
Hussien Mohammed Jumaah
CABMLecturer in internal medicine
Mosul College of Medicine
2016

learning-topics
Clinical biochemistryand metabolism
There is a worldwide trend towards increased use of
laboratory-based diagnostic investigations, and biochemical investigations in particular.In the health-care systems of developed countries, it has been estimated that 60–70% of all critical decisions taken in regard to patients, and over 90% of data stored in electronic medical records systems, involve a laboratory service or result.
BIOCHEMICAL INVESTIGATIONS
There are three broad reasons why a clinician mayrequest a biochemical laboratory investigation:
• to screen an asymptomatic subject for the presence
of disease
• to assist in diagnosis of a patient’s presenting
complaint
• to monitor changes in test results, as a marker of
disease progression or response to treatment.
Contemporary medical practice has become increasingly
reliant on laboratory investigation, and in particular,
on biochemical investigation.
This has been associated with extraordinary improvements in the analytical capacity and speed of laboratory instrumentation and the following operational trends:
• Large central biochemistry laboratories feature
extensive use of automation and information
technology. Specimens are transported from clinical
areas to the laboratory using high-speed transport
systems (such as pneumatic tubes) and identified
with machine-readable labels (such as bar codes).
Laboratory instruments have been miniaturised and
integrated with robot transport systems to enable
multiple rapid analyses of a single sample.
Statistical process control techniques are used to assure the quality of analytical results, and increasingly to monitor other aspects of the laboratory, such as the time taken to complete the analysis (‘turn-around time’).
• Point-of-care testing (POCT) brings selected laboratory analytical systems into clinical areas, to the patient’s bedside or even connected to an individual patient. These systems allow the clinician to receive results almost instantaneously for immediate treatment of the patient, although often with less precision or at greater cost than using a central laboratory.
• The diversity of analyses has widened considerably
with the introduction of many techniques borrowed
from the chemical or other industries .
Good medical practice involves the appropriate
ordering of laboratory investigations and correct interpretation of test results. Many laboratoryinvestigations can be subject to variability arising from
incorrect patient preparation (for example, in the fasting
or fed state), timing of sample collection (for example,
in relation to diurnal variation of hormone levels, or
dosage regimens for therapeutic agents), analytical
factors (for example, serum versus plasma; use of the
correct anticoagulant, or POCT versus central analysis)
or artefact (for example, taking a venous sample proximal
to the site of an intravenous infusion).
It is therefore important for clinical and laboratory staff to communicate effectively and for clinicians to follow local recommendations concerning collection and transport of samples in the appropriate container and with appropriate labelling.
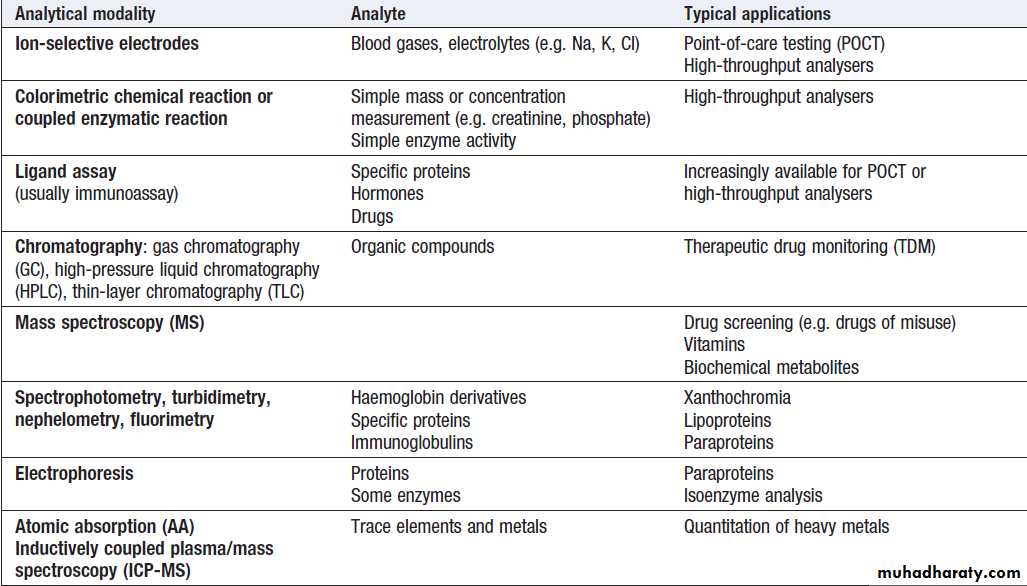

Inherited and somatic cell mutations
Genetic polymorphismsVariations in rates of drug metabolism
Microbial diagnosis
Range of analytical modalities used in the clinical biochemistry laboratory
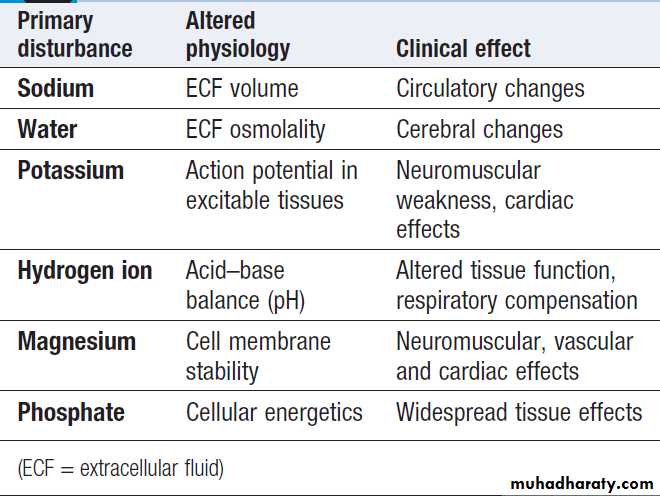
Manifestations of disordered water,
electrolyte and acid–base statusWater and electrolyte distribution
The following basic concepts are relevant to understandingthe origin, consequences and therapy of many of
the fluid and electrolyte disturbances discussed.
In a typical adult male, total body water (TBW) is
approximately 60% of body weight (somewhat more for
infants and less for women). For an average individual, TBW is about 40 L. Approximately 25 L is located inside
cells (the intracellular fluid or ICF), while the remaining
15 L is in the extracellular fluid (ECF) compartment.
Most of the ECF (approximately 12 L) is interstitial
fluid, which is within the tissues but outside cells, whereas the remainder (about 3 L) is in the plasma compartment.
Figure illustrates some of the major differences
in composition between the main body fluid compartments.
The dominant cation in the ICF is potassium, while the dominant cation in the ECF is sodium. Phosphates
and negatively charged proteins constitute the
major intracellular anions, while chloride and, to a lesser
extent, bicarbonate dominate the ECF anions.
An important difference between the plasma and interstitial compartments of the ECF is that only plasma contains significant concentrations of protein.
The major force maintaining the difference in cation
concentration between the ICF and ECF is the sodium–potassium pump (Na,K-activated ATPase), which is
present in all cell membranes. Maintenance of the cation
gradients across cell membranes is essential for many
cell processes, including the excitability of conducting
tissues such as nerve and muscle. The difference in
protein content between the plasma and the interstitial
fluid compartment is maintained by the impermeability
of the capillary wall to protein. This protein concentration
gradient (the colloid osmotic, or oncotic, pressure
of the plasma) contributes to the balance of forces across
the capillary wall that favour fluid retention within the
plasma compartment.
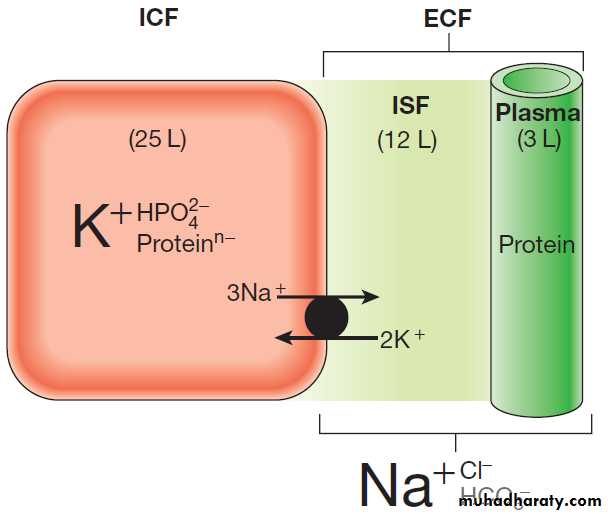
Fig. Normal distribution of body water and electrolytes.
Schematic representation of volume (L = litres) and composition (dominantionic species only shown) of the intracellular fluid (ICF) and extracellular
fluid (ECF) in a 70 kg male. The main difference in composition between
the plasma and interstitial fluid (ISF) is the presence of appreciable
concentrations of protein in the plasma but not the ISF.
Investigation of water and electrolytes
The most common biochemical test in plasma is calledthe urea and electrolytes (U&E) test in some parts of the
world, and the electrolytes/urea/creatinine (EUC) in
others.
A guide to its interpretation is shown in Box . Because the blood consists of both intracellular (red cell) and extracellular (plasma) components, it is important to avoid haemolysis during or after collection of the sample, which causes contamination of the plasma compartment by intracellular elements, particularly potassium.
Blood should not be drawn from an arm into
which an intravenous infusion is being given, to avoidcontamination by the infused fluid. Repeated measurements
of plasma electrolytes are frequently necessary
when abnormalities have been detected and corrective
therapy instituted.
Since the kidney maintains the constancy of body fluids by adjusting urine volume and composition, it is frequently helpful to obtain a sample of urine (‘spot’ specimen or 24-hour collection) at the time of blood analysis. An example of the use of urine biochemistry is given for the differential diagnosis of hyponatraemia in Box.
How to interpret ‘U&Es’ (urea and electrolytes) or ‘EUC’ (electrolytes/urea/creatinine) results
Na+ (sodium)
• Largely reflects reciprocal changes in body water content
• See ‘Hypernatraemia’ and ‘Hyponatraemia’ .
K+ (potassium)
• May reflect K shifts in and out of cells
• Low levels usually mean excessive losses (gastrointestinal or renal)
• High levels usually mean renal dysfunction
• See ‘Hypokalaemia’ and ‘Hyperkalaemia’.
Cl− (chloride)
• Generally changes in parallel with plasma Na
• Low in metabolic alkalosis
• High in some forms of metabolic acidosis
HCO3− (bicarbonate)
• Abnormal in acid–base disorders
Urea
• Increased with a fall in glomerular filtration rate (GFR), reduced renal perfusion or urine flow rate, and in high protein intake or catabolic states
Creatinine
• Increased with a fall in GFR, in individuals with high muscle mass, and with some drugs
DISORDERS OF SODIUM BALANCE
Functional anatomy and physiology of renal sodium handlingSince the great majority of the body’s sodium content is
located in the ECF, total body sodium is a principal determinant of ECF volume. Regulation of sodium excretion by the kidney is crucially important in maintaining normal
ECF volume, and plasma volume, in the face of wide
variations in sodium intake, which typically may range
between 50 and 250 mmol/day. The GFR is 125 mL/min (equivalent to 180 L/day) in a typical adult. Over 99% of this filtered fluid is reabsorbed into the blood in the peritubular capillaries during its passage through successive segments of the nephron
Nephron segments
At least four different functional segments of the
nephron can be defined in terms of their mechanism for
sodium reabsorption .
Proximal tubule
This is responsible for the reabsorption of some 65% of
the filtered sodium load. Filtered sodium in the luminal fluid enters the cell via several sodium transporters in the apical membrane that couple sodium transport to the entry of glucose, amino acid, phosphate and other organic molecules. Entry of sodium into the tubular cells at this site is also linked to secretion of H+ ions, through the sodium–hydrogen exchanger (NHE-3).
Intracellular H+ ions are generated within tubular cells from the breakdown of carbonic acid, which is produced from carbon dioxide and water under the influence of carbonic anhydrase.
Large numbers of Na,K-ATPase pumps are present on the basolateral membrane of tubular cells that transport sodium from the cells into the blood. In addition,
a large component of the transepithelial flux of
sodium, water and other dissolved solutes occurs
through the gaps between the cells (the ‘shunt’ pathway). Overall, fluid and electrolyte reabsorption is almost isotonic in this segment, as water reabsorption is matched
very closely to sodium fluxes.
The loop of Henle
The thick ascending limb of the loop of Henle reabsorbs a further 25% of the filtered sodium but is impermeable to water, resulting in dilution of the luminal fluid. Again, the primary driving force for sodium reabsorption is the Na,K-ATPase on the basolateral cell membrane. Some of the potassium accumulated inside the cell recirculates across the apical membrane back into the lumen through a specific potassium channel (ROMK), providing a continuing supply of potassium to match the high concentrations of sodium and chloride available in the lumen.Early distal tubule
Some 6% of filtered sodium is reabsorbed in the earlydistal tubule (distal convoluted tubule) , again driven by the activity of the basolateral Na,K-ATPase. In this segment, entry of sodium into the cell from the luminal fluid is via a sodium–chloride co-transport carrier (NCCT). This segment is also impermeable to water, resulting in further dilution of the luminal fluid.
There is no significant transepithelial flux of potassium in this segment, but calcium is reabsorbed from the luminal fluid through a calcium channel.
Late distal tubule and collecting ducts
The late distal tubule and cortical collecting duct are
anatomically and functionally continuous .
Here, sodium entry from the luminal fluid occurs via
the epithelial sodium channel (ENaC). This sodium flux into the tubular cells is balanced by secretion of potassium and hydrogen ions into the lumen and by reabsorption of chloride ions. Potassium is accumulated in the cell by the basolateral Na,K-ATPase, and passes into the luminal fluid down its electrochemical gradient, through an apical potassium channel (ROMK).
Chloride ions pass largely between cells. Hydrogen ion
secretion is mediated by an H+-ATPase located on theluminal membrane of the intercalated cells. This part of the nephron has a variable permeability to water, depending on the availability of antidiuretic hormone (ADH, or vasopressin) in the circulation. All ion transport processes in this segment are stimulated by the steroid hormone aldosterone, which can increase sodium reabsorption in this segment to a maximum of 2–3% of the filtered sodium load. <1% of sodium reabsorption occurs in medullary collecting duct, where it is inhibited by atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP).
Regulation of sodium transport
A large number of interrelated mechanisms serve tomaintain whole body sodium balance and hence ECF
volume by matching urinary sodium excretion to sodium
intake . Important sensing mechanisms include volume
receptors in the atria and the intrathoracic veins,
as well as pressure receptors located in the central arterial
tree (aortic arch and carotid sinus) and the afferent
arterioles within the kidney. A further afferent signal is
generated within the kidney itself; the enzyme renin is
released from specialised smooth muscle cells in the
walls of the afferent and efferent arterioles, at the point
where they make contact with the early distal tubule (at
the macula densa) to form the juxtaglomerular apparatus.
Renin release is stimulated by:
• reduced perfusion pressure in the afferent arteriole
• increased sympathetic nerve activity
• decreased sodium chloride concentration in the
distal tubular fluid.
Renin released into the circulation activates the effector
mechanisms for sodium retention, which are components
of the renin–angiotensin–aldosterone (RAA) system). Renin acts on the peptide substrate, angiotensinogen (manufactured in the liver), producing angiotensin I in the circulation. This in turn is cleaved by angiotensin-converting enzyme (ACE) into angiotensin II, largely in the pulmonary capillary bed.
Angiotensin II has multiple actions: it stimulates proximal tubular sodium reabsorption and release of
aldosterone from the zona glomerulosa of the adrenal cortex, and causes vasoconstriction of small arterioles.
Aldosterone amplifies sodium retention by its action on
the cortical collecting duct. The net effect is to restore
ECF volume and blood pressure towards normal,
thereby correcting the initiating hypovolaemic stimulus.
The sympathetic nervous system also increases
sodium retention, both through haemodynamic mechanisms
(afferent arteriolar vasoconstriction and GFR
reduction) and by direct stimulation of proximal tubular
sodium reabsorption.
Other humoral mediators, such as the natriuretic peptides, inhibit sodium reabsorption, contributing to natriuresis during periods of sodium and volume excess. Hypovolaemia also has haemodynamic effects that reduce GFR and alter the peritubular physical forces around the proximal tubule, thereby decreasing sodium excretion. Conversely, increased renal perfusion in hypervolaemia and hypertension results in a compensatory increase in sodium excretion.
