1
Fifth stage
Pediatrics
Lec 3
Dr. Athl
1/3/2017
Diabetic Keto Acidosis
DKA is the end result of metabolic abnormalities resulting from a severe deficiency of
insulin or insulin effectiveness, resulting in:
:
DKA occur in the following conditions
1. 20-40% of children with new onset diabetes.
2. Omitted insulin doses.
3. Unsuccessful management of an intercurrent illness.
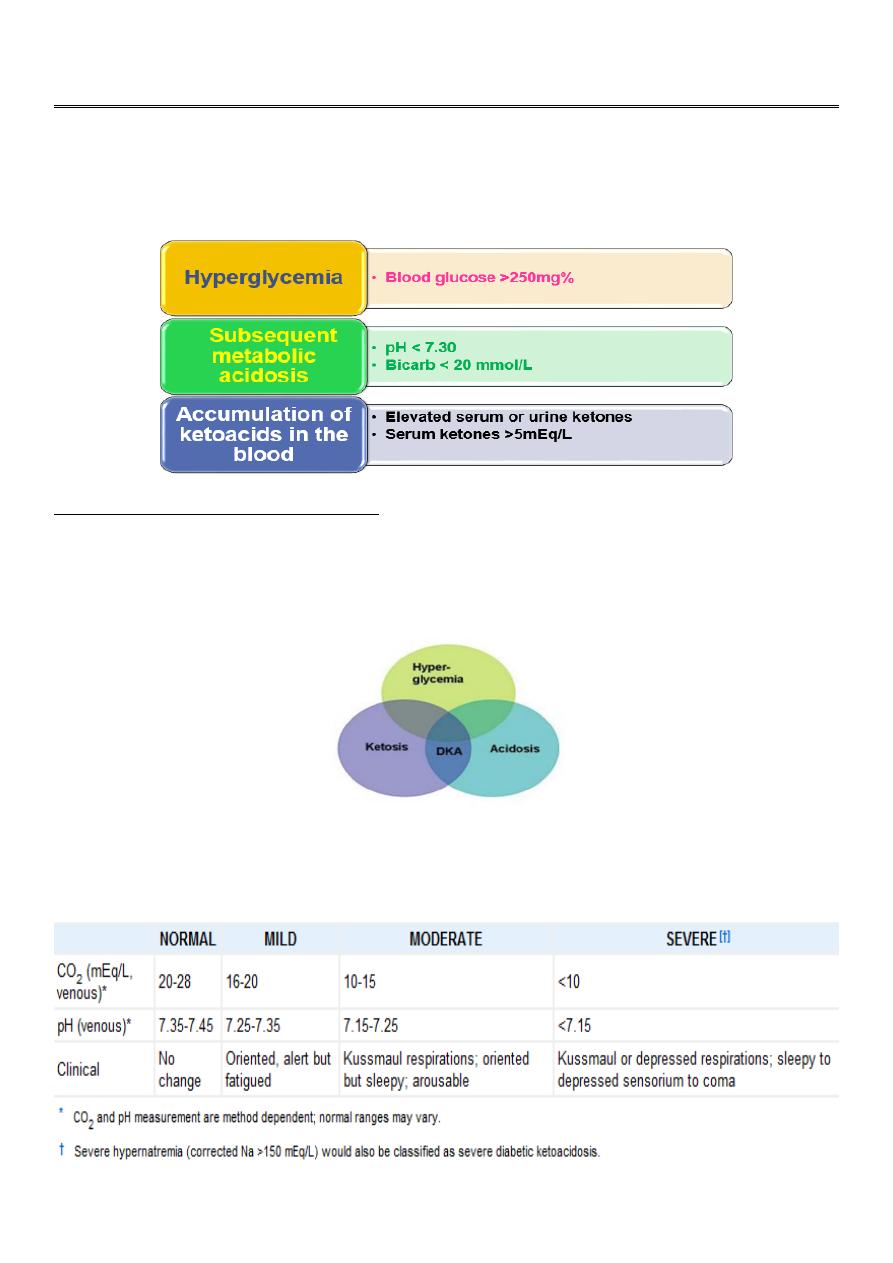
Classification
According to the severity, DKA can be classified into mild, moderate & severe DKA.
