): colostomy + pull-through
In 1st stage then closure of stoma in 2nd stage.
One stage (for selected cases).
Anorectal malformation
The baby born with absence of anal orifice (imperforate anus), agenesis of anal canal with variable development of sphincteric muscle & it is often associated with fistulous connection to the urinary tract in male or genital tract in female with high possibility of associated anomalies.
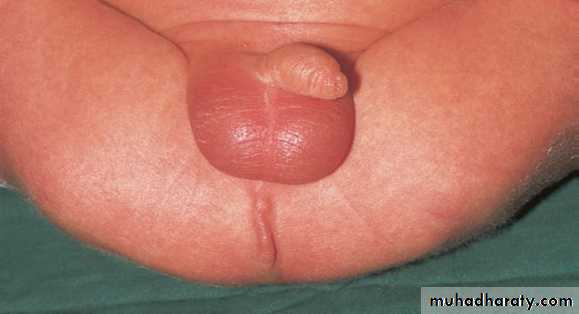

Classification :-
Male :-Imperforate anus with cutaneous perineal fistula.
Imperforate anus with Recto-urethral fistula (most common).
Imperforate anus with recto-vesical fistula (bladder neck).
Imperforate anus without fistula.
Rectal atresia.
Female :-
Imperforate anus with cutaneous perineal fistula.
Imperforate anus with recto-vestibular vestibular fistula (most common).
Imperforate anus without fistula.
Rectal atresia.
Cloaca (common orifice for urethra, vagina, & rectum).
Incidence 1 in 5000 live birth, no specific cause, some genetic predisposition.
These anomalies classified into high & low types based on the position of the terminal bowel in relation to the levator ani or pelvic floor in order to decide the management and predict the final outcome.Low types are easily treated with an excellent functional prognosis.
High types are difficult to manage & often associated with other anomalies, and have a poor functional prognosis.
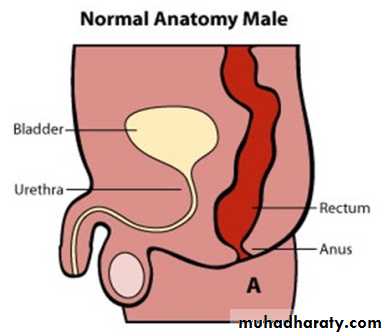
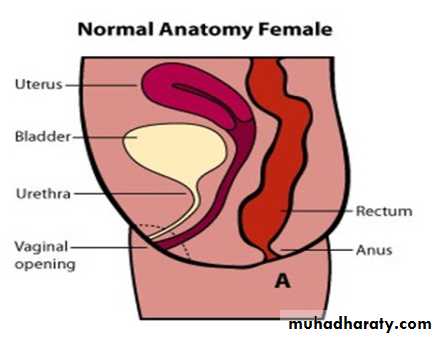
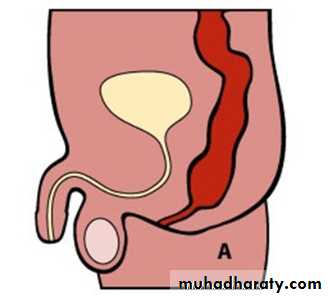
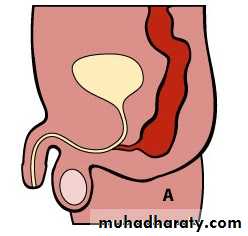
Male female Perineal fistula urethral fistula
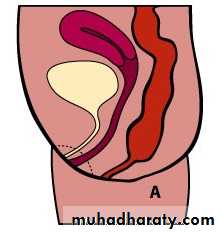
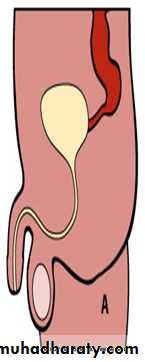
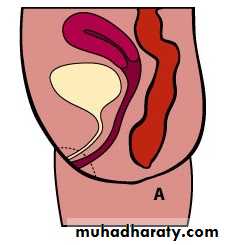
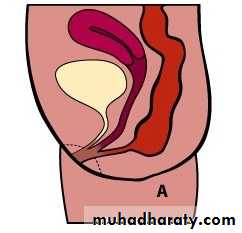
Vestibular fistula recto-vesical fistula imp.anus without fistula cloaca
Associated anomalies :-
Most babies with anorectal malformations have one or more other associated abnormalities that affect other systems. Some of them are simple, incidental findings but others, such as cardiovascular defects, may be life threatening.