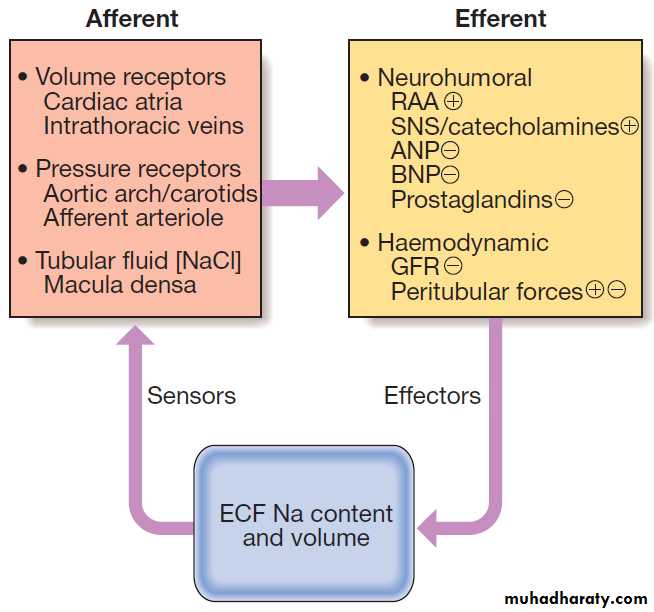
Fig. Mechanisms involved in the regulation of sodium
transport. (ANP = atrial natriuretic peptide; BNP = brain natriuretic peptide; ECF = extracellular fluid;GFR = glomerular filtration rate;
RAA = renin–angiotensin–aldosterone system; SNS = sympathetic nervous system. + indicates an effect to stimulate Na reabsorption and hence
reduce Na excretion, while _ indicates an effect to inhibit Na reabsorption and hence increase Na excretion)
Sodium depletion
Aetiology and clinical assessmentSodium depletion can occur occasionally under extreme
environmental conditions due to inadequate intake of salt, but it is much more commonly due to pathological
losses of sodium-containing fluids .
Loss of whole blood, as in acute haemorrhage, is also an obvious cause of hypovolaemia, and elicits the same mechanisms for the conservation of sodium and water. Although plasma sodium concentration may not be reduced if salt and water are lost in equal proportions, a number of other parameters are altered during appropriate renal, hormonal and haemodynamic responses to hypovolaemia.
During the early stages of hypovolaemia, GFR is maintained while urinary flow rate is reduced as a consequence of activation of sodium and water-retaining mechanisms in the nephron.
Thus, plasma creatinine, which reflects GFR, may be relatively normal, but the plasma urea concentration is typically elevated, since urea excretion is affected by both GFR and urine flow rate.
Plasma uric acid may also rise, reflecting activation of compensatory proximal tubular reabsorption. With avid retention of sodium and water, the urine osmolality increases while the urine sodium concentration falls. Under these circumstances, sodium excretion may fall to less than 0.1% of the filtered sodium load.
Management
Has two main components:
• treat the cause where possible
• replace the salt and water deficits, and provide
ongoing maintenance requirements, usually by IV
fluid replacement when depletion is severe.
Intravenous fluid therapy
The choice of fluid and the rate of administration depend on the clinical circumstances. In the absence of normal oral intake (as in a fasting or post-operative), maintenance quantities of fluid, sodium and potassium should be provided. In prolonged periods of fasting (more than a few days), attention also needs to be given to providing sufficient caloric and nutritional intake to prevent excessive
catabolism of body energy stores.
The choice of intravenous fluid therapy in the treatment
of significant hypovolaemia relates to the conceptsin Figure . If fluid containing neither sodium
nor protein is given, it will distribute in the body fluid
compartments in proportion to the normal distribution
of total body water. Thus, giving 1 L of 5% dextrose will
contribute relatively little (approximately 3/40 of the
infused volume) towards expansion of the plasma
volume. This makes 5% dextrose ineffective at restoring
the circulation and perfusion of vital organs. Intravenous infusion of an isotonic saline solution, on the other hand, results in more effective expansion of the extracellular fluid, although a minority of the infused volume (some 3/15) will contribute to plasma volume.
Carrying this reasoning further, it might be expected
that a solution containing plasma proteins would be
largely retained within the plasma, thus maximally
expanding the circulating fluid volume and improving
tissue perfusion. However, recent clinical studies have
not shown any overall advantage of infusions containing
albumin in the treatment of acute hypovolaemia. Resuscitation fluids containing synthetic colloids such as carbohydrate polymers should not be used in the acute resuscitation of acute hypovolaemia since they offer no benefit over crystalloids and are associated with increased mortality.
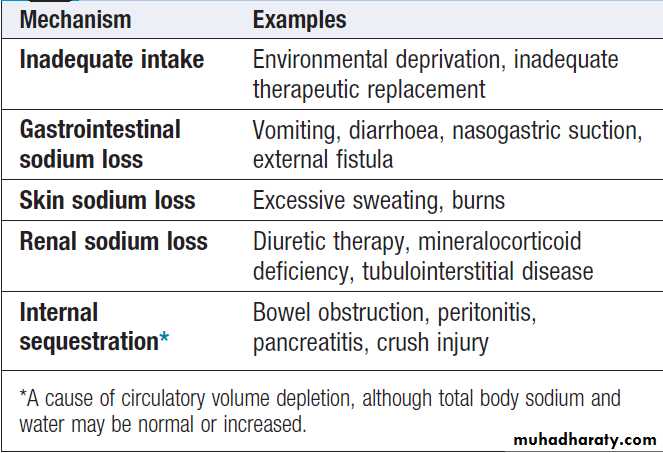
Causes of sodium and water depletion
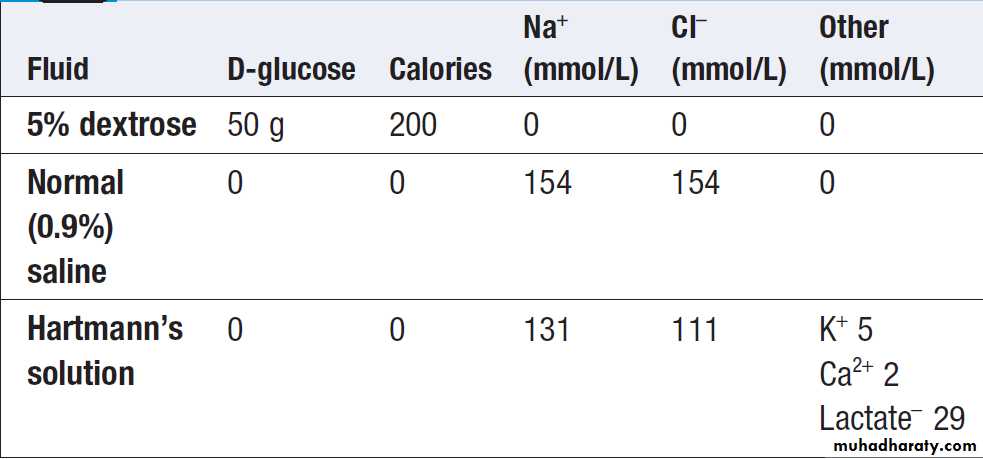
Composition of some isotonic intravenous fluids
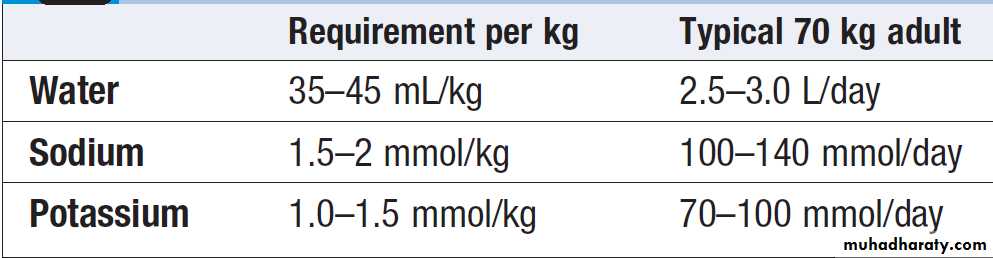
Basic daily water and electrolyte requirements
Step 1: assess clinical volume status
• Examine patient for signs of hypovolaemia or hypervolaemia
• Check daily weight change
Step 2: review fluid balance chart
• Check total volumes IN and OUT on previous day (IN–OUT is positive by ~400 mL in normal balance, reflecting insensible fluid losses of ~800 mL and metabolic water generation of ~400 mL)
• Check cumulative change in daily fluid balance over previous 3–5 days
• Correlate chart figures with weight change and clinical volume status to estimate net fluid balance
Step 3: assess ongoing pathological process
• Check losses from gastrointestinal tract and surgical drains
• Estimate increased insensible losses (e.g. in fever) and internal sequestration (‘third space’)
How to assess fluid and electrolyte balance in hospitalised patients
Step 4: check plasma U&Es
• Check plasma Na as marker of relative water balance• Check plasma K as a guide to extracellular K balance
• Check HCO3 as a clue to acid–base disorder
• Check urea and creatinine to monitor renal function
Step 5: prescribe appropriate IV fluid replacement therapy
• Replace basic water and electrolytes each day
• Allow for anticipated oral intake and pathological fluid loss
• Adjust amounts of water (if IV, usually given as isotonic 5% dextrose), sodium and potassium according to plasma electrolyte results
How to assess fluid and electrolyte balance in hospitalised patients
– cont’d

Albumin infusions in hypovolaemia
Sodium excessAetiology and clinical assessment
In patients with normal cardiac and renal function,
excessive intakes of salt and water are compensated for
by increased excretion and do not lead to clinically
obvious features of sodium and water overload.
However, patients with cardiac, renal or hepatic disease frequently present with signs and symptoms of sodium
excess .This does not always involve an increase in circulating blood volume, since the excess fluid often leaks out of the capillaries to expand the interstitial compartment of the ECF, especially in diseases like nephrotic syndrome and chronic liver disease that cause hypoalbuminaemia.
Peripheral oedema is the most common physical sign
of ECF volume expansion .The three most common systemic disorders associated with sodium and fluid overload are cardiac failure, cirrhosis and nephrotic syndrome.
In each of these, sodium retention is largely a secondary response to circulatory insufficiency.
The pathophysiology is different in renal failure,
when the primary cause of volume expansion is the
profound reduction in GFR impairing sodium and water
excretion, and secondary tubular mechanisms are of
lesser importance.
Mechanism Examples
Impaired renal function Primary renal disease
Primary hyperaldosteronism* Conn’s syndrome
Secondary hyperaldosteronism
Congestive cardiac failureCirrhotic liver disease
Nephrotic syndrome
Protein-losing enteropathy
Malnutrition
Idiopathic/cyclical oedema
Renal artery stenosis*
Causes of sodium and water excess
*Conditions in this table other than primary hyperaldosteronism and renal
artery stenosis are typically associated with generalised oedema
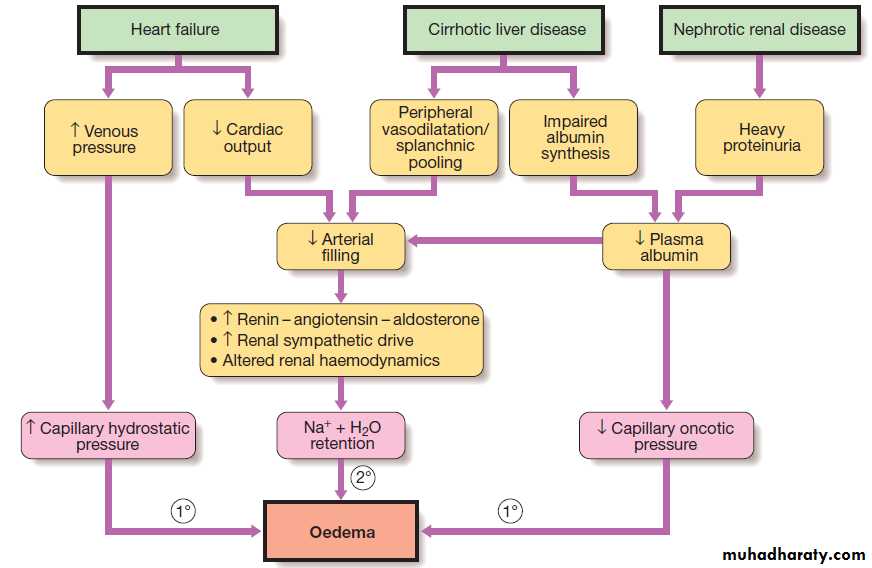
Secondary mechanisms causing sodium excess and oedema in cardiac failure, cirrhosis and nephrotic syndrome. Primary renal retention of Na and water may also contribute to oedema formation when GFR is significantly reduced
Management
The management of ECF volume overload involves a
number of components:
• specific treatment directed at the underlying cause,
such as ACE inhibitors in heart failure and
corticosteroids in minimal change nephropathy
• restriction of dietary sodium (to 50–80 mmol/day)
to match the diminished excretory capacity
• treatment with diuretics.
Diuretic therapy
Diuretics are important in the treatment of conditions ofECF expansion due to salt and water retention and in
hypertension .They act by inhibiting sodium reabsorption at various locations along the nephron .
Their potency and adverse effects relate to their mechanism and site of action.
Mechanisms of action
In the proximal tubule, carbonic anhydrase inhibitorssuch as acetazolamide inhibit the intracellular production
of H+ ions, thereby reducing the fraction of sodium
reabsorption that is exchanged for H+ by the apical membrane sodium–hydrogen exchanger.
These drugs have limited usefulness, however, since only a small fraction of proximal sodium reabsorption uses this
mechanism, and much of the sodium that is not
reabsorbed can be reabsorbed by downstream segments
of the nephron. In the thick ascending limb of the loop of Henle, loop diuretics such as furosemide inhibit sodium reabsorption by blocking the action of the apical membrane Na,K,2Cl co-transporter. Because this segment reabsorbs a large fraction of the filtered sodium, these drugs are potent diuretics.
In the early distal tubule, thiazides inhibit sodium
reabsorption by blocking the sodium–chloride cotransporter in the apical membrane. Since this segmentreabsorbs a much smaller fraction of the filtered sodium,
these are less potent than loop diuretics, but are widely
used in the treatment of hypertension and less severe
oedema.
All diuretic drugs acting in the proximal, loop and early distal segments cause excretion not only of sodium (and with it water), but also of potassium. This occurs largely as a result of delivery of increased amounts of sodium to the late distal/cortical collecting ducts, where sodium reabsorption is associated with excretion of potassium, and is amplified if circulating aldosterone levels are high.
By contrast, drugs acting to inhibit sodium reabsorption in the late distal/cortical collecting duct segment are associated with reduced potassium secretion, and are described as ‘potassium-sparing’. One target of drug action in this segment is the apical sodium channel in the principal cells , which is blocked by drugs such as amiloride and triamterene. Another is the mineralocorticoid receptor, to which binding of aldosterone is blocked by spironolactone and eplerenone. This secretory process may be impaired in chronic renal failure and chronic liver failure, leading to resistance to diuretics.
Osmotic diuretics act independently of specific transport
mechanism. They are freely filtered at the glomerulusbut are not reabsorbed by any part of the tubular
system. They retain fluid osmotically within the tubular
lumen and limit the extent of sodium reabsorption in
multiple segments.
Mannitol is the most commonly used osmotic diuretic. It is given by intravenous infusion to achieve short-term diuresis in conditions such as cerebral oedema.
Clinical use of diuretics
In the selection of a diuretic drug, the following principles should be observed:
• Use the minimum effective dose.
• Use for as short a period of time as necessary.
• Monitor regularly for adverse effects.
The choice determined by the required potency, the presence of coexistent conditions, and the anticipated side-effect profile. Since most drugs from these classes are sulphonamides, there is a relatively high incidence of hypersensitivity reactions, and occasional idiosyncratic side-effects. The disturbances in potassium, magnesium and acid–base balance are in the opposite direction, and there is a tendency to metabolic acidosis, especially when renal function is impaired.
Diuretic resistance is encountered under a variety of
circumstances, including impaired renal function, activationof sodium-retaining mechanisms, impaired oral
bioavailability (for example, in patients with gastrointestinal disease) and decreased renal blood flow. In these circumstances, short-term intravenous therapy
with a loop-acting agent such as furosemide may be
useful. Combinations of diuretics administered orally
may also increase potency.
Either a loop or a thiazide drug can be combined with a potassium-sparing drug, and all three classes can be used together for short periods, with carefully supervised clinical and laboratory monitoring.
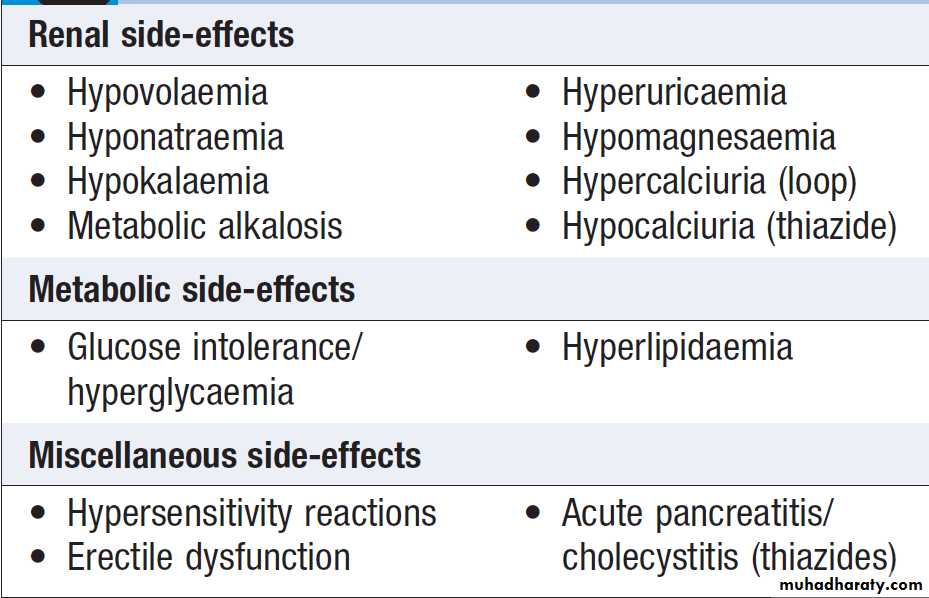
Adverse effects of loop-acting and thiazide diuretics
DISORDERS OF WATER BALANCE
Daily water intake can vary from about 500 mL to
several litres a day. While a certain amount of water is
lost through the stool, sweat and the respiratory tract
(‘insensible losses’, approximately 800 mL/day), and
some water is generated by oxidative metabolism (‘metabolic water’, approximately 400 mL/day), the kidneys are chiefly responsible for adjusting water excretion to maintain constancy of body water content and body fluid osmolality (reference range 280–295 mmol/kg).