2
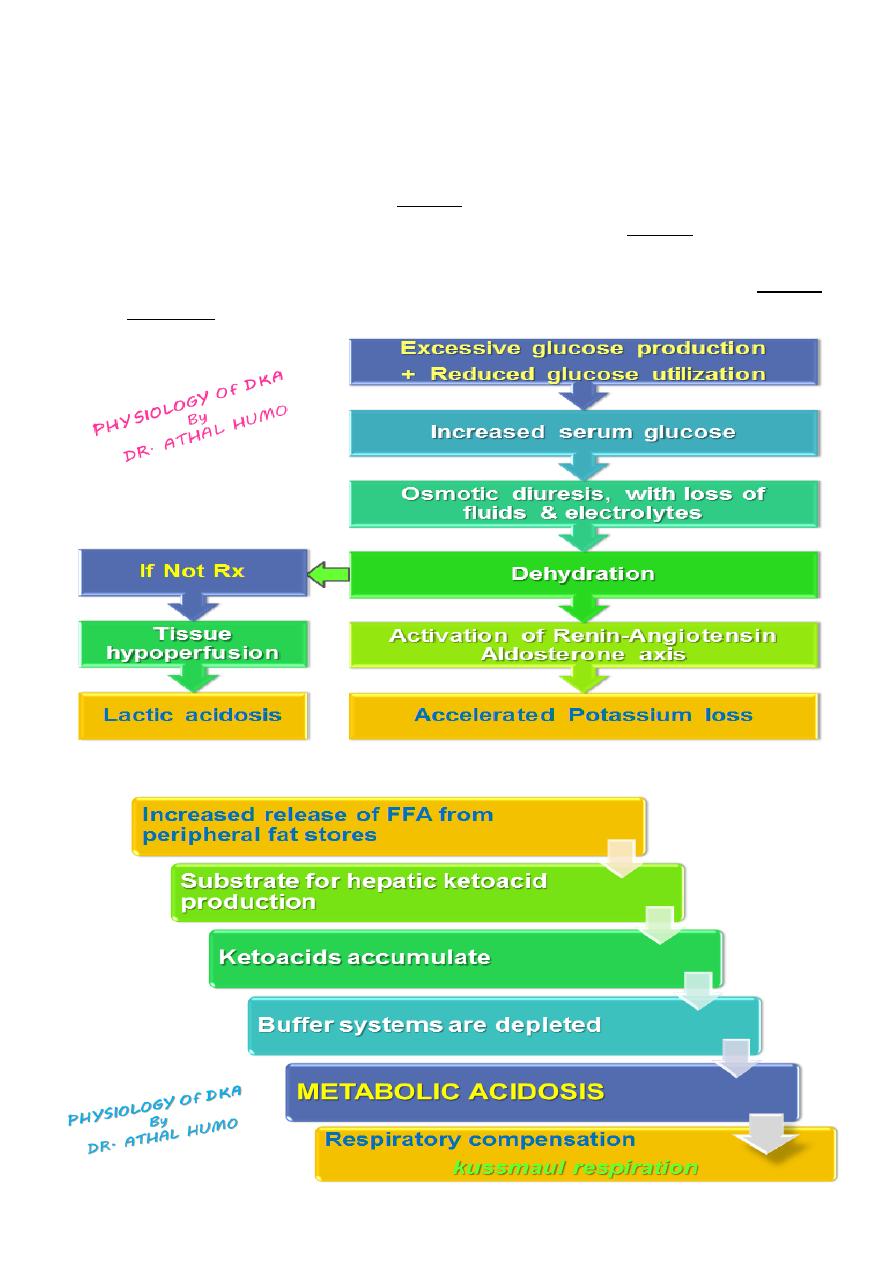
Physiology of DKA
Diabetic ketoacidosis is a superfasted state in which the body’s tissues are robbed of
their normal energy substrate Glucose.