VACTERL association:
V – Vertebral anomaliesA – Anal atresia
C – Cardiovascular anomalies
T – Tracheoesophageal fistula
E – Esophageal atresia
R – Renal (kidney) and/or radial anomalies
L – Limb defects
Diagnosis & management :-
The diagnosis of absent anal orifice is usually obvious by inspection of perineum & external genitalia but such anomaly may be missed & not discovered until abdominal distension begins or the baby not passing meconium. The initial assessment of neonate with imperforate anus is to answer the followings :-
Q / at which level the rectum is ended in relation to sphincter muscle?
Q / where is the fistula ?
Q/ can the anomaly be corrected by single perineal surgery or need initial colostomy then definitive surgery later on?
Q / is there any associated anomalies needs immediate or subsequent correction?
Investigations :-
(Invertogram) or (prone cross table lateral film): after 24 hr from birth , the baby is held upside down(invertogram) or placed in prone genu-pectroral position for 3 min., then lateral view X-ray is taken centered on greater trochanter & P-C line (pubis-coccyx) with marker applied at the natal cleft on the site of anal dimple. If the gas shadow in rectum is proximal to P-C line = high type imperforate anus & need initial colostomy while if the gas shadow in rectum is distal to P-C line = low type imperforate anus & no need for colostomy, just perineal anoplasty. Or we measure the bowel – skin distance, if more than 2 cm = high type need colostomy, if less than 2 cm = low type , no colostomy is needed just perineal anoplasty.
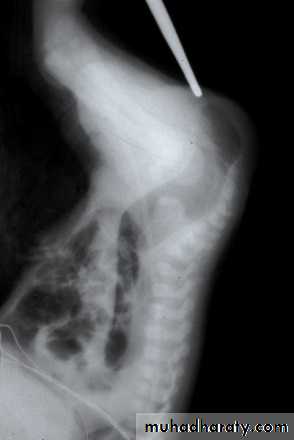
Low type high type meconium per urethra
Examination of urine in male: passage of meconium per urethra (meconuria) or air per urethra (pneumatuira) indicate high type impeforate anus with recto-urethral fistula or recto-vesical fistula that need initial colostomy.
Plane X-ray showing presence of gas in viscera other than GIT like bladder (recto-vesical fistula) ,or huge colonic gas shadow occupying half the width of abdomen in Congenital Pouch Colon syndrome.
Dye study of the fistula: to distinguish between recto-vestibular & ano-vestibular fistula.
Ultrasound: perineal ultrasound for measuring the distance between skin & rectal pouch as alternative to invertogram.
C.T Scan.
NO decision should be made about doing colostomy or primary anorectoplasty before age of 24 hr. because there should be significant intraluminal pressure for meconium to be forced through fistula orifice.( passing meconium through fistula orifice in perineum indicate low type , while passing meconium with the urine indicate high type).
Radiological evaluation should not be done before age of 24 hr. because the rectum is still collapsed & it requires significant intraluminal pressure to overcome the muscle tone surrounding distal rectum. So during the 1st 24 hr. after birth the baby should receive I.V.F , antibiotics , investigated for associated anomalies by Cardiac Echo , continuity of esophagus , renal ultrasound , spinal ultrasound , X-ray of the lumbar spine & sacrum.
Colostomy :-
Divided descended colostomy is ideal for anorectal malformation :-
For complete diversion of stool.
Permit doing distal colostogram to give us the detailed anatomy of the defect before anorectoplasty.
Better protection of the anorectoplasty repair.
Loop colostomy is not preferable :-
Because it permit passage of stool from proximal stoma into distal bowel then through fistula into urinary tract leading to UTI.
It causes collection of stool in distal rectal pouch , fecal impaction , dilatation of rectum , irreversible hypomotility.
Colostomy prolapse is more common in loop type.