Functional anatomy and physiology of renal water handling
While regulation of total ECF volume is largely achievedthrough renal control of sodium excretion, mechanisms also exist to allow for the excretion of a ‘pure’ water load when water intake is high, and for retention of water when access is restricted. These functions are largely achieved by the loop of Henle and the collecting ducts. The counter-current configuration of flow in adjacent limbs of the loop, involves osmotic movement of water from the descending limbs and reabsorption of solute from neighbouring ascending limbs, to set up a gradient of osmolality from isotonic (like plasma) in the renal cortex to hypertonic (around 1200 mmol/kg) in the inner medulla.
Further changes in the urine osmolality on passage
through the collecting ducts depend on the circulatinglevel of antidiuretic hormone (ADH), which is released
by the posterior pituitary gland under conditions
of increased plasma osmolality or hypovolaemia.
• When water intake is high and plasma osmolality is
normal or low-normal, ADH levels are suppressed
and the collecting ducts remain impermeable to water. The luminal fluid osmolality remains low, resulting in the excretion of a dilute urine (minimum osmolality approximately 50 mmol/kg in a healthy young person).
• When water intake is restricted and plasma
osmolality is high, or in the presence of plasma
volume depletion, ADH levels rise.
This causes water permeability of the collecting ducts to
increase through binding of ADH to the V2 receptor, which enhances collecting duct water. This results in osmotic reabsorption of water along the entire length of the collecting duct, with maximum urine osmolality approaching that in the medullary tip (up to 1200 mmol/kg).
In summary, for adequate dilution of the urine there
must be:
• adequate solute delivery to the loop of Henle and
early distal tubule
• normal function of the loop and early distal tubule
• absence of ADH in the circulation.
If any of these processes is faulty, water retention and
hyponatraemia may result.
Conversely, to achieve concentration of the urine there must be:
• adequate solute delivery to the loop of Henle• normal function of the loop of Henle
• ADH release into the circulation
• ADH action on the collecting ducts.
Failure of any of these steps may result in inappropriate
water loss and hypernatraemia.
Presenting problems in disorders of water balance
Disturbances in body water balance, in the absenceof changes in sodium balance, alter plasma sodium concentration and hence plasma osmolality. When extracellular osmolality changes abruptly, water flows rapidly across cell membranes with resultant cell swelling
(during hypo-osmolality) or shrinkage (during hyperosmolality).
Cerebral function is very sensitive to such volume changes, particularly brain swelling during hypo-osmolality, which can lead to an increase in intracerebral pressure and reduced cerebral perfusion.
Hyponatraemia
Aetiology and clinical assessment
(plasma Na < 135 mmol/L) is common, often asymptomatic but can also be associated with profound disturbances of cerebral function, manifesting as anorexia, nausea, vomiting, confusion, lethargy, seizures and coma. The likelihood of symptoms occurring is related more to the speed at which electrolyte abnormalities develop rather than their severity. When plasma Osmolality falls rapidly, water flows into cerebral cells, which become swollen and ischaemic. However, when develops gradually, cerebral neurons have time to respond by reducing intracellular osmolality, through excreting potassium and reducing synthesis of intracellular organic osmolytes .
Artefactual causes of hyponatraemia should also be considered in the presence of severe hyperlipidaemia or hyperproteinaemia, when the aqueous fraction of the plasma specimen is reduced because of the volume occupied by the macromolecules (although this artefact is dependent on the assay technology).
Transient hyponatraemia may also occur due to osmotic shifts of water out of cells during hyperosmolar states caused by acute hyperglycaemia or by mannitol infusion. Hyponatraemia with hypovolaemia
Patients who have hyponatraemia in association with a
sodium deficit (‘depletional hyponatraemia’) have clinical
features of hypovolaemia and low urinary sodium (< 30 mmol/L) and elevated plasma renin activity.
Hyponatraemia with euvolaemia
dilutional hyponatraemia have no major disturbance of body sodium content and are clinically euvolaemic. Excess body water may be the result of abnormally high intake, either orally (primary polydipsia) or as a result of infused fluids (as intravenous dextrose solutions, or by absorption of sodium-free bladder irrigation fluid after prostatectomy). Water retention also occurs in the syndrome of inappropriate secretion of ADH (SIADH). In this condition, an endogenous source of ADH (either cerebral or tumour-derived) promotes water retention by the kidney in the absence of an appropriate physiological stimulus . Clinical diagnosis requires the patient to be euvolaemic, with no evidence of cardiac, renal or hepatic disease potentially associated with hyponatraemia.Other non-osmotic stimuli that cause release of ADH (pain, stress, nausea) should also be excluded. Urine osmolality, which should physiologically be maximally dilute (approximately 50 mmol/kg) in the face of low plasma osmolality, is higher than at least 100 mmol/kg and
indeed is typically higher than the plasma osmolality.
The urine sodium concentration is typically high
(> 30 mmol/L), consistent with euvolaemia and lack
of compensatory factors promoting sodium retention. Hyponatraemia with hypervolaemia
In this situation, excess water retention is associated
with sodium retention and volume expansion, as in
heart failure, liver disease or kidney disease.
Investigations
Plasma and urine electrolytes and osmolality are usually the only tests required to classify the hyponatraemia. Doubt about clinical signs of ECF volume may be resolved with measurement of plasma renin activity. Measurement of ADH is not generally helpful in distinguishing between these categories of hyponatraemia. This is because ADH is activated both in hypovolaemic states and in most chronic hypervolaemic states, as the impaired circulation in those disorders activates ADH release through non-osmotic mechanisms. Indeed, these disorders may have higher circulating ADH levels than patients with SIADH.
The only disorders listed in Box in which ADH is suppressed are primary polydipsia and iatrogenic water intoxication, where the hypo-osmolar state inhibits ADH release from the pituitary.
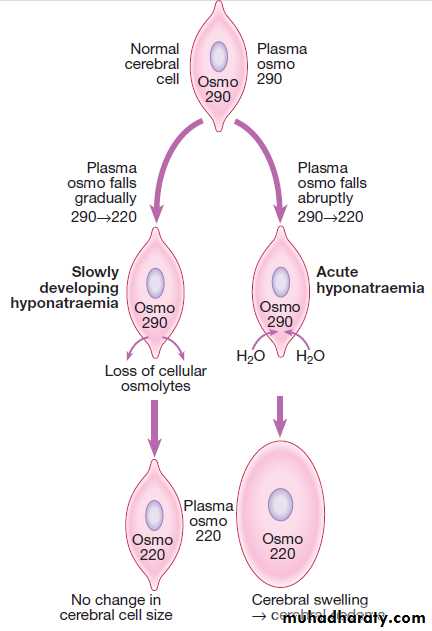
Hyponatraemia and the brain. Numbers represent
osmolality (osmo) in mmol/kg.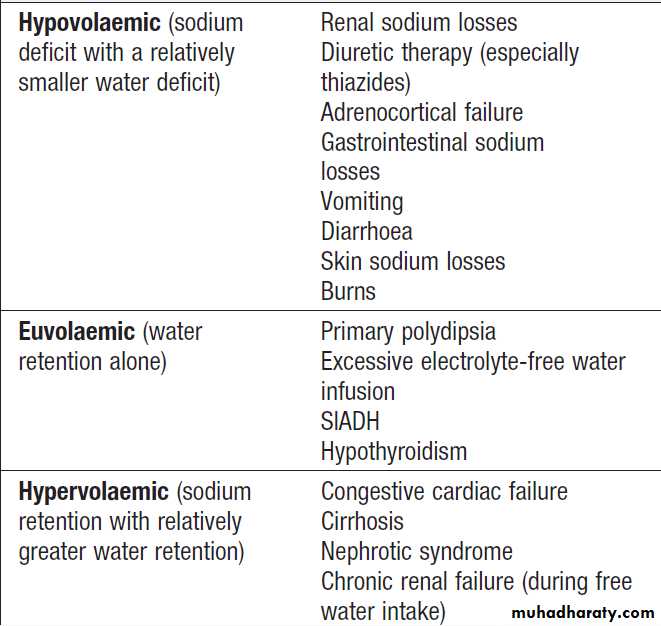

Causes of hyponatraemia

Diagnosis
• Low plasma sodium concentration (typically < 130 mmol/L)• Low plasma osmolality (< 270 mmol/kg)
• Urine osmolality not minimally low (typically > 150 mmol/kg)
• Urine sodium concentration not minimally low (> 30 mmol/L)
• Low-normal plasma urea, creatinine, uric acid
• Exclusion of other causes of hyponatraemia
• Appropriate clinical context (above)
Syndrome of inappropriate antidiuretic hormone secretion (SIADH): causes and diagnosis
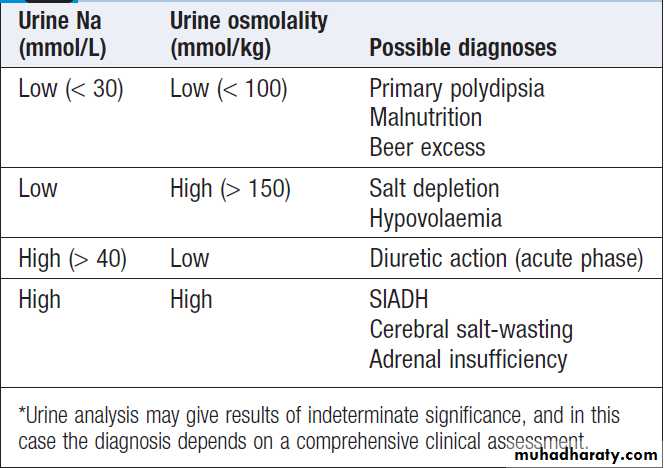
Urine Na and osmolality in the differential
diagnosis of hyponatraemia*Management
The treatment of hyponatraemia is critically dependenton its rate of development, severity and cause. If has developed rapidly (over hours to days), and there are signs of cerebral oedema such as obtundation or convulsions, sodium levels should be restored to normal rapidly by infusion of hypertonic (3%) sodium chloride. Initial bolus of 100 mL, which may be repeated once or twice over the initial hours of observation, depending on the neurological response and rise in plasma sodium.
On the other hand, rapid correction of hyponatraemia
that has developed slowly (over weeks to months) can
be hazardous, since brain cells adapt to slowly developing hypo-osmolality by reducing the intracellular osmolality, thus maintaining normal cell volume .
Under these conditions, an abrupt increase in extracellular osmolality can lead to water shifting out of neurons, abruptly reducing their volume and causing them to detach from their myelin sheaths. The resulting ‘myelinolysis’ can produce permanent structural and functional damage to mid-brain structures, and is generally fatal. The rate of correction should not exceed 10 mmol/L/day, and an even slower rate is safer. The underlying cause should be treated. Patients with dilutional hyponatraemia generally respond to fluid restriction in the range of 600–1000 mL/day, accompanied where possible by withdrawal of the precipitating stimulus (such as drugs causing SIADH).
If the response of plasma sodium is inadequate, treatment with demeclocycline (600–900 mg/day) may be of value by enhancing water excretion, through its inhibitory effect on responsiveness to ADH in the collecting duct.
An effective alternative for SIADH is oral urea therapy (30–45 g/ day), which provides a solute load to promote water excretion. Where available, oral vasopressin receptor antagonists such as tolvaptan may be used to block the ADH-mediated component of water retention in a range of hyponatraemic conditions. Hypervolaemic hyponatraemia need treatment of the underlying condition, cautious use of diuretics in conjunction with strict fluid restriction. Potassium-sparing diuretics may be useful in this where there is secondary hyperaldosteronism.
Hypernatraemia
Aetiology and clinical assessmentJust as hyponatraemia represents a failure of the mechanisms for diluting the urine during free access to water, so hypernatraemia (plasma Na > 148 mmol/L) reflects inadequate concentration of the urine in the face of
restricted water intake. This can be due to failure to
generate an adequate medullary concentration gradient
(low GFR states, loop diuretic therapy), but more commonly
it is due to failure of the ADH system, either because of pituitary damage (central or ‘cranial’ diabetes insipidus, p.) or because the collecting duct cells are unable to respond to circulating ADH (nephrogenic diabetes insipidus).
Patients with hypernatraemia generally have reduced
cerebral function, either as a primary problem or as a
consequence of the hypernatraemia itself, which results
in dehydration of neurons and brain shrinkage. In the
presence of an intact thirst mechanism and preserved
capacity to obtain and ingest water, hypernatraemia
may not progress very far. If adequate water is not
obtained, dizziness, confusion, weakness and ultimately
coma and death can result.

Management
Treatment depends on both the rate of development and the underlying cause. If the condition has developed rapidly, neuronal shrinkage may be acute and relatively rapid correction may be attempted. This can be achieved by infusing an appropriate volume of IV fluid (isotonic 5% dextrose or hypotonic 0.45% saline) at an initial rate of 50–70 mL/hour. However, in older, institutionalized patients it is more likely that the disorder has developed slowly, and extreme caution should be exercised in lowering plasma sodium to avoid the risk of cerebral oedema. Where possible, the underlying cause should also be addressed . Elderly, predisposed to both hypo and hypernatraemia,and a high index of suspicion is appropriate in elderly with recent alterations in behavior.
Hyponatraemia and hypernatraemia in old age
DISORDERS OF POTASSIUM BALANCE
Potassium is the major intracellular cation, and plays an important part in generating the resting membrane potential and allowing the propagation of the action potential that is crucial to normal functioning of nerve, muscle and cardiac tissues.Changes in the distribution of potassium between the
ICF and ECF compartments can alter plasma potassium
concentration, without any overall change in total body
potassium content.
Potassium is driven into the cells by extracellular alkalosis and by a number of hormones, including insulin, catecholamines (through the β2 receptor) and aldosterone. Any of these factors can produce hypokalaemia.
Where as extracellular acidosis, lack of insulin, and insufficiency or blockade of catecholamines or aldosterone can cause hyperkalaemia due to efflux of potassium from the intracellular compartment.
In the steady state, the kidneys excrete some 90% of the
daily intake of potassium, typically 80–100 mmol/day.
Potassium is freely filtered at the glomerulus; around
65% is reabsorbed in the proximal tubule and a further
25% in the thick ascending limb of the loop of Henle.
Little potassium is transported in the early distal tubule
but a significant secretory flux of potassium into the
urine occurs in the late distal tubule and cortical collecting
duct to ensure that the amount removed from the
blood is proportional to the ingested load.
Movement of potassium from blood to lumen is dependent
on active uptake across the basal cell membrane by
the Na,K-ATPase, followed by diffusion of potassium
through a luminal membrane potassium channel
(ROMK) into the tubular fluid. A number of factors influence the rate of potassium secretion. Luminal influences include the rate of sodium delivery and fluid flow through the late distal tubule and cortical collecting ducts. This is a major factor responsible for the increased potassium loss that accompanies diuretic treatment.
Agents interfering with the generation of the negative luminal potential also impair potassium secretion, and this is the basis of reduced potassium secretion associated with potassium-sparing diuretics such as amiloride.
Factors acting on the blood side of this tubular segment include plasma potassium and pH, such that hyperkalaemia and alkalosis both enhance potassium secretion directly. However, the most important factor in the acute and chronic adjustment of potassium secretion to match metabolic potassium load is aldosterone. As shown in Figure, a negative feedback relationship exists between the plasma potassium concentration and aldosterone.
In addition to its regulation by the renin–angiotensin system , aldosterone is released from the adrenal cortex in direct response to an elevated plasma potassium. Aldosterone then acts on the kidney to enhance potassium secretion, hydrogen secretion and sodium reabsorption, in the late distal tubule and cortical collecting ducts.
The resulting increased excretion of potassium maintains potassium within a narrow range (3.3–4.7 mmol/L). Factors that reduce angiotensin II levels may indirectly affect potassium balance by blunting the rise in aldosterone that would otherwise be provoked by hyperkalaemia.
This accounts for the increased risk of hyperkalaemia
during therapy with ACE inhibitors and related drugs.