And the body resorts to catabolism of glycogen, protein and fat for energy :
Glycogen is broken down to form glucose (glycogenolysis)
Protein is catabolized to amino acids which are converted to glucose
(gluconeogenesis)
Fats are broken down to free fatty acids, which are converted in the liver to glucose
or ketoacids (ketogenesis)
3
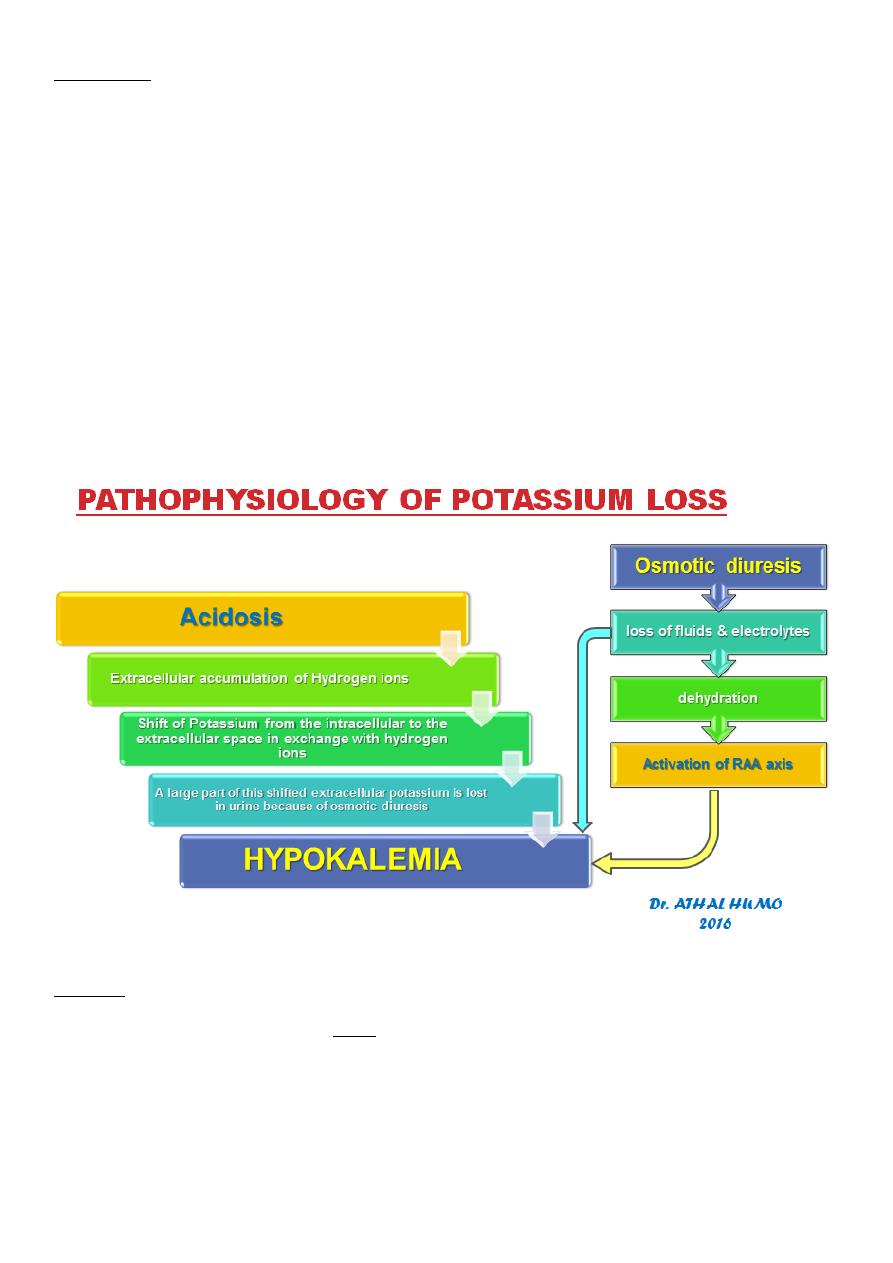
Potassium
The most characteristic disturbance in electrolytes is total body potassium loss.
K+ is largely an intracellular ion.
Both lack of insulin (catabolic predominance) and acidosis cause a shift of K+
extracellularly.
High urinary losses of K+ occur due to:
compartmental shift.
osmotic diuresis.
kaliuretic effect of hyperaldosteronism.
This loss is not mirrored in serum potassium levels, which may be low, within the
normal range, or even high.
Serum K+ levels are usually normal, even when total body K+ is depleted, because:
The compartmental shift of K+ from inside to outside the cell
Only extracellular K+ is measured
Sodium
Initial serum sodium may be ‘low’ for several reasons:
Depletion secondary to urinary losses / vomiting
Hyperlipidemia displaces sodium in the most frequently used laboratory assay,
factitiously lowering sodium values.