Transvers or right sided colostomy is not preferable :-
Because there will be long segment of defunctional distal colon, atrophied colon & colostomy diarrhea.
The mechanical cleaning of distal colon before definitive repair is much more difficult.
If there is large fistula with the urethra, there will be significant absorption of the urine in the colon before it escape through distal stoma leading to metabolic acidosis.
Mid or lower sigmoidostomy is not preferable :-
Because it will interfere with the mobilization of rectum during definitive repair of high type imperforate anus.
Operative repair of ano-rectal malformations:-
Simple anal cut-back.
V-Y anoplasty.
Anal transposition.
Limited Posterior Ano-Rrectoplasty PARP.
Anterior Sagittal Ano-Rrectoplasty ASARP.
Posterior Sagittal Ano-Rectoplasty PSARP.
Inguinal hernia
Embryology:-The processus vaginalis is a peritoneal diverticulum that extends through internal inguinal ring & dragged with the testis into the scrotum. The portion of the processus vaginalis that enveloping the testis is called tunica vaginalis. The remainder of the processus vaginalis in the inguinal canal is obliterated. But in 20% of population the processus vaginalis may remain patent asymptomatically throughout the life.
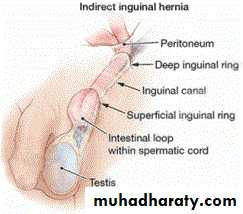
Anatomy:-
The inguinal hernia comprises of:Covering: layers of inguinal canal (ext.spermatic fasica, cremasteric muscle & fascia, int.spermatic fascia).
Sac: a protruding pouch of peritoneum consist of mouth, neck, body & fundus.
Content: small & large bowel, ovaries, fallopian tubes, omentum, part of urinary bladder, appendix, Meckel's diverticulum).
The hernia sac is formed from patent processus vaginalis & lies antero-medial to spermatic cord structures. The sac may be thin or thick depending on age of the patient, duration of hernia, & whether incarceration has occurred or not.
Epidemiology:-
Nearly all inguinal hernia in children is congenital indirect type (99%).
The direct one is rare less than 1% & they believed to be acquired. It is more common in connective tissue diseases & conditions with increased intra-abdominal pressure or post inguinal surgery.
Incidence of indirect inguinal hernia is 1-2 per 100 live births.
Inguinal hernia repair is the most common surgery performed by pediatric surgeons.
Male: female = 9: 1
60% in the right side, 25% in the left side, & 15 % bilateral.
More in premature than full term.
Inguinal hernia in children is a high risk hernia, 60% risk of incarceration in the first 6 months of age.
Timing of repair: we recommend repair of inguinal hernia soon after diagnosis.
90 % of complications can be avoided if hernia repairs done within 1 month of diagnosis.
Presentation:-
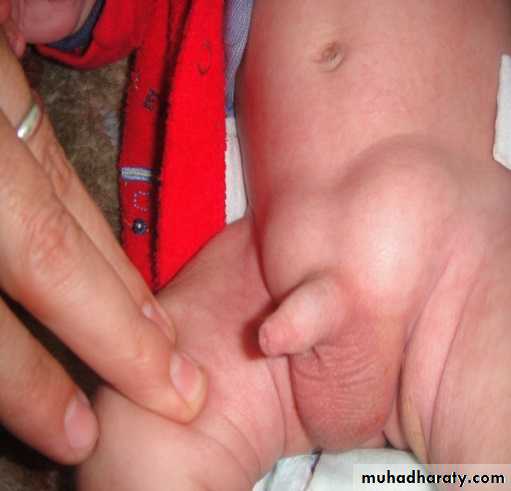
Intermittent swelling overlying external inguinal ring usually painless, evident during crying, coughing, straining & may descend to scrotum. It reduced spontaneously during sleeping & relaxation or can be reduced manually by upward & posterior pressure directly on the mass. 30% of inguinal hernias have the first presentation as incarcerated hernia. If the parents gave suggestive history but on examination there is no hernia, palpation of thickened cord compared with the other side & sensation of silk glove sign (double layers of empty hernia sac) can help in the diagnosis. Or simply you can repeat the examination again when bulging appears. For older children, ask them to stand, jump up & down to reveal the hidden inguinal hernia. The opposite side should be examined & both testes confirmed to be in the scrotum.