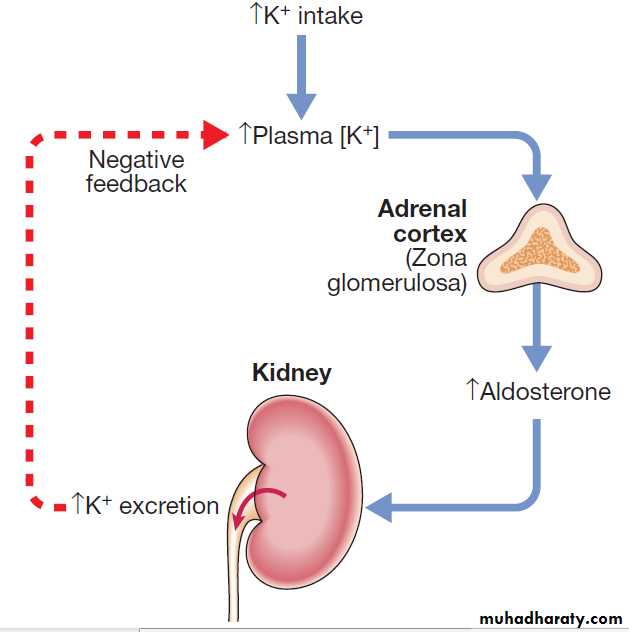
Feedback control of plasma potassium concentration.
Presenting problems in disorders of potassium balanceHypokalaemia
Aetiology and clinical assessment
Patients with mild hypokalaemia (plasma K 3.0– 3.3 mmol/L) are generally asymptomatic, but more profound reductions in plasma potassium often lead to muscular weakness and associated tiredness. Ventricular ectopic beats or more serious arrhythmias may occur and the arrhythmogenic effects of digoxin may be potentiated. Typical ECG changes occur, affecting the T wave in particular . Functional bowel obstruction may occur due to paralytic ileus. Long-standing hypokalaemia causes renal tubular damage (hypokalaemic nephropathy) and interferes with the tubular response to ADH (acquired nephrogenic diabetes insipidus), resulting in polyuria and polydipsia.
Redistribution of potassium into cells should be considered, since correction of the factors involved may be sufficient to correct the plasma concentration. An inadequate intake of potassium can contribute to
hypokalaemia but is unlikely to be the only cause, except
in extreme cases. Generally, hypokalaemia is indicative
of abnormal potassium loss from the kidney or the GIT. When there is no obvious clinical clue to which pathway is
involved, measurement of urinary potassium is helpful; if the kidney is the route of potassium loss, the urine potassium is high (> 30 mmol/day), whereas if potassium is being lost through the GIT, the kidney retains potassium, resulting in a lower urinary potassium (< 20 mmol/day).
It should be noted, however, that if gastrointestinal fluid loss is also associated with hypovolaemia, activation of
the renin–angiotensin–aldosterone system may occur,
causing increased loss of potassium in the urine.
Renal causes of hypokalaemia can be divided into
those with and those without hypertension.
Hypokalaemia in the presence of hypertension may be due to increased aldosterone secretion in Conn’s syndrome
or a genetic defect affecting sodium channels in the distal nephron (Liddle’s syndrome).
Excessive intake of liquorice or treatment with carbenoxolone may result in a similar clinical picture, due to inhibition of the renal 11βHSD2 enzyme, which inactivates cortisol in peripheral tissues.
If blood pressure is normal or low, hypokalaemia can
be classified according to the associated change inacid–base balance. Inherited defects in tubular transport
should be suspected when hypokalaemia occurs in association with alkalosis, provided that diuretic use has
been excluded. One such disease is Bartter’s syndrome,
in which sodium reabsorption in the thick ascending limb of Henle is defective. The clinical and biochemical features are similar to chronic treatment with furosemide.
In Gitelman’s syndrome there is a loss-of-function mutation affecting the NCCT transporter in the early distal tubule. The clinical and biochemical features are similar to chronic
thiazide treatment.
Note that while both Bartter’s and Gitelman’s syndromes characterised by hypokalaemia and hypomagnesaemia, urinary calcium excretion is increased in Bartter’s but decreased in Gitelman’s syndrome, analogous to the effects of the loop and thiazide diuretics, respectively, on calcium transport. When hypokalaemia is due to wasting through the GIT, the cause is usually obvious clinically. In some cases, when there is occult induction of vomiting, the hypokalaemia is characteristically associated with metabolic alkalosis, due to loss of gastric acid. If, however, potassium loss has occurred through the surreptitious use of aperients, the hypokalaemia is generally associated with metabolic acidosis.
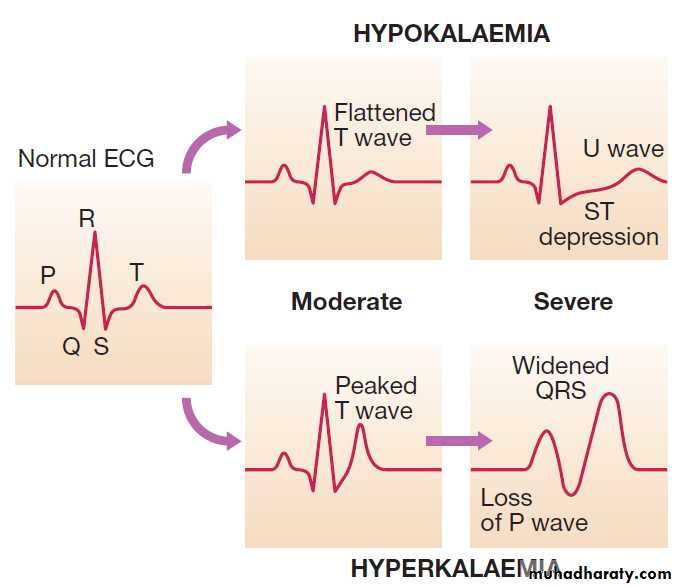
The ECG in hypokalaemia and hyperkalaemia.
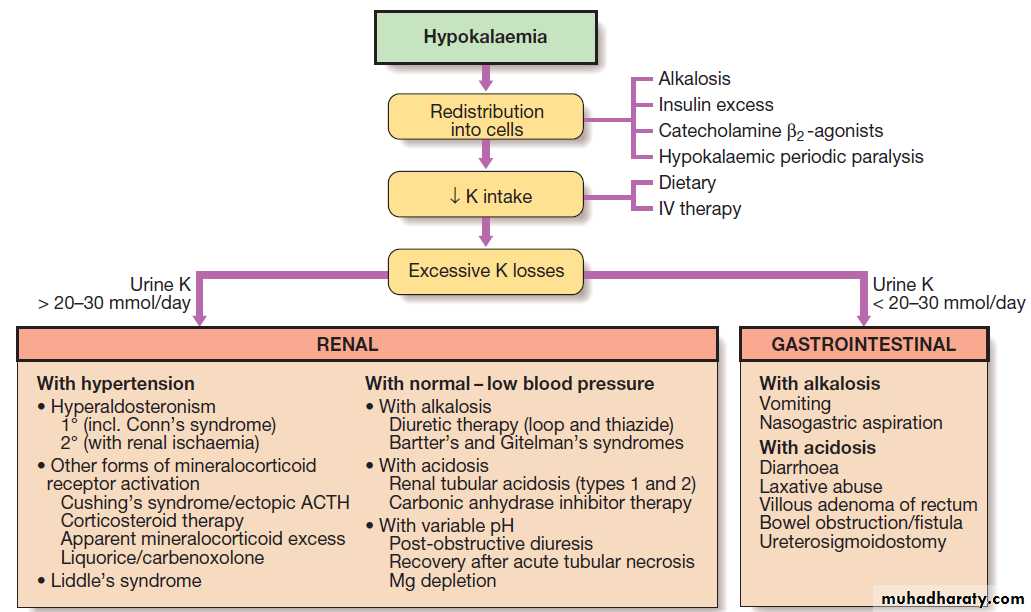
Diagnostic decision tree for hypokalaemia.
InvestigationsMeasurement of plasma electrolytes, bicarbonate, urine
potassium , plasma calcium and magnesium. If the diagnosis remains unclear, plasma renin should be measured. Levels are low in primary hyperaldosteronism and other forms of mineralocorticoid excess, but raised in other causes of hypokalaemia. Many such cases are associated with metabolic alkalosis, and in this setting the measurement of urine chloride concentration can be helpful. A low urine chloride (< 30 mmol/L) is characteristic of vomiting (chloride is lost in HCl in the vomit), while a urine chloride > 40 mmol/L suggests diuretic therapy or a tubular disorder such as Bartter’s or Gitelman’s syndrome.
Management
Treatment of hypokalaemia involves first determiningthe cause and then correcting this where possible. If the
problem is mainly one of redistribution of potassium
into cells, reversal of this (for example, correction of
alkalosis) may be sufficient to restore plasma potassium .
In most cases, however, some form of potassium replacement will be required. This can generally be achieved with slow-release potassium chloride tablets, but in more acute circumstances intravenous potassium chloride may be necessary. The rate of administration depends on the severity and the presence of cardiac or neuromuscular complications, but should generally not exceed 10 mmol of potassium per hour.
In patients with severe, life-threatening hypokalaemia, the concentration of potassium in the infused fluid may be increased to 40 mmol/L if a peripheral vein is used, but higher concentrations must be infused into a large ‘central’
vein with continuous cardiac monitoring.
In the less common situation where hypokalaemia occurs in the presence of systemic acidosis, alkaline salts of potassium, such as potassium bicarbonate, can
be given by mouth. If magnesium depletion is also present,
replacement of magnesium may also be required for
hypokalaemia to be corrected since low cell magnesium
can enhance the mechanism for tubular potassium secretion,
causing ongoing urinary losses.
In some circumstances, potassium-sparing diuretics, such as amiloride, can assist in the correction of hypokalaemia, hypomagnesaemia and metabolic alkalosis, especially when loop or thiazide diuretics are the underlying cause.
Hyperkalaemia
Aetiology and clinical assessmentHyperkalaemia can present with progressive muscular
weakness, but sometimes there are no symptoms until
cardiac arrest occurs. The typical ECG changes are
shown in Figure. Hyperkalaemia may occur either because of redistribution of potassium between the ICF and ECF in the presence of systemic acidosis, or when the circulating levels of insulin, catecholamines and aldosterone are reduced or when the effects of these hormones are blocked or because intake exceeds excretion. It is also important to remember that hyperkalaemia can also be artefactual due to in vitro haemolysis of blood specimens.
Impaired excretion of potassium into the urine may
be associated with a reduced GFR, as in acute kidneyinjury or chronic kidney disease.
Hyperkalaemia can also develop when tubular potassium
secretory processes are impaired, even if the GFR
is well maintained. In some cases, this is due to low
levels of aldosterone, as occurs in Addison’s disease or
with ACE inhibitor therapy. Another cause is hyporeninaemic hypoaldosteronism where the renin–angiotensin system is inactivated. This condition typically occurs in association with diabetic nephropathy with neuropathy, and is thought to be due to impaired β-adrenergic stimulation of renin release.
Other causes include angiotensin receptor antagonists, NSAIDs and β-blocking drugs. In another group of conditions, tubular potassium secretion is impaired as the result of aldosterone resistance. This can occur in tubulointerstitial diseases, following renal transplantation; during treatment with potassium-sparing diuretics. In all conditions of aldosterone deficiency or aldosterone
resistance, hyperkalaemia may be associated with
acid retention, giving rise to the pattern of hyperkalaemic
distal (‘type 4’) renal tubular acidosis.
Investigations
Measurement of electrolytes, creatinine and bicarbonate,when combined with clinical assessment, usually provides
the explanation for hyperkalaemia. In aldosterone
deficiency, plasma sodium concentration is characteristically low, although this can occur in many causes of hyperkalaemia. Addison’s disease should be excluded unless there is an obvious alternative diagnosis.
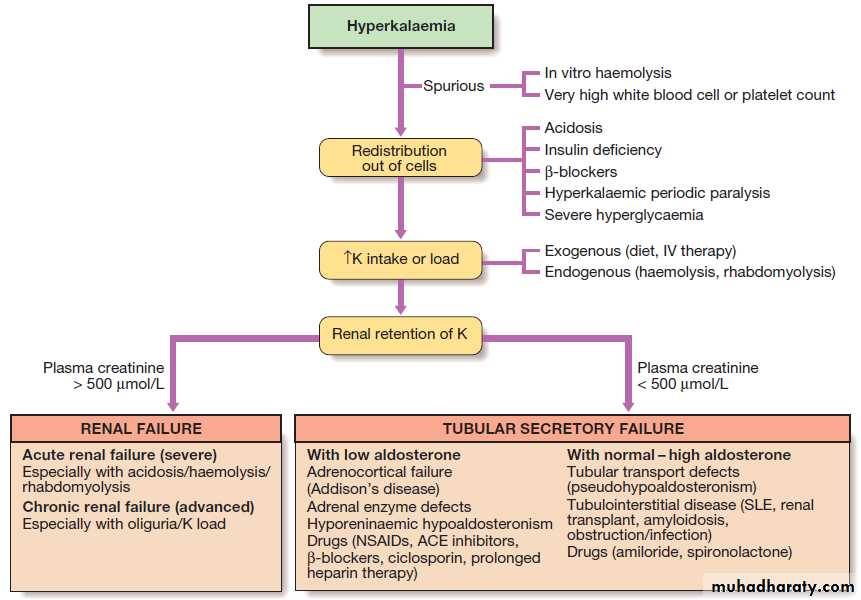
Diagnostic decision tree for hyperkalaemia. Creatinine of 500 μmol/L = 5.67 mg/dL.
ManagementDepends on its severity and the rate of development. In the absence of neuromuscular symptoms or ECG changes, reduction of potassium intake and correction of underlying abnormalities may be sufficient. However, in acute and/or severe hyperkalaemia (plasma K > 6.5–7.0 mmol/L) more urgent measures must be taken .
If ECG changes are present, the first step should be infusion of 10 mL 10% calcium gluconate to stabilize conductive tissue membranes (calcium has the opposite effect to potassium on conduction of an action potential). Measures to shift potassium from the ECF to the ICF should also be taken, as they generally act rapidly and may avert arrhythmias.
Ultimately, a means of removing potassium from the body is generally necessary.
When renal function is reasonably preserved, loop diuretics (accompanied by intravenous saline if hypovolaemia is present) may be effective; in established renal failure, ion-exchange resins acting through the gastrointestinal tract and urgent dialysis may be required.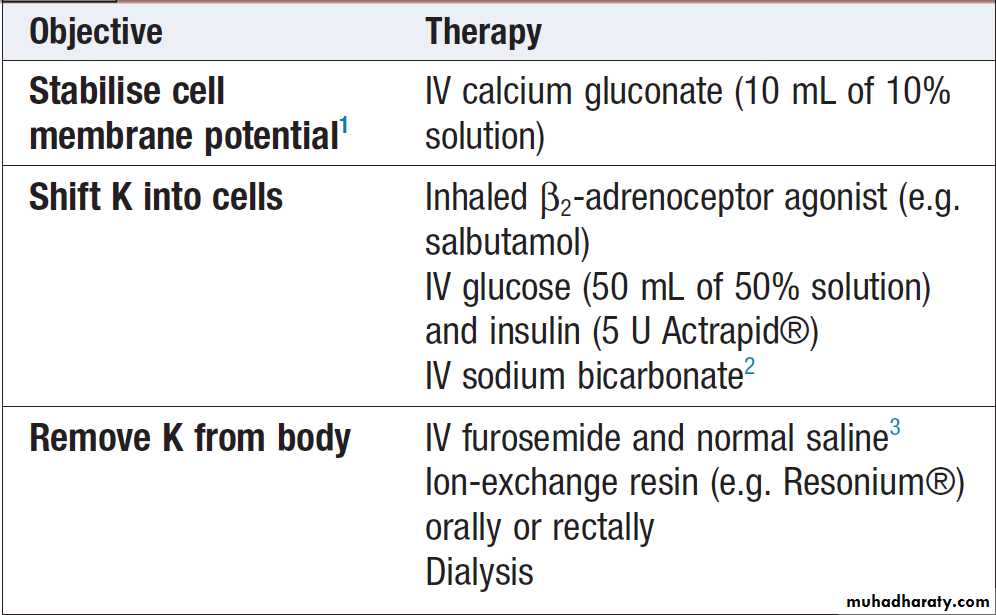
Treatment of severe hyperkalaemia

DISORDERS OF ACID–BASE BALANCE
The pH of the arterial plasma is normally 7.40, corresponding to a H+ concentration of 40 nmol/L. Anincrease in H + concentration corresponds to a decrease
in pH. Under normal circumstances, H + concentrations
do not vary outside the range of 36–44 nmol/L (pH
7.44–7.36), but abnormalities of acid–base balance occur
in a wide range of diseases.
Functional anatomy and physiology of
acid–base homeostasisA variety of physiological mechanisms maintain pH of
the ECF within narrow limits. The first is the action of
blood and tissue buffers, of which the most important
involves reaction of + ions with bicarbonate to form
carbonic acid, which, under the influence of the enzyme
carbonic anhydrase (CA), dissociates to form CO2 and
water:
This buffer system is important because bicarbonate
is present at relatively high concentration in ECF (21–
28 mmol/L), and two of its key components are under
physiological control: CO2 by the lungs, and bicarbonate
by the kidneys. These relationships are illustrated in
Figure (a form of the Henderson–Hasselbalch equation).