Hyperglycemia
High serum osmolarilty
Water driven from Intra to Extracellular
space
Dilutional hyponatremia

4
For each 100mg% increase of serum glucose above 100mg%, there is an expected
decrease of about 1.6mEq/L in measured sodium.
The ‘true’ serum sodium level can be calculated as:
[Na] + Glucose-100 x 1.6
100
The sodium should increase by about 1.6 mmol/L for each 100mg/dL decline in glucose.
If the corrected value is >150 mmol/L, severe hypernatremic dehydration may be
present and may require slower fluid replacement.
Declining sodium may indicate excessive water accumulation & risk of cerebral edema.
Electrolytes losses continue for several hours during therapy until catabolic state is
reversed & diuresis is controlled.
Even though Sodium deficit may be repaired within 24 hours, intracellular Potassium &
Phosphate may not be completely restored for several days.
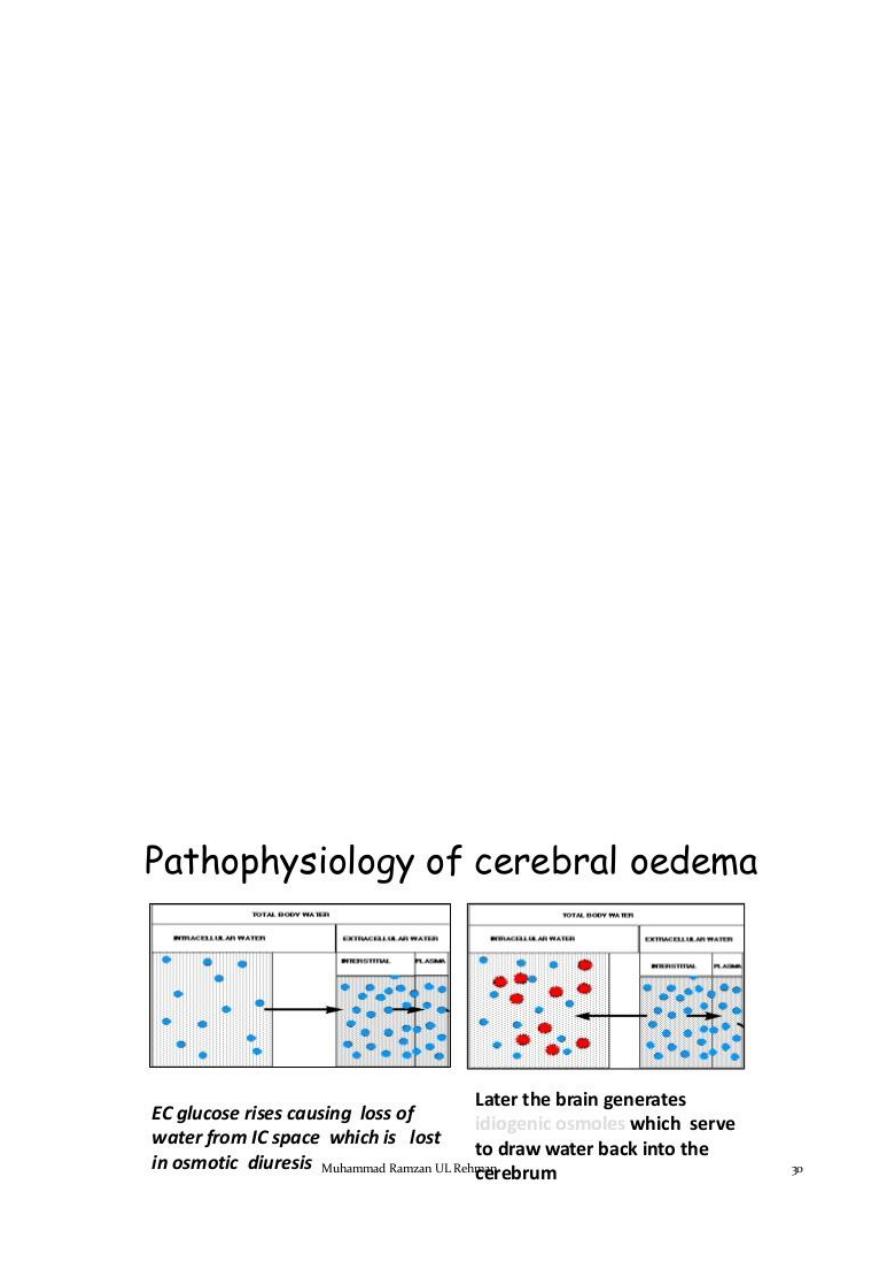
The combined effects of serum hyperosmolarity, dehydration, and acidosis ; result in
increased osmolarity in brain cells clinically manifests as an altered consciousness.