Incarcerated hernia: omentum or a loop of bowel becomes trapped in the hernia sac & not reduced by manipulation.
Strangulated hernia: ischemia to the contents of hernia sac.
Obstructed hernia: hernia causing intestinal obstruction.
In these conditions, the infant presented with crying, tense tendered swelling in the groin, irreducible without impulse on crying, colicky abdominal pain, vomiting, abdominal distension, constipation. if ischemia developed, there will be edema, redness, hotness, indurations overlying the lump & signs of peritonitis.
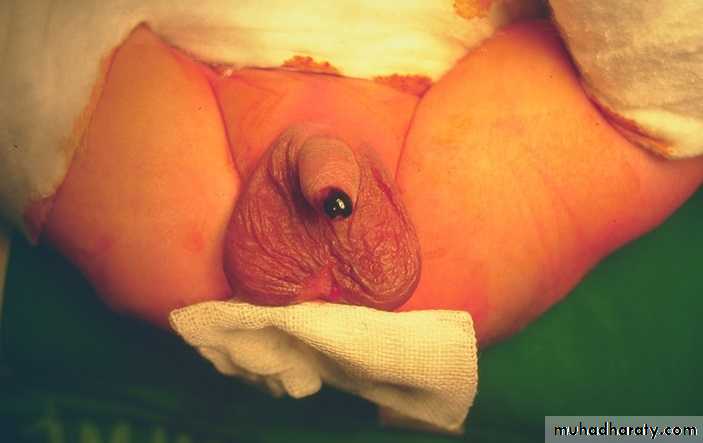
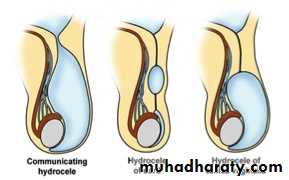
Hydrocele :- is a collection of peritoneal fluid in the tunica vaginalis around the testes due to patent processus vaginalis.
Types:-
Non-communicated: fluid just in the tunica vaginalis without communication with the peritoneal cavity.
Communicated: fluid in the tunica & processus vaginalis communicated with the peritoneal cavity, the same as hernia.
Hydrocele of the cord (male), hydrocele of canal of Nuck (female): encysted fluid.
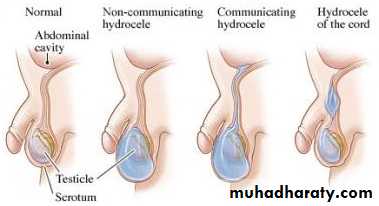
Unilateral or bilateral hydrocele are common in the first few months of life, they are usually small – moderate in size, asymptomatic & have strong tendency to resolve spontaneously in the first year of life, it require no treatment just observation by the family & surgery is indicated only if hydrocele is not resolved after the age of 1 year or if it is very large or increasing in size that causing pressure on the testis.
Communicated hydrocele can be differentiated from inguinal hernia by the following features:-
Cystic.
Irreducible.
Trans-illuminable.
No impulse on crying.
Difficult to separate it from the testis.
Can get above its proximal limits.
Surgery:-
Surgical repair of inguinal hernia should be done once they diagnosed because of the high risk of complications & it is better to repair inguinal hernia for a premature baby before discharging him from neonatal care unit. While the usual infantile hydrocele does not need surgery in the first year of life. The surgical procedure for both hernia & hydrocele is the same & done as outpatient surgery (discharged on the same day) except for premature baby who need observation in hospital for 24 hr after repair because of risk of apnea after G.A (general anesthesia).
For irreducible inguinal hernia, manual reduction (taxis procedure) is usually successful in reducing the herniated viscera in about 80% of cases but still surgical repair is required later on electively. This is usually done by:-
Sedation (diazepam, midazolam, chloral hydrate).
Elevation of the lower half of the body.
Application of ice packs on the swelling to reduce edema.
Experienced doctor push the mass upward & posteriorly by one hand with the other hand supporting the roof of inguinal canal.