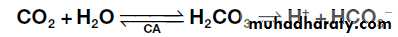
Respiratory compensation for acid–base disturbances
can occur quickly. In response to acid accumulation, pHchanges in the brainstem stimulate ventilatory drive,
serving to reduce PCO2 and increase pH .
Conversely, systemic alkalosis leads to inhibition of ventilation, causing a rise in PCO2 and reduction in pH.
The kidney provides a third line of defence against disturbances of arterial pH. When acid accumulates due to chronic respiratory or metabolic (non-renal) causes, the kidney has the long-term capacity to enhance urinary excretion of acid, effectively increasing the plasma bicarbonate.
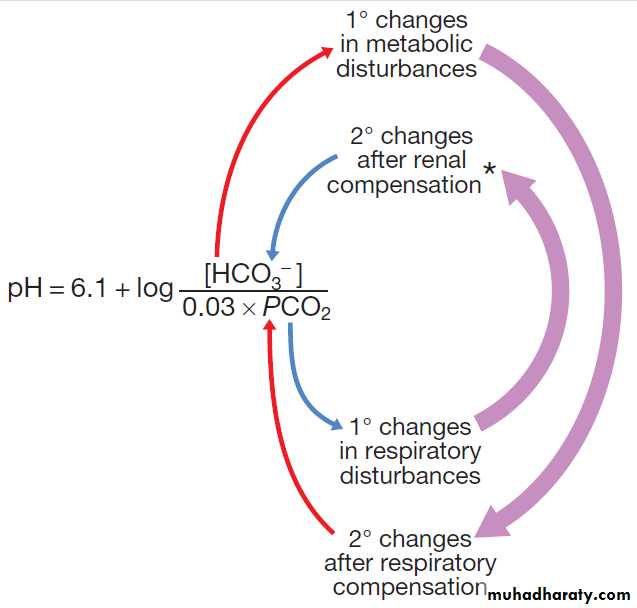
Fig-Relationship between pH, PCO2 (in mmHg) and plasma bicarbonate concentration (in mmol/L).
*Note that changes in HCO3 − concentration are also part of the renal correction for sustained metabolic acid–base disturbances as long as the kidney itself is not the cause of the primary disturbance.
Renal control of acid–base balance
Regulation of acid–base balance occurs at several sitesin the kidney. The proximal tubule reabsorbs some 85%
of the filtered bicarbonate ions, through the mechanism
for H+ secretion illustrated in Figure . This is dependent on the enzyme carbonic anhydrase both in the cytoplasm of the proximal tubular cells and on the luminal surface of the brush border membranes.
Distal nephron segments have an important role in
determining net acid excretion by the kidney.
In the intercalated cells of the cortical collecting duct and the outer medullary collecting duct cells, acid is secreted
into the lumen by an H+-ATPase. This excreted acid is
generated in the tubular cell from the hydration of CO2
to form carbonic acid, which dissociates into an H+ ion
secreted luminally, and a bicarbonate ion that passes
across the basolateral membrane into the blood. The
secreted H+ ions contribute to the reabsorption of any
residual bicarbonate present in the luminal fluid, but
also contribute net acid for removal from the body, bound to a variety of urinary buffers, of which phosphate
and ammonia are the most important.
Filtered phosphate (HPO4 2−) combines with H+ in the distal tubular lumen to form dihydrogen phosphate
(H2PO4−), which is excreted in the urine with sodium. Ammonia (NH3) is generated within tubular cells by deamination of the amino acid glutamine by the enzyme glutaminase.
The NH3 then reacts with secreted H+ in the tubular lumen
to form ammonium (NH4 +), which becomes trapped in
the luminal fluid and is excreted with chloride ions.
These two mechanisms remove approximately 1 mmol/kg of hydrogen ions from the body per day,
which equates to the non-volatile acid load arising from
the metabolism of dietary protein. The slightly alkaline
plasma pH of 7.4 (H+ 40 nmol/L) that is maintained
during health can be accounted for by the kidney’s
ability to generate an acidic urine (pH typically 5–6), in
which the net daily excess of metabolic acid produced
by the body can be excreted.
Presenting problems in disorders of acid–base balance
Patients with disturbances of acid–base balance may
present clinically either with the effects of tissue malfunction
due to disturbed pH (such as altered cardiac and CNS function), or with secondary changes in respiration as a response to the underlying metabolic change (such as Kussmaul respiration during metabolic acidosis). The clinical picture is often dominated by the cause of the acid–base change, such as uncontrolled diabetes mellitus or primary lung disease. Frequently the acid–base disturbance only becomes evident when the venous plasma bicarbonate concentration is noted to be abnormal, or when a full arterial blood gas analysis shows abnormalities in pH, PCO2 or bicarbonate.
The ‘base excess’ or ‘base deficit’ can also be
calculated from these data. This is the difference betweenthe patient’s bicarbonate level and the normal bicarbonate, measured in vitro with the PCO2 adjusted to
5.33 kPa (40 mmHg). Calculation of the base excess or
deficit is particularly useful in patients with combined
respiratory and metabolic disorders .
In metabolic disturbances, respiratory compensation
is almost immediate, so that the predicted compensatorychange in PCO2 is achieved soon after the onset of the
metabolic disturbance. In respiratory disorders, on the
other hand, a small initial change in bicarbonate occurs
as a result of chemical buffering of CO2, largely within
red blood cells, but over days and weeks the kidney
achieves further compensatory changes in bicarbonate
concentration as a result of long-term adjustments in
acid secretory capacity. When the clinically obtained
acid–base parameters do not accord with the predicted
compensation shown, a mixed acid–base disturbance
should be suspected.
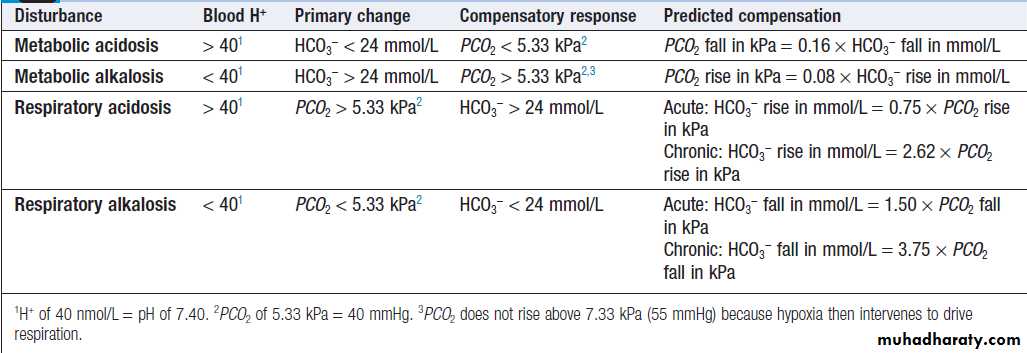
Principal patterns of acid–base disturbance
Metabolic acidosisAetiology and assessment
Metabolic acidosis occurs when an acid other than
carbonic acid (due to CO2 retention) accumulates in
the body, resulting in a fall in the plasma bicarbonate.
The pH fall that would otherwise occur is blunted by
hyperventilation, resulting in a reduced PCO2.
If the kidneys are intact and the primary cause of acidosis is not renal in origin, the kidney can gradually increase
acid secretion over days to weeks and restore a new
steady state.
Two patterns of metabolic acidosis are recognised,
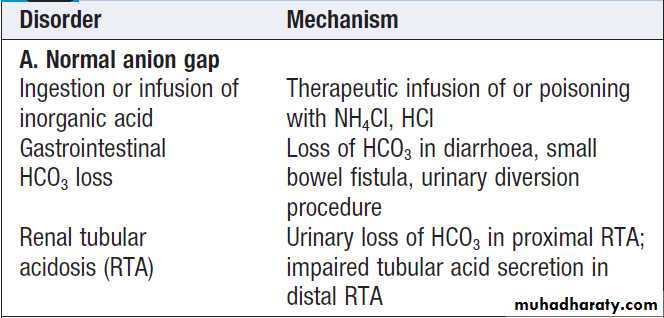
depending on the nature of the accumulating acid:• In pattern A, when a mineral acid such as hydrochloric acid accumulates, or when there is a primary loss of bicarbonate buffer from the ECF, there is no addition to the plasma of a new acidic anion. In this case, the ‘anion gap’ (calculated as the difference between the main measured cations (Na+ + K+) and the anions (Cl− + HCO3 −)) is normal, since the plasma chloride increases to replace the depleted bicarbonate levels. This ‘gap’ is normally
around 12–16 mmol/L (12–16 meq/L) and is made
up of anions such as phosphate, sulphate and multiple negative charges on plasma protein molecules.
Normal anion gap metabolic acidosis (pattern A) is usually due either to bicarbonate loss in diarrhoea, where the clinical diagnosis is generally obvious, or to renal tubular acidosis (see below).
• In pattern B, an accumulating acid is accompanied
by its corresponding anion, which adds to theunmeasured anion gap, while the chloride concentration remains normal. The cause is usually apparent from associated clinical features such as uncontrolled diabetes mellitus, renal failure or shock, or may be suggested by associated symptoms, such as visual complaints in methanol
poisoning. It is noteworthy that a number of causes of increased anion gap acidosis are associated with alcoholism, including starvation ketosis, lactic acidosis and intoxication by methanol or ethylene glycol.
Lactic acidosis
The diagnosis of lactic acidosis can be confirmed by themeasurement of plasma lactate, which is increased over
the normal maximal level of 2 mmol/L (20 mg/dL) by
as much as tenfold. Two types of lactic acidosis have
been defined:
• type 1, due to tissue hypoxia and peripheral
generation of lactate, as in patients with circulatory
failure and shock
• type 2, due to impaired metabolism of lactate, as in
liver disease or by a number of drugs and toxins,
including metformin, which inhibit lactate metabolism
Renal tubular acidosis
Renal tubular acidosis (RTA) should be suspected whenthere is a hyperchloraemic acidosis with a normal anion
gap and no evidence of gastrointestinal disturbance.
The urine pH is inappropriately high (> 5.5) in the
presence of systemic acidosis. RTA can be caused by a
defect in one of three processes: impaired bicarbonate
reabsorption in the proximal tubule (proximal RTA);
impaired acid secretion in the late distal tubule or
cortical collecting duct intercalated cells (classical distal
RTA); or impaired sodium reabsorption in the late distal
tubule or cortical collecting duct, which is associated
with reduced secretion of both potassium and H+ ions
(hyperkalaemic distal RTA).

Accumulation of ketones1 with hyperglycaemia
Accumulation of ketones without hyperglycaemiaShock, liver disease, drugs Accumulation of organic acids
Accumulation of salicylate2Accumulation of formate
Accumulation of glycolate, oxalate1Ketones include acid anions acetoacetate and β-hydroxybutyrate 2Salicylate poisoning is also associated with respiratory alkalosis due to direct ventilatory stimulation.
Causes of metabolic acidosis
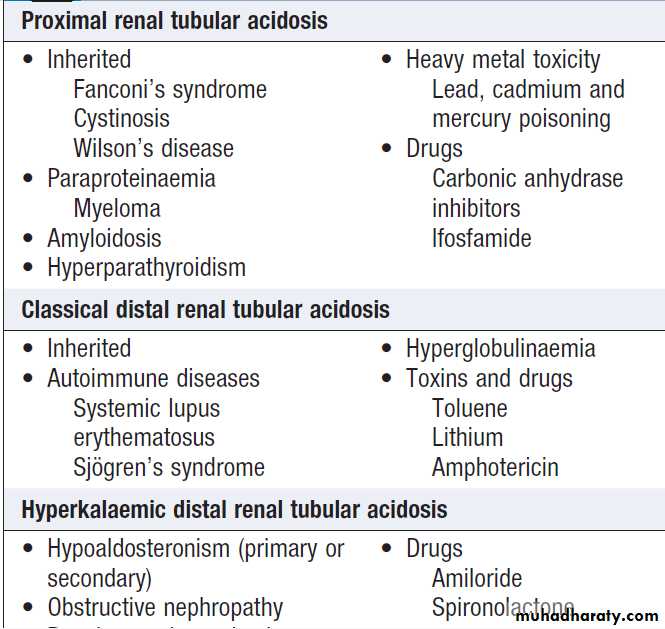

Causes of renal tubular acidosis
The inherited forms of RTA are due to mutations in the genes that regulate acid or bicarbonate transport in the renal tubules (Fig.). However, RTA is often an acquired disorder and in these circumstances the metabolicacidosis may serve as an early clue to the underlying
diagnosis. Sometimes distal RTA is ‘incomplete’ and the plasma bicarbonate concentration is normal under resting conditions. However, in incomplete distal RTA the urine pH
fails to fall below 5.3 after an acid challenge test, involving the ingestion of ammonium chloride sufficient to
lower the plasma bicarbonate.
A number of features allow differentiation of types
of RTA. Proximal RTA is frequently associated withurinary wasting of amino acids, phosphate and glucose
(Fanconi’s syndrome), as well as bicarbonate and potassium.
Patients with this disorder can lower the urine pH
when the acidosis is severe and plasma bicarbonate
levels have fallen below 16 mmol/L since distal H+
secretion mechanisms are intact. In the classical form of
distal RTA, however, acid accumulation is relentless and
progressive, resulting in mobilisation of calcium from
bone and consequent osteomalacia with hypercalciuria, renal stone formation and nephrocalcinosis. Potassium
is also lost in classical distal RTA, while it is retained in
hyperkalaemic distal RTA.
Management
The first step in management of metabolic acidosis is to
identify and correct the underlying cause when possible
(see Box). This may involve controlling diarrhoea,
treating diabetes mellitus, correcting shock, stopping
drugs that might cause the condition, or using dialysis to
remove toxins. Since metabolic acidosis is frequently
associated with sodium and water depletion, resuscitation
with intravenous fluids is often needed. Use of
intravenous bicarbonate in this setting is controversial.
Because rapid correction of acidosis can induce hypokalaemia or a fall in plasma ionised calcium, the use of bicarbonate infusions is best reserved for situations
where the underlying disorder cannot be readily corrected
and acidosis is severe (H+ > 100 nmol/L, pH
< 7.00) or associated with evidence of tissue dysfunction.
The acidosis in RTA can sometimes be controlled by
treating the underlying cause , but supplements of sodium and potassium bicarbonate are usually also necessary in types 1 and 2 RTA to achieve a target plasma bicarbonate level of above 18 mmol/L and normokalaemia. In type 4 RTA, loop diuretics, thiazides or fludrocortisone (as appropriate to the underlying diagnosis) may be effective in correcting the acidosis and the hyperkalaemia.
Metabolic alkalosis
Aetiology and clinical assessmentMetabolic alkalosis is characterised by an increase in the
plasma bicarbonate concentration and the plasma pH.
There is a compensatory rise in PCO2 due to hypoventilation but this is limited by the need to avoid hypoxia. The causes classified by the accompanying changes in ECF volume. Hypovolaemic metabolic alkalosis is the most common pattern. This can be caused by sustained vomiting, in which acid-rich fluid is lost directly from the body, or by treatment with loop diuretics or thiazides. In the case of sustained vomiting, loss of gastric acid is the immediate cause of the alkalosis, but several factors act to sustain or amplify this in the context of volume depletion.
Loss of sodium and fluid leads to hypovolaemia and secondary hyperaldosteronism, triggering
proximal sodium bicarbonate reabsorption and
additional acid secretion by the distal tubule. Hypokalaemia occurs due to potassium loss in the vomitus
and by the kidney as the result of secondary hyperaldosteronism, and itself is a stimulus to acid secretion.
Additionally, the compensatory rise in PCO2 further
enhances tubular acid secretion. The net result is sustained
metabolic alkalosis with an inappropriately acid
urine, which cannot be corrected until the deficit in
circulating volume has been replaced.
Normovolaemic (or hypervolaemic) metabolic alkalosis
occurs when bicarbonate retention and volumeexpansion occur simultaneously. Classical causes
include primary hyperaldosteronism (Conn’s syndrome,
Cushing’s syndromeand corticosteroid therapy). Occasionally, overuse of antacid salts for treatment of dyspepsia produces a similar pattern.
Clinically, apart from manifestations of the underlying
cause, there may be few symptoms or signs related
to alkalosis itself. When the rise in systemic pH is abrupt, plasma ionised calcium falls and signs of increased neuromuscular irritability, such as tetany, may develop.
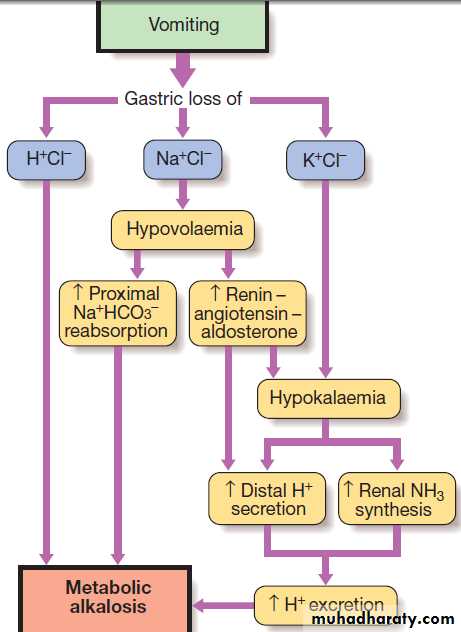
Generation and maintenance of metabolic alkalosis
during prolonged vomiting. Loss of H+Cl− generates metabolic
alkalosis, which is maintained by renal changes.