Presentation
The acidosis and ketosis of DKA creates an ileus (in particular beta hydroxybutyrate),
causing patients to develop nausea and vomiting, that consequently aggravate fluid
and electrolyte loss already existing in DKA. Occasionally the ileus will produce pain
severe enough to raise concern for an acute abdomen.
The ketosis (Acetone ) can give patients a fruity breath.
As the DKA becomes more severe, the patients will develope lethargy that can
progress to coma.

5
Lab Abnormalities and Diagnosis
Serum glucose is elevated >250mg/dL
Serum bicarbonate <20 mmol/L
Arterial pH <7.3 , depending on the severity of the acidosis.
Elevated serum osmolarity
Urine analysis : Glucosuria + Ketones
Blood Urea Nitrogen (BUN) may be elevated due to dehydration.
Complete Blood Counts: may reveal possible infectious etiology
Treatment of DKA
Aims of the treatment:
1. Restore normal hemodynamic status
2. Restore normal acid-base balance
3. Correct blood glucose level
Restore perfusion by giving fluids, which will increase glucose use in the periphery,
restore GFR, and reverse the progressive acidosis.
Stop ketogenesis by giving insulin, which will reverse proteolysis and lipolysis, and
stimulate glucose uptake and processing, normalize blood glucose, and reverse acidosis.
Correct electrolyte losses by electrolyte supplementation.
Avoid the complications of treatment, including intracerebral complications,
hypoglycemia, and hypokalemia.
I.
Correction of dehydration:
It is prudent to approach any child in any hyperosmotic state with cautious rehydration
due to the risk of cerebral edema.
Therefore, patients should not be allowed oral fluids until rehydration is well
progressed and significant electrolyte shifts are no longer likely. All fluid intake and
output should be closely monitored.
Calculation of fluid deficits using clinical signs is difficult in children with DKA because
intravascular volume is better maintained in the hypertonic state.
The fluid deficit considered to be 8.5% of body weight ;
Deficit= (8.5/100)x Body weight x 1000= 85ml/kg
Maintenance (24 hr) = 100 mL/kg (for the 1st 10 kg) + 50 mL/kg (for the 2nd 10 kg) +
25 mL/kg (for all remaining kg)
This protocol corrects a deficit of 85ml/kg (8.5% dehydration) for all patients in the first
24 hours.

6
Children with mild DKA rehydrate earlier & can be switched to oral intake, whereas
those with severe DKA and a greater volume deficit require 30-36 hours with this
protocol.
The rehydration start by giving a bolus of 20ml/kg of normal saline or ringer lactate over
a short time (20min-1hr) & can be repeated . This ensures quick volume expansion.
Subsequent fluid is hypotonic (like glucose saline ) to repair the free water deficit, to
allow intracellular rehydration. Usually to start with it when blood glucose reaches
250mg/dl or less.
“Sodium”
The corrected sodium is usually normal or slightly elevated and indicates moderate
hypernatremic dehydration.
If the corrected value is > 150 mmol/L, severe hypernatremic dehydration may be
present and may require slower fluid replacement.
The sodium should steadily increase with therapy. Declining sodium may indicate
excessive free water accumulation and the risk of cerebral edema
“Potassium”
During treatment of DKA, the insulin that is given, as well as correction of the acidosis
that occurs, both cause potassium to move intracellularly. Because of this hypokalemia
is a potentially fatal complication during treatment of DKA.
Therefore, unless the patient is hyperkalemic or anuric, potassium is added to the
intravenous fluids at the beginning of the second hour of therapy; otherwise it is added
as soon as urine output is established or the hyperkalemia abates.
If the patient presents with hypokalemia, potassium replacement should be initiated
immediately.
Most patients will require 40 mEq/L of potassium in the replacement fluids (given as 20
mEq/L Kphos & 20 mEq/L Kac)
If K <3 mEq/L, give:
0.5 to 1.0 mEq/kg as oral K solution
OR increase IV K to 80 mEq/L, adjusted based on serum potassium levels
measured every 1 to 2 hours.