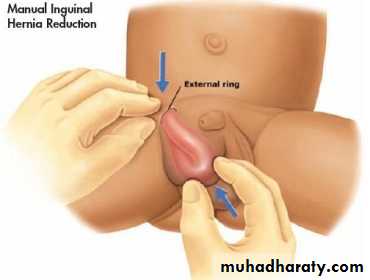
If not reduced after several attempts of manual reduction, urgent surgery is indicated.
Complications of inguinal hernia:-
Incarceration, intestinal obstruction, strangulation (ischemic necrotic bowel).
Testicular atrophy due to compression of testicular blood vessels by hernia sac if prolong.
Ischemia & gangrene of the testis if there is acute severe testicular compression.
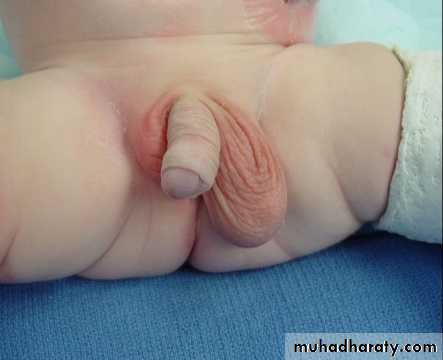
Undescended testes
The second most common problem in pediatric surgery after inguinal hernia.Risk of UDT:-
Infertility: in bilateral more than in unilateral.
Malignancy: the risk for inguinal canal UDT is 1%, & for intra- abdominal UDT is 5%.
Testicular torsion.
Testicular trauma.
Infection.
Psychosocial effect.
Incidence: 3% of fullterm, 33% of preterm. The majority will descend in the first year of life so the incidence at the age of 1 year will be 1%. Their descent after 1 year age is unlikely.
True UDT: is a testis that stopped anywhere along the normal pathway of descent in retroperitoneum between kidney & scrotum.
Ectopic testis: is a testis that left the normal pathway of descent & fixed in abnormal position (in inguinal region, perineum, femoral, contralateral scrotum).
Retractile testis: is normally descended testis that pulled up into inguinal canal as a result of hyperactive cremasteric reflex.
Iatrogenic UDT (ascending testis): is previously normal descended testis & then have been trapped in scar tissue of previous inguinal surgery.
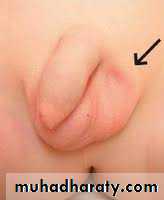
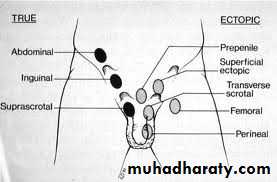
Classification:-
Palpable & non palpable UDT.
Intra -abdominal UDT & inguinal canal UDT.
The intra-abdominal UDT either with opened ring or with closed ring (internal inguinal ring).
Absent or vanishing testis is called monorchia.
Absent both testes is called anorchia.
Diagnosis:-
The cremasteric reflex is weak in the 1st 2 year of life so if the testis was in the scrotum at birth then become impalpable in scrotum, it is retractile testis rather than UDT.
Gentle physical examination in warm room, supine position, & squatting position.
The epsilateral hemiscrotum is atrophic with little rougal folds.
In case of monorchia, there will be hypertrophy of the solitary testis.
Gentle pressure on mid abdomen may help to push the intra-abdominal testis into inguinal canal.
Bilateral UDT can be differentiated from anorchia by HCG stimulation test.
Investigation:-
Ultrasound
CT scan
MRI
Laparoscopy which has 95% sensitivity for locating a testis or confirming its absence.
Treatment:-
Hormonal therapy for UDT is controversial.Buserline (LH-releasing hormone agonist).
Pregnil ( HCG-analoque).
Timing of surgical repair:-
At the age of 1 year.
Earlier if associated with inguinal hernia.
Orchiopexy : fixation of testis in the subdartous pouch of hemiscrotum.
Orchiectomy: if the testis is abnormally soft tissue, rudimentary or in case of impossible orchiopexy especially in post pubertal intra- abdominal testis because of risk of malignancy.
(((((((((((((((((((((((((((((((((((((((((((((End))))))))))))))))))))))))))))))))))))))))))))))))))

7th of March 2017