Management
Metabolic alkalosis with hypovolaemia can be correctedby intravenous infusions of 0.9% saline with potassium
supplements. This reverses the secondary hyperaldosteronism and allows the kidney to excrete the excess alkali in the urine.
In metabolic alkalosis with normal or increased
volume, treatment should focus on management of the
underlying endocrine cause.
Respiratory acidosis
Respiratory acidosis occurs when there is accumulationof CO2 due to type II respiratory failure . This
results in a rise in the PCO2, with a compensatory
increase in plasma bicarbonate concentration, particularly
when the disorder is of long duration and the
kidney has fully developed its capacity for increased
acid excretion.
This acid–base disturbance can arise from lesions anywhere along the neuromuscular pathways from the brain to the respiratory muscles that result in impaired ventilation.
It can also arise during intrinsic lung disease if there
is significant mismatching of ventilation and perfusion.
Clinical features are primarily those of the underlying
cause of the respiratory disorder such as paralysis, chest
wall injury or chronic obstructive lung disease, but the
CO2 accumulation may itself lead to drowsiness that
further depresses respiratory drive.
Management involves correction of causative factors
where possible, but ultimately ventilatory support may
be necessary.
Respiratory alkalosis
Respiratory alkalosis develops when there is a periodof sustained hyperventilation, resulting in a reduction
of PCO2 and increase in plasma pH. If the condition
is sustained, renal compensation occurs, such that
tubular acid secretion is reduced and the plasma bicarbonate falls.
Respiratory alkalosis is usually of short duration, occurring in anxiety states or as the result of overvigorous assisted ventilation. It can be prolonged in the context of pregnancy, pulmonary embolism, chronic liver disease, and ingestion of certain drugs such as salicylates that directly stimulate the respiratory centre in the brainstem.
Clinical features are those of the underlying cause but
agitation associated with perioral and digital tinglingmay also occur, due to a reduction in ionised calcium
concentrations because of increased binding of calcium
to albumin as the result of the alkalosis. In severe cases,
Trousseau’s sign and Chvostek’s sign may be positive,
and tetany or seizures may develop .
Management involves correction of identifiable
causes, reduction of anxiety, and a period of rebreathing
into a closed bag to allow CO2 levels to rise.
Mixed acid–base disorders
It is not uncommon for more than one disturbance of acid–base metabolism to be present at the same time : for example, respiratory acidosis due to narcotic overdose with metabolic alkalosis due to vomiting. In these situations, the arterial pH will represent the net effect of all primary and compensatory changes. Indeed, the pH may be normal, but the presence of underlying acid–base disturbances can be gauged from concomitant abnormalities in the PCO2 and bicarbonate concentration. In assessing these disorders, all clinical influences on the patient’s acid–base status should be identified .
DISORDERS OF DIVALENT ION METABOLISM
The present section excludes discussion of calcium disorders.Functional anatomy and physiology of magnesium metabolism
Like potassium, magnesium is mainly an intracellular cation. It is important to the function of many enzymes, including the Na,K-ATPase, and can regulate both potassium and calcium channels. Its overall effect is to stabilise excitable cell membranes. Renal handling of magnesium involves filtration of free plasma magnesium at the glomerulus (about 70% of the total) with extensive reabsorption (50–70%) in the loop of Henle, and other parts of the proximal and distal renal tubule. Magnesium reabsorption is also enhanced by parathyroid hormone (PTH).
Presenting problems in disorders of magnesium metabolism Hypomagnesaemia
Aetiology and clinical assessmentHypomagnesaemia exists when plasma magnesium
concentrations are below the reference range of 0.75–
1.0 mmol/L (1.5–2.0 meq/L). This is usually a reflection
of magnesium depletion , which can be caused by excessive magnesium loss from the gastrointestinal
tract (notably in chronic diarrhoea) or the kidney (during prolonged use of loop diuretics).
Excessive alcohol ingestion can cause magnesium depletion through both gut and renal losses. Some inherited tubular transport disorders, such as Gitelman’s syndrome, can also result in urinary magnesium wasting.
Hypomagnesaemia is frequently associated with
hypocalcaemia, probably because magnesium is required for the normal secretion of PTH in response to a fall in serum calcium, and because hypomagnesaemia causes end-organ resistance to PTH. The clinical features of hypomagnesaemia and hypocalcaemia are similar in that tetany, cardiac arrhythmias (notably torsades de pointes, central nervous excitation and seizures, vasoconstriction and hypertension may all occur. Magnesiumdepletion is also associated (through uncertain mechanisms)
with hyponatraemia and hypokalaemia, which
may contribute to some of the clinical manifestations.
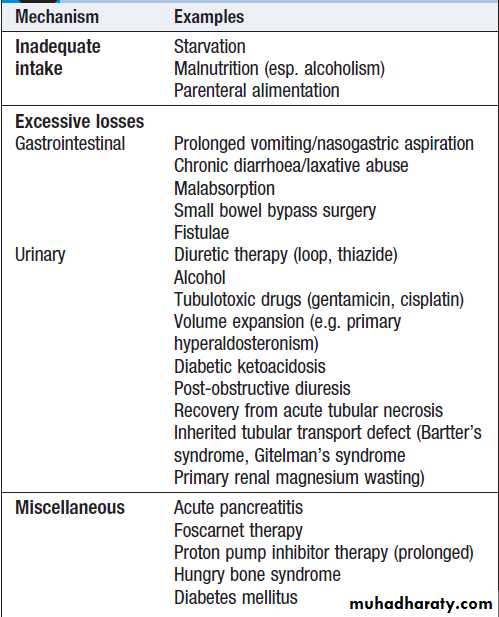
Causes of hypomagnesaemia
ManagementThe underlying cause should be identified and treated
where possible. When symptoms are present, the treatment
of choice is intravenous magnesium chloride at a
rate not exceeding 0.5 mmol/kg in the first 24 hours.
When intravenous access is not available, magnesium
sulphate can be given intramuscularly. Oral magnesium
salts have limited effectiveness due to poor absorption
and may cause diarrhoea.
If hypomagnesaemia is due to diuretic treatment, adjunctive use of a potassium sparing agent can also help by reducing magnesium loss into the urine.
Hypermagnesaemia
This is a much less common than hypomagnesaemia.Predisposing conditions include AKI , CKD, and adrenocortical insufficiency. Precipitated in patients at risk by an increased intake of magnesium, or through the use of magnesium-containing antacids, laxatives and enemas. Clinical features include bradycardia, hypotension, reduced consciousness and respiratory depression.
Management involves ceasing all magnesium containing drugs and reducing dietary magnesium intake, and promoting urinary magnesium excretion using a loop diuretic with IV hydration. Calcium gluconate may be given intravenously to ameliorate cardiac effects. Dialysis may be necessary in patients with poor renal function.
Functional anatomy and physiology of phosphate metabolism
Inorganic phosphate (mainly present as HPO4 2−) is intimately involved in cell energy metabolism, intracellular
signalling and bone and mineral balance .The
normal plasma concentration is 0.8–1.4 mmol/L (2.48–4.34 mg/dL). It is freely filtered at the glomerulus and approximately 65% is reabsorbed by the proximal tubule, via an apical sodium–phosphate co-transport carrier. A further 10–20% is reabsorbed in the distal tubules, leaving a fractional excretion of some 10% to pass into the urine, usually as H2PO4 −. Proximal reabsorption is decreased by PTH, fibroblast growth factor 23 (FGF-23), volume expansion, osmotic diuretics and glucose infusion.
Presenting problems in disorders of phosphate metabolism
HypophosphataemiaThe causes of hypophosphataemia are shown in Box .
Phosphate may redistribute into cells during periods of increased energy utilisation (such as refeeding after a period of starvation) and during systemic alkalosis. However, severe hypophosphataemia usually represents an overall body deficit due to either inadequate intake or absorption through the gut, or excessive renal losses, most notably in primary hyperparathyroidism or as the result of acute plasma volume expansion, osmotic diuresis and diuretics acting on the proximal renal tubule.
Less common causes include inherited defects of proximal sodium–phosphate co-transport and tumour-induced osteomalacia due to ectopic production of the hormone FGF-23 .
The clinical manifestations of phosphate depletion
are wide-ranging, reflecting the involvement of phosphate
in many aspects of metabolism. Defects appear in
the blood (impaired function and survival of all cell
lines), skeletal muscle (weakness, respiratory failure),
cardiac muscle (congestive cardiac failure), smooth
muscle (ileus), central nervous system (decreased consciousness, seizures and coma) and bone (osteomalacia
in severe prolonged hypophosphataemia).
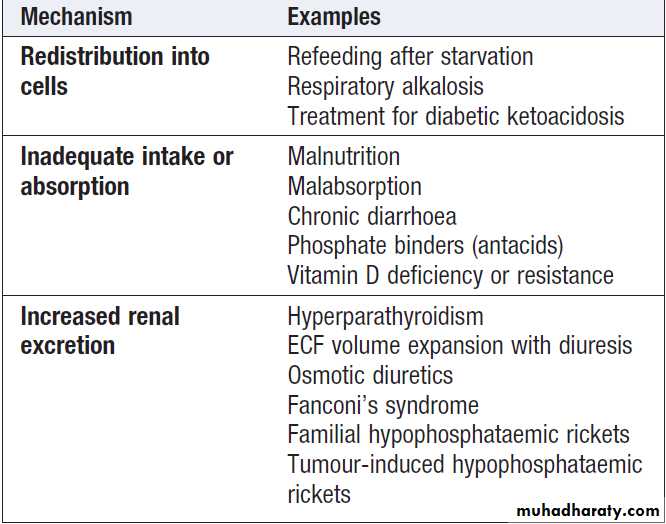
Causes of hypophosphataemia
Managementinvolves administering oral phosphate supplements and high-protein/high-dairy dietary supplements that are rich in naturally occurring phosphate.
Intravenous treatment with sodium or potassium phosphate
salts can be used in critical situations, but there is a risk of precipitating hypocalcaemia and metastatic calcification.
Hyperphosphataemia
Usually the result of AKI or CKD. Phosphate excretion is also reduced in hypoparathyroidism and pseudohypoparathyroidism . Redistribution from cells into the plasma can also be a contributing factor in the ‘tumour lysis’ syndrome and in catabolic states. Phosphate accumulation is aggravated if the patient takes phosphate-containing preparations or inappropriate vitamin D therapy. The clinical features relate to hypocalcaemia and metastatic calcification, particularly in CKD with tertiary hyperparathyroidism (a high calcium–phosphate product occurs).If renal function is normal, normal saline should be given to promote phosphate excretion.In renal failure should be treated with dietary phosphate restriction and the use of oral phosphate binders.DISORDERS OF AMINO ACID METABOLISM
Phenylketonuria (PKU)Autosomal recessive disorder caused by loss-of-function mutations in the PAH gene, which encodes phenylalanine hydroxylase, an enzyme required for degradation of phenylalanine.
As a result, phenylalanine accumulates at high
levels in the neonate’s blood, causing mental retardation.
The diagnosis of PKU is almost always made by routine neonatal screening .
Treatment involves life-long adherence to a low-phenylalanine diet. Early and adequate dietary treatment prevents major mental retardation, although there may still be a slight reduction in IQ.
Homocystinuria
Autosomal recessive disorder caused by loss-of-function mutations in the CBS gene, causes cystathionine beta–synthase deficiency and accumulation of homocysteine and methionine in the blood. Many cases are diagnosed through newborn screening programmes.Clinical manifestations are wide-ranging , involve the eyes (ectopia lentis – displacement of the lens), mental retardation, delayed development,seizures,psychiatric disturbances), skeleton (resembling Marfan’s syndrome, and osteoporosis), vascular system (thrombotic lesions of arteries and veins) and skin (hypopigmentation).
Treatment is dietary, involving a methionine restricted,
cystine-supplemented diet, and large doses of pyridoxine.
DISORDERS OF CARBOHYDRATE METABOLISM
The most common disorder of carbohydrate metabolism
is diabetes mellitus, which is discussed.
There are also some rare inherited defects.
Galactosaemia
Galactosaemia is caused by loss-of-function mutations
in the GALT gene, which encodes galactose-1-phosphate
uridyl transferase. It is usually inherited as an autosomal
recessive disorder. The neonate is unable to metabolise galactose, one of the hexose sugars contained in lactose. Vomiting or diarrhoea usually begins within a few days of ingestion of milk, and the neonate may become jaundiced. Failure to thrive is the most common presentation.
The classic form of the disease results in hepatomegaly, cataracts and mental retardation, and fulminant infection with Escherichia coli is a frequent complication.
Treatment involves life-long avoidance of
galactose- and lactose-containing foods.
The widespread inclusion of galactosaemia in
newborn screening programmes has resulted in the
identification of a number of milder variants.
Glycogen storage diseases
(GSD, or glycogenoses) result from an inherited defect in one of the many enzymes responsible for the formation or breakdown of glycogen, a complex carbohydrate that can be broken down quickly to release glucose during exercise or between meals.
There are several major types of GSD, which are classified
by a number, by the name of the defective enzymeor eponymously after the physician who first described
the condition (Box). Most forms of GSD are inherited
as autosomal recessive disorders.
A diagnosis of GSD is made on the basis of the symptoms, physical examination and results of biochemical tests. Occasionally, a muscle or liver biopsy is required to confirm the enzyme defect. Different types of GSD present at different ages, and some may require life-long modifications of diet and lifestyle.
Glycogen storage diseases
DISORDERS OF COMPLEX LIPID METABOLISMComplex lipids are key components of the cell membrane
that are normally catabolised in organelles
called lysosomes. The lysosomal storage diseases are a
heterogeneous group of disorders caused by loss-offunction
mutations in various lysosomal enzymes, resulting in an inability to break down complex glycolipids or other intracellular macromolecules. These disorders have diverse clinical manifestations, typically including mental retardation. Some can be treated with enzyme replacement therapy, while others (such as Tay–
Sachs disease) can be prevented through community participation in genetic carrier screening programmes.
Lysosomal storage diseases
DISORDERS OF BLOOD LIPIDS AND LIPOPROTEINSThe three most important classes of lipid are cholesterol,
which is composed of hydrocarbon rings; triglycerides
(TG), which are esters composed of glycerol linked to
three long-chain fatty acids; and phospholipids, which
are composed of a hydrophobic ‘tail’ consisting of two
long-chain fatty acids linked through glycerol to a
hydrophilic head containing a phosphate group. Phospholipids are present in cell membranes and are important signalling molecules.
Despite their poor water solubility, lipids need to
be absorbed from the gastrointestinal tract and transported throughout the body.
This is achieved by incorporating lipids within lipoproteins. Plasma cholesterol and TG are clinically important because they are major treatable risk factors for cardiovascular disease, whilst severe hypertriglyceridaemia also predisposes to acute pancreatitis.
Functional anatomy, physiology and investigation of lipid metabolism
Lipids are transported and metabolised by apolipoproteins, which combine with lipids to form spherical or disc-shaped lipoproteins, consisting of a hydrophobic core and a less hydrophobic coat.The structure of some apolipoproteins also enables them to act as enzyme co-factors or cell receptor ligands. Variations in lipid and apolipoprotein composition result in distinct classes of lipoprotein that perform specific metabolic functions.
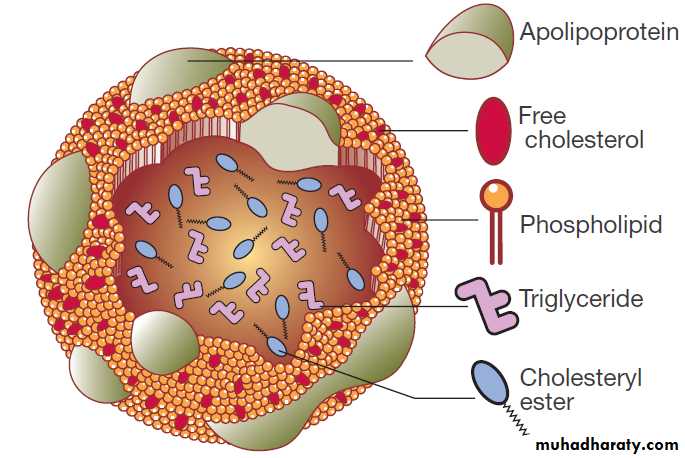
Structure of lipoproteins.
Processing of dietary lipidEnterocytes lining the gut extract monoglyceride and free fatty acids from micelles and re-esterify them into TG, which are combined with a truncated form of apolipoprotein B (Apo B48) as it is synthesised.
Intestinal cholesterol derived from dietary and biliary sources is also absorbed via a specific intestinal
membrane transporter termed NPC1L1. This produces
chylomicrons containing TG and cholesterol ester
that are secreted basolaterally into lymphatic lacteals
and carried to the circulation through the thoracic duct.
Upon entering the blood stream, nascent chylomicrons
are modified by further addition of apolipoproteins.
Chylomicron TG are hydrolysed by lipoprotein lipase
located on the endothelium of tissue capillary beds. This
releases fatty acids that are used locally for energy production or stored as TG in muscle or fat. The residual
‘remnant’ chylomicron particle is avidly cleared by lowdensity lipoprotein (LDL)-receptors in the liver, which
recognise Apo E on the remnant lipoproteins.
Complete absorption of dietary lipids takes about 6–10 hours, so chylomicrons are usually undetectable in the plasma after a 12-hour fast.
The main dietary determinants of plasma cholesterol
concentrations are the intake of saturated and transunsaturated fatty acids, which reduce LDL-receptoractivity (see below). Dietary cholesterol has surprisingly
little effect on fasting cholesterol levels. Plant sterols
and drugs that inhibit cholesterol absorption are effective
because they also reduce the re-utilisation of biliary
cholesterol. The dietary determinants of plasma TG concentrations are complex since excessive intake of carbohydrate, fat or alcohol may all contribute to increased plasma TG by different mechanisms.
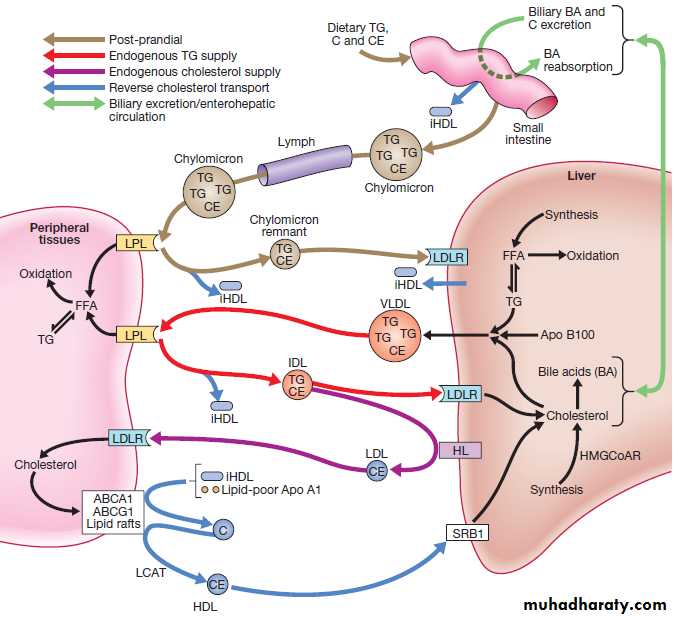
Fig. Absorption, transport and storage of lipids. Pathways of lipid transport are shown; in addition, cholesterol ester transfer protein
exchanges triglyceride and cholesterol ester between VLDL/chylomicrons and HDL/LDL, and free fatty acids released from peripheral lipolysis can be taken up in the liver. (ABCA1/ABCG1 = ATP-binding cassette A1/G1; Apo = apolipoprotein; BA = bile acids; C = cholesterol; CE = cholesterol ester; FFA = free
fatty acids; HDL = mature high-density lipoprotein; HL = hepatic lipase; HMGCoAR = hydroxy-methyl-glutaryl-coenzyme A reductase; IDL = intermediatedensity lipoprotein; iHDL = immature high-density lipoprotein; LCAT = lecithin cholesterol acyl transferase; LDL = low-density lipoprotein; LDLR = low-density lipoprotein receptor (Apo B100 receptor); LPL = lipoprotein lipase; SRB1 = scavenger receptor B1; TG = triglyceride;
VLDL = very low-density lipoprotein)
Endogenous lipid synthesis
In the fasting state, the liver is the major source of plasmalipids . The liver may acquire lipids by uptake, synthesis or conversion from other macronutrients.
These lipids are transported to other tissues by
secretion of very low-density lipoproteins (VLDL),
which are rich in TG but differ from chylomicrons in that
they contain full-length Apo B100. Following secretion
into the circulation, VLDL undergo metabolic processing
similar to that of chylomicrons. Hydrolysis of VLDL
TG releases fatty acids to tissues and converts VLDL into
‘remnant’ particles, referred to as intermediate-density
lipoproteins (IDL).
Most IDL are rapidly cleared by LDL receptors in the liver, but some are processed by hepatic lipase, which converts the particle to an LDL by removing TG and most materials other than Apo B100, and free and esterified cholesterol.
The LDL particles act as a source of cholesterol
for cells and tissues . LDL cholesterol is internalised by receptor-mediated endocytosis via the LDL receptor. Delivery of cholesterol via this pathway down-regulates further expression of the LDL receptor gene and reduces the synthesis and activity of the rate-limiting enzyme for cholesterol synthesis, HMGCoA reductase. This negative feedback loop, together with the modulation of cholesterol esterification, controls the intracellular free cholesterol level
within a narrow range.
Reverse cholesterol transport
Peripheral tissues are further guarded against excessive
cholesterol accumulation by HDL. Lipid-poor Apo A1 (derived from the liver, intestine and the outer layer of chylomicrons and VLDL) accepts cellular cholesterol and phospholipid from a specific membrane transporter known as the ATP-binding cassette A1 (ABCA1). This produces small HDL that are able to accept more free cholesterol from cholesterol-rich regions of the cell membrane known as ‘rafts’ via another membrane transporter (ABCG1). The cholesterol that has been accepted by these small HDL is esterified by lecithin cholesterol acyl transferase (LCAT), thus maintaining an uptake gradient and remodelling the particle into a mature spherical HDL.
These HDL release their cholesterol to the liver and
other cholesterol-requiring tissues via the scavengerreceptor B1 (SRB1).
The cholesterol ester transfer protein (CETP) in
plasma allows transfer of cholesterol from HDL or LDL
to VLDL or chylomicrons in exchange for TG. When TG
is elevated, the action of CETP may reduce HDL cholesterol
and remodel LDL into ‘small, dense’ LDL particles that appear to be more atherogenic in the blood vessel wall.
Animal species that lack CETP are resistant to atherosclerosis
Lipids and cardiovascular disease
Plasma lipoprotein levels are major modifiable riskfactors for cardiovascular disease. Increased levels of atherogenic lipoproteins (especially LDL, but also IDL,
lipoprotein(a) and possibly chylomicron remnants) contribute to the development of atherosclerosis .
Increased plasma concentration and reduced diameter
favour subendothelial accumulation of these lipoproteins.
Following chemical modifications such as oxidation,
Apo B-containing lipoproteins are no longer cleared
by normal mechanisms.
They trigger a self-perpetuating inflammatory response during which they are taken up by macrophages to form foam cells, a hallmark of atherosclerotic lesions. These processes also have an adverse effect on endothelial function.
Conversely, HDL removes cholesterol from the tissues to the liver, where it is metabolised and excreted in bile. HDL may also counteract some components of the inflammatory response, such as the expression of vascular adhesion molecules by the endothelium. Consequently, low HDL cholesterol levels, which are often associated with TG elevation, also predispose to atherosclerosis.
Lipid measurement
Abnormalities of lipid metabolism most commonlycome to light following routine blood testing. Measurement
of plasma cholesterol alone is not sufficient for
comprehensive assessment. Levels of total cholesterol
(TC), triglyceride (TG) and HDL cholesterol (HDL-C)
need to be obtained after a 12-hour fast to permit accurate calculation of LDL cholesterol (LDL-C) according to
the Friedewald formula
LDL-C = TC − HDL-C − (TG/2.2) mmol/L; or
LDL-C = TC − HDL-C − (TG/5) mg/dL.
The formula becomes unreliable when TG levels exceed 4 mmol/L (350 mg/dL).
Other risk markers, such as NHDL-C (non-HDL-C, calculated as the difference between TC and HDL-C levels) or Apo B,
may assess risk of cardiovascular disease more accurately
than LDL-C when TG levels are increased.
Furthermore, non-fasting samples are often used to guide
therapeutic decisions since they are unaffected in terms
of TC and measured LDL-C, albeit that they differ from
fasting samples in terms of TG, HDL-C and, to some
extent, calculated LDL-C. Consideration must be given
to confounding factors, such as recent illness, after
which cholesterol, LDL and HDL levels temporarily
decrease in proportion to severity.
Elevated levels of TG are common in obesity, diabetes
and insulin resistance and are frequently
associated with low HDL and increased ‘small, dense’
LDL. Under these circumstances, LDL-C may underestimate
risk. This is one situation in which measurement
of Apo B may provide additional useful information.
Presenting problems in disorders of lipids
Lipid measurements performed for the following reasons:
• screening for primary or secondary prevention of
cardiovascular disease
• investigation of patients with clinical features of
lipid disorders and their relatives
• monitoring of response to diet, weight control and
medication.
Aetiology and clinical assessment
The first step is to consider the effect of other diseases and drugs .Overt or subclinical hypothyroidism may cause hypercholesterolaemia, and so measurement of thyroid function is warranted in most cases, even in the absence of typical symptoms and signs. Once secondary causes are excluded, primary lipid abnormalities may be diagnosed. Primary lipid abnormalities can be classified according to the predominant lipid problem: hypercholesterolaemia, hypertriglyceridaemia or mixed hyperlipidaemia. Althoughsingle-gene disorders are encountered in all three categories, most cases are due to multiple-gene (polygenic) loci interacting with environmental factors.
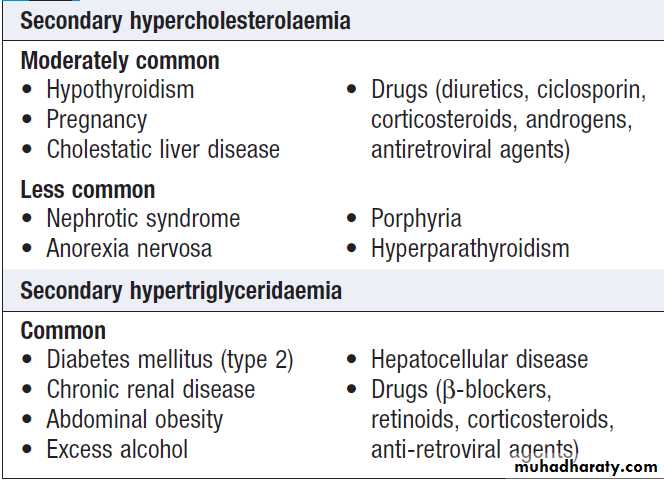
Causes of secondary hyperlipidaemia
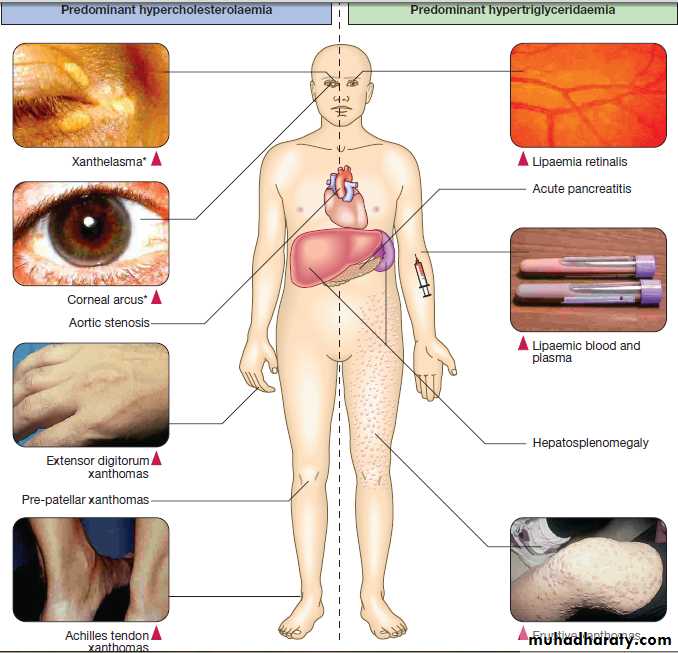
Clinical manifestations of hyperlipidaemia. *Note that xanthelasma and corneal arcus may be non-specific, especially in later life.
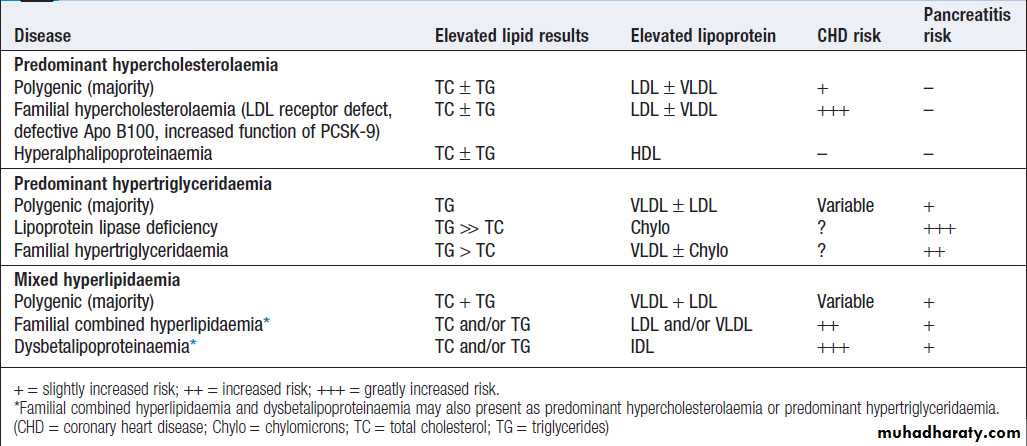
Classification of hyperlipidaemia
Predominant hypercholesterolaemiaPolygenic hypercholesterolaemia is the most common
cause of a mild to moderate increase in LDL-C. Physical signs, such as corneal arcus and xanthelasma, may be found in this as well as other forms of lipid disturbance .The risk of cardiovascular disease is proportional to the degree of LDL-C (or Apo B) elevation, but is modified by other major risk factors, particularly low HDL-C.
Familial hypercholesterolaemia (FH) causes moderate
to severe hypercholesterolaemia and has a prevalence
of at least 0.2% in most populations. It is usually caused by a loss-of-function mutation in the LDL receptor gene, which results in an autosomal dominant pattern of inheritance.
Affected subjects suffer from severe hypercholesterolaemia and premature cardiovascular disease. FH may be accompanied by xanthomas of the Achilles or extensor digitorum tendons , which are strongly suggestive of FH. The onset of corneal arcus before age 40 is also suggestive of this condition. Identification of an index case of FH (the first case of FH in a family) should trigger genetic and biochemical screening of other family members, which is a cost-effective method for case detection.
Affected individuals should be managed from childhood . Homozygous FH may occur in populations in which
there is a ‘founder’ effect or consanguineous marriage,
resulting in more extensive xanthomas and precocious
cardiovascular disease in childhood.
Hyperalphalipoproteinaemia refers to increased levels of HDL-C. In the absence of an increase in LDL-C, this condition does not cause cardiovascular disease, so it should not be regarded as pathological.
Familial combined hyperlipidaemia, and dysbetalipoproteinaemia, may present with the pattern of
predominant hypercholesterolaemia.

Familial hypercholesterolaemia in adolescence
Predominant hypertriglyceridaemiaPolygenic hypertriglyceridaemia is the most common
cause of a raised TG level . Other common causes include excess alcohol intake, medications (such as β-blockers and retinoids), type 2 diabetes, impaired glucose tolerance, central obesity or other manifestations of insulin resistance and impaired absorption of bile acids. It is often accompanied reduced HDL-C, both of which may contribute to cardiovascular risk. Excessive intake of alcohol or dietary fat, or other exacerbating factors may precipitate a massive increase in TG levels, which, if they exceed 10 mmol/L (880 mg/dL), may pose a risk of acute pancreatitis.
Inherited forms of hypertriglyceridaemia also occur. It often presents in childhood and is associated with episodes of acute abdominal pain and pancreatitis. In common with other causes of severe hypertriglyceridaemia, it may result in hepatosplenomegaly, lipaemia retinalis and eruptive xanthomas.
Familial hypertriglyceridaemia may also be inherited
in a dominant manner. This disorder may be associated with high levels of TG that predispose to
cardiovascular disease and pancreatitis.
Familial combined hyperlipidaemia, and dysbetalipoproteinaemia, may present with the pattern of predominant hypertriglyceridaemia.
Mixed hyperlipidaemia
The term ‘mixed’ usually implies the presence of hypertriglyceridaemia, as well as an increase in LDL or IDL. Treatment of massive hypertriglyceridaemiamay improve TG faster than cholesterol, thus temporarily mimicking mixed hyperlipidaemia.
Usually polygenic and, often occurs in association with type 2 diabetes, impaired glucose tolerance, central obesity or other manifestations of insulin resistance .
Both components of mixed hyperlipidaemia may contribute to the risk of cardiovascular disease.
Dysbetalipoproteinaemia (also referred to as type 3
hyperlipidaemia, broad-beta dyslipoproteinaemia or
remnant hyperlipidaemia) involves accumulation of
roughly equimolar levels of cholesterol and TG.
It is caused by homozygous inheritance of the Apo E2 allele.
In conjunction with other exacerbating factors,
such as obesity and diabetes, it leads to accumulation of
atherogenic IDL and chylomicron remnants. Premature
cardiovascular disease is common and it may also result
in the formation of palmar xanthomas, tuberous xanthomas or tendon xanthomas.
Management of dyslipidaemia
Lipid-lowering therapies have a key role in the secondaryand primary prevention of cardiovascular diseases.
Assessment of absolute risk, treatment of all modifiable risk factors and optimisation of lifestyle, especially diet and exercise, are central to management in all cases.
Patients with the greatest absolute risk of cardiovascular
disease will derive the greatest absolute benefit
from treatment. Public health organisations recommend
thresholds for the introduction of lipid-lowering therapy
based on the identification of patients in very high-risk
categories, or those calculated to be at high absolute risk.
In general, patients who have CVD , diabetes mellitus, CKD , familial hypercholesterolaemia or an absolute risk of cardiovascular disease of > 20% in the ensuing 10 years are arbitrarily regarded as having
sufficient risk to justify drug treatment.
Public health organisations also recommend target
levels for patients receiving drug treatment.
High-risk patients should aim for HDL-C > 1 mmol/L (38 mg/dL) and fasting TG < 2 mmol/L (180 mg/ dL), whilst target levels for LDL-C 2.0 mmol/L (76 mg/dL) or less. Total cholesterol should be < 5 mmol/L (190 mg/dL) during treatment, and < 4 mmol/L (150 mg/dL) in high-risk patients and in secondary prevention of CVD.
Non-pharmacological management
• reduce intake of saturated and trans-unsaturated fatto less than 7–10% of total energy
• reduce intake of cholesterol to < 250 mg/day
• replace sources of saturated fat and cholesterol with
alternative foods, such as lean meat, low-fat dairy
products, polyunsaturated spreads and low glycaemic index carbohydrates.
• reduce energy-dense foods such as fats , soft drinks, and increasing activity and exercise to maintain or lose weight. • increase consumption of cardioprotective and
nutrient-dense foods, such as vegetables, unrefined
carbohydrates, fish, pulses, nuts, legumes, fruit, etc.
• adjust alcohol consumption, reducing intake if
excessive or if associated with hypertension,hypertriglyceridaemia or central obesity
• achieve additional benefits with supplementary
intake of foods containing lipid-lowering nutrients
such as n-3 fatty acids, dietary fibre and plant sterols.
The response to diet is usually apparent within 3–4
weeks. Even minor weight loss can substantially reduce cardiovascular risk, especially in centrally obese patients .
All other modifiable cardiovascular risk factors should be assessed and treated.
If possible, Intercurrent drug treatments that adversely affect the lipid profile should be replaced.
Pharmacological management
Predominant hypercholesterolaemia
Statins. These reduce cholesterol synthesis by inhibiting
the HMGCoA reductase enzyme. The reduction in
cholesterol synthesis up-regulates activity of the LDL
receptor, which increases clearance of LDL and its precursor, IDL, resulting in a secondary reduction in LDL
synthesis. Statins reduce LDL-C by up to 60%, reduce
TG by up to 40% and increase HDL-C by up to 10%. They also reduce the concentration of intermediate
metabolites such as isoprenes, which may lead to other
effects such as suppression of the inflammatory response.
There is clear evidence of protection against total and
coronary mortality, stroke and cardiovascular events.
Statins are generally well tolerated and serious side effects are rare (< 2%). Liver function test abnormalities and muscle problems, such as myalgia, asymptomatic increase in CK, myositis and, infrequently, rhabdomyolysis, are the most common. Side-effects are more likely in patients who are elderly, debilitated or receiving other drugs that interfere with statin degradation, which usually involves cytochrome P450 3A4 or glucuronidation.

Benefits of using statins to treat hypercholesterolaemia
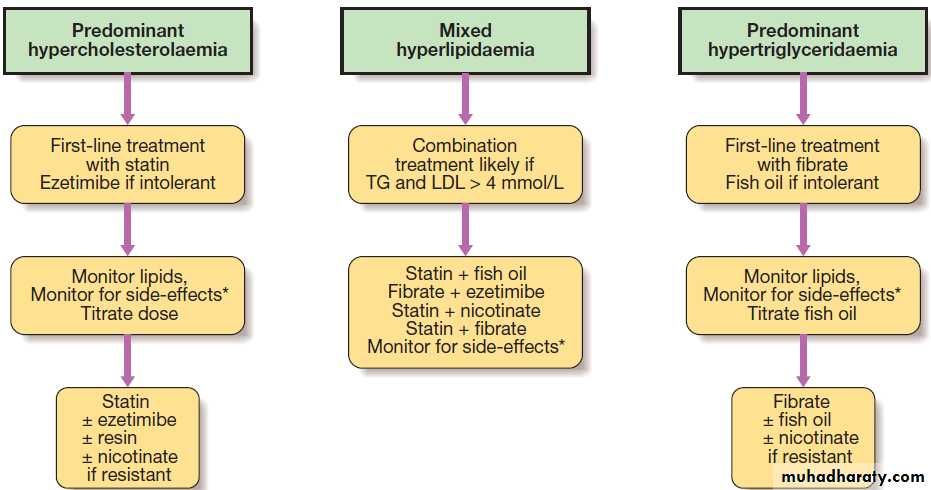
Drug treatment of hyperlipidaemia. *Interrupt treatment if CK is more than 5–10 times the upper limit of normal, or if elevated with muscle symptoms, or if ALT is more than 2–3 times the upper limit. To convert TG in mmol/L to mg/dL, multiply by 88.
To convert LDL-C in mmol/L to mg/dL, multiply by 38.
Cholesterol absorption inhibitors. The only licensed
drug in this class is ezetimibe, which inhibits activity ofthe intestinal mucosal transporter NPC1L1 that absorbs
dietary and biliary cholesterol. Depletion of hepatic cholesterol up-regulates hepatic LDL receptor activity. This
mechanism of action is synergistic with the effect of statins. Monotherapy with the standard 10 mg/day dose reduces LDL-C by 15–20%. Slightly greater (17–25%) incremental LDL-C reduction occurs when ezetimibe is added to statins. Ezetimibe is well tolerated, and evidence of a beneficial effect on cardiovascular disease endpoints may be inferred from a trial of combination therapy with simvastatin. Plant sterol supplemented foods, which also reduce cholesterol absorption, lower LDL-C by 7–15%.
Bile acid sequestering resins. Drugs in this class include
colestyramine, colestipol and colesevelam. These preventthe reabsorption of bile acids, thereby increasing de novo bile acid synthesis from hepatic cholesterol. As with ezetimibe, the resultant depletion of hepatic cholesterol up-regulates LDL receptor activity and reduces LDL-C in a manner that is synergistic with the action of statins. Resins reduce LDL-C and modestly increase HDL-C, but may increase TG. They are safe but may interfere with bioavailability of other drugs. Colesevelam has fewer gastrointestinal effects than older preparations that are less well tolerated. Development of specific inhibitors of the intestinal bile acid transporter may further improve tolerability of this class of agent.
Nicotinic acid. Pharmacological doses reduce peripheral
fatty acid release with the result that VLDL and LDLdecline whilst HDL-C increases. Randomised clinical
trials have been inconsistent regarding effects on atherosclerosis and cardiovascular events.
Side-effects include flushing, gastric irritation, liver function disturbances, and exacerbation of gout and hyperglycaemia.
Slow release formulations and low-dose aspirin may reduce flushing.
Combination therapy with the prostaglandin D2 receptor inhibitor laropiprant to reduce flushing further is being evaluated.
Combination therapy. In many patients, treatment of
predominant hypercholesterolaemia can be achieved by
diet plus the use of a statin in sufficient doses to achieve
target LDL-C levels. Patients who do not reach LDL
targets on the highest tolerated statin dose, or who are
intolerant of statins, may receive ezetimibe, plant sterols,
nicotinic acid or resins. Ezetimibe and resins are safe and
effective in combination with a statin, but
nicotinic acid with a statin requires a greater caution because the risk of side-effects is slightly increased.
Predominant hypertriglyceridaemia
Predominant hypertriglyceridaemia can be treated withone of the TG-lowering drugs .
Fibrates. These stimulate peroxisome proliferator activated
receptor (PPAR) alpha, which controls the expression
of gene products that mediate the metabolism of TG
and HDL. As a result, synthesis of fatty acids, TG and
VLDL is reduced, whilst that of lipoprotein lipase, which
catabolises TG, is enhanced. In addition, production of
Apo A1 and ATP binding cassette A1 is up-regulated,
leading to increased reverse cholesterol transport via
HDL. Consequently, fibrates reduce TG by up to 50%and increase HDL-C by up to 20%, but LDL-C variable.
Fewer large-scale trials have been conducted with
fibrates than with statins and the results are less conclusive,
but reduced rates of cardiovascular disease
have been reported with fibrate therapy in the subgroup
of patients with low HDL-C levels and elevated TG.
Fibrates are usually well tolerated but share a similar side-effect profile to statins. In addition, they may increase the risk of cholelithiasis and prolong the action of anticoagulants.
Accumulating evidence suggests that they may
also have a protective effect against diabetic microvascular complications.
Highly polyunsaturated long-chain n-3 fatty acids.
These include eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which comprise approximately 30% of the fatty acids in fish oil. EPA and DHA are potent inhibitors of VLDL TG formation. Intakes ofgreater than 2 g n-3 fatty acid (equivalent to 6 g of most
forms of fish oil) per day lower TG in a dose-dependent
fashion. Up to 50% reduction in TG may be achieved
with 15 g fish oil per day. Fish oil fatty acids have also been shown to inhibit platelet aggregation and improve arrhythmia in animal models. Dietary and pharmacological trials suggest that n-3 fatty acids may reduce mortality from CHD . Fish oils appear to be safe and well tolerated.
Patients with predominant hypertriglyceridaemia
who do not respond to lifestyle intervention can betreated with fibrates, fish oil or nicotinic acid, depending
on individual response and tolerance. If target levels are
not achieved, the fibrates and fish oil or nicotinic acid
can be combined. Massive hypertriglyceridaemia may
require more aggressive limitation of dietary fat intake
(< 10–20% energy as fat). Any degree of insulin deficiency should be corrected because insulin is required
for optimal activity of lipoprotein lipase. The initial
target for patients with massive hypertriglyceridaemia
is TG < 10 mmol/L (880 mg/dL), to reduce the risk of
acute pancreatitis.
Mixed hyperlipidaemia
Mixed hyperlipidaemia can be difficult to treat.
Statins alone are less effective first-line therapy once fasting TG exceeds around 4 mmol/L (350 mg/dL). Fibrates are first-line therapy for dysbetalipoproteinaemia, but they may not control the cholesterol component in other forms of mixed hyperlipidaemia.
Combination therapy
is often required. Effective combinations include: statin
plus fish oil when TG is not too high; fibrate plus
ezetimibe; statin plus nicotinic acid; or statin plus fibrate.
The risk of myopathy is increased with gemfibrozil, but
fenofibrate is relatively safe in this regard.
Monitoring of therapy
The effect of drug therapy should be assessed after6 weeks (12 weeks for fibrates). At this point, it is prudent
to review side-effects, lipid response (see target levels
above), CK and liver function tests. During longer-term
follow-up, compliance with drug treatment, diet and
exercise should be assessed, with monitoring of weight,
blood pressure and lipid levels. The presence of cardiovascular symptoms or signs should be noted and absolute CV risk assessed periodically.
It is not necessary to perform routine checks of CK and liver function unless symptoms occur, or if statins are used in combination with fibrates, nicotinic acid or other drugs that may interfere with their clearance.
If myalgia or weakness occurs in association with CK elevation over 5–10 times the upper limit of normal, or if sustained ALT elevation more than 2–3 times the upper limit of normal occurs that is not accounted for by fatty liver , treatment should be discontinued and alternative therapy sought.
The principles of the management of dyslipidaemia can be applied broadly, but the objectives of treatment in the elderly and the safety of pharmacological therapy in pregnancy warrant special consideration.

Management of hyperlipidaemia in old age

Dyslipidaemia in pregnancy
DISORDERS OF HAEM SYNTHESIS – THE PORPHYRIASThe porphyrias are a group of disorders caused by
inherited abnormalities in the haem biosynthetic
pathway , due to partial enzyme deficiencies with a dominant mode of inheritance.
Classified as either hepatic or erythropoietic, depending on whether the major site of excess porphyrin production is in the liver or in the red cell. Environmental factors are important in disease expression in some forms. In the most common of these conditions, porphyria cutanea tarda (PCT), these include alcohol, iron accumulation, exogenous oestrogens and exposure to various chemicals. Many cases are associated with hepatitis C infection and this should always be screened for on presentation.
Clinical features
Fall into two broad categories, photosensitivity and acute neurovisceral syndrome.Photosensitive skin manifestations, attributable to excess production and accumulation of porphyrins in the skin, cause pain, erythema, bullae, skin erosions, hirsutism and hyperpigmentation, and occur predominantly on areas of the skin that are exposed to sunlight. The skin also becomes sensitive to damage from minimal trauma. The other pattern of presentation is with an
Acute neurological syndrome.
This presents with acute abdominal pain together with features of autonomic dysfunction such as tachycardia, hypertension and constipation.
Neuropsychiatric manifestations, hyponatraemia due to
inappropriate ADH release , and an acute neuropathy may also occur .
The neuropathy is usually motor and may, in severe cases, progress to respiratory failure.
There is no proven explanation for the episodic
nature of the attacks in porphyria, which can relapse and
remit or follow a prolonged and unremitting course.
Sometimes, specific triggers can be identified, such as
alcohol, fasting, or drugs such as anticonvulsants, sulphonamides, oestrogen and progesterone. The oral contraceptive pill is a common precipitating factor. In a
significant number, no precipitant can be identified.
Diagnosis
The diagnosis relied on the pattern of the porphyrins and porphyrin precursors found in blood, urine and faeces . This is a straightforward diagnosis at clinical presentation when the metabolites are significantly elevated, but this is not always the case in asymptomatic individuals. More recently, measurement of the enzymes that are deficient in the various porphyrias has provided further diagnostic information (for example, PBG deaminase activity in red blood cells to diagnose acute intermittent porphyria). However, there is often considerable overlap between enzyme activities in affected and normal subjects.Metabolite excretory patterns are always grossly
abnormal during an acute attack. A normal metabolite profile under these circumstances effectively excludes porphyria. Metabolites usually remain abnormal for long periods after an acute attack, and in some individuals never return to normal. The diagnosis is not so straightforward in patients who are in remission, or in asymptomatic individuals with a positive family history. Neurological porphyria rarely manifests before puberty, nor can it be readily diagnosed from metabolite patterns after the menopause. In these circumstances, genetic testing for disease-specific mutations can now clarify the situation.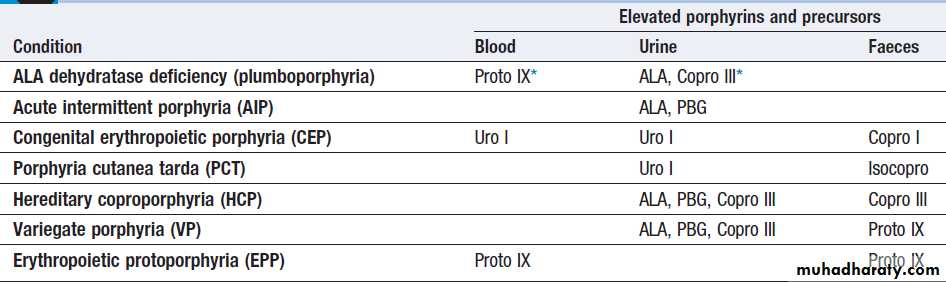
Diagnostic biochemical findings in the porphyrias
Management
Management of neurovisceral attacks, includes avoidance of any agents known to precipitate acute porphyria. Specific management includes intravenous glucose, as provision of 5000 kilojoules per day can, in some cases, terminate acute attacks through a reduction in ALA synthetase activity, leading to reduced ALA synthesis.
More recently, administration of haem (in various forms such as haematin or haem arginate) has been shown to reduce metabolite excretory rates, relieve pain and accelerate recovery. Cyclical acute attacks in women sometimes respond to suppression of the menstrual cycle using gonadotrophin-releasing hormone analogues. In rare intractable to treatment, liver transplantation has been effective.
The primary goal is to avoid sun exposure and skin trauma. Barrier sun creams containing zinc or titanium oxide are the most effective products. New colourless creams containing nanoparticle formulations have improved patient acceptance. Beta-carotene is used in some patients with erythropoietic porphyria with some efficacy. Afamelanotide, a synthetic analogue of α-MSH), has also been shown to provide protection in erythropoietic protoporphyria. In porphyria cutanea tarda, a course of venesections to remove iron can result in long-lasting clinical and biochemical remission, especially if exposure to identified precipitants, such as alcohol or oestrogens, is reduced. Alternatively, a prolonged course of low-dose chloroquine therapy is also effective.