If patient tolerate oral feeding potassium can given orally.
“Phosphate”
Clinical studies have not shown benefit from phosphate replacement during the
treatment of DKA.
Although phosphate replacement should be given if the level drops less than 1 mg/dL.

7
In the absence of severe hypophosphatemia, phosphate may be provided by giving half
of the potassium replacement as potassium phosphate.
II.Correction of hyperglycemia:
Insulin treatment is begun at the beginning of the second hour of therapy, with regular
monitoring of blood glucose level.
It is given as a continuous intravenous infusion of regular insulin at a rate of 0.1 U/kg/h.
Short or rapid acting insulin injected intramuscularly every 2 hours can be used.
III.Correction of acidosis:
Once insulin is infused at appropriate rates, ketoacid production stops, the bicarbonate
buffers are regenerated leading to correction of acidosis without the need for sodium
bicarbonate adminstration.
Urine testing for ketone bodies is not reliable because correction of acidosis leads to
conversion of betahydroxybutyric acid to acetoacetic acid which gives a positive result
on such test.
Hyperglycemia is corrected well before the correction of acidosis. Therefore, even after
normal glucose levels are reached, insulin is still required to control fatty acid release.
The dose of insulin should remain at 0.1U/kg/h until the acidosis resolves (pH >7.3,
bicarbonate > 18).
Clinical trials have failed to show any benefit of bicarbonate use during the treatment of
DKA. Potential risks of bicarbonate therapy include paradoxical CNS acidosis and
exacerbation of hypokalemia. Bicarbonate treatment has also been associated with
cerebral edema, the most common cause of mortality for children with DKA.
Therefore, bicarbonate treatment should only be considered in cases of extreme
acidosis, such as:
Those with pH less than 6.9
When the acidosis may impair cardiovascular stability.
As treatment of life-threatening hyperkalemia.
If bicarbonate is felt to be necessary, 1 to 2 mmol/kg (added to 0.45% saline) should be
given over 1 to 2 hours.
Monitoring
Vital signs and mental status are monitored frequently, at least every hour. If the
patient is markedly obtunded, NGT should be placed to decrease the risk of
aspiration.
The balance of total fluid intake and fluid output should also be calculated each
hour.
Serum glucose and pH should be measured hourly.
8
Serum electrolytes and urine ketones measured every 2 to 3 hours.
The goal for correction of hyperglycemia is a fall of 100 mg/dL per hour. The
persistence of severe hyperglycemia suggests inadequate rehydration (or incorrectly
mixed insulin), while too rapid a fall may be an indicator of too rapid a rate of
rehydration.
Transition to oral intake & sc insulin
Criteria for this transition include:
normal sensorium.
normal vital signs.
an ability to tolerate oral intake, with no emesis.
serum sodium 135-145mEq/L
resolution of the acidosis reflected by:
o serum PH > 7.3.
o serum bicarbonate >15mEq/L.
o normal anion gap.
The IV insulin infusion is stopped & the 1st dose of insulin 0.2-0.4unit/kg is given s.c &
repeated every 6 hrs for the next 24-48hrs.
Cerebral Edema (Imp.)
Cerebral edema is responsible for the majority of deaths related to DKA in children,
and significant neurologic morbidity persists in many of the survivors.
While it typically presents 4 to 12 hours after treatment is begun, it can present later,
or earlier, including before treatment is initiated.

9
Risk factors for cerebral edema
The cause of cerebral edema in DKA is not known, but there are some risk factors
attributed to cerebral edema. These include:
young age.
Rapid fluid replacement & insulin infusion.
Injudicious use of bicarbonate therapy.
Severe presentation:
pH < 7.2
lower serum bicarbonate concentration
higher serum glucose concentration
higher blood urea nitrogen concentration
hypernatremia
Clinical features
severe headache.
sudden deterioration in mental status.
bradycardia (or a sudden, persistent drop in heart rate not attributable to improved
hydration).
Hypertension.
depressed respiration
cranial nerve dysfunction.
posturing, and seizures
incontinence.
Papilledema is a late sign.
Treatment
The 1st thing to be excluded is hypoglycemia.
Mannitol 0.5g/kg is infused over 30-60 min & can repeated if necessary.
Fluid restricted to 2/3 maintenance & the total correction should made over 48-
72hrs.
If the patient requires intubation, hyperventilation should be avoided, as it has been
shown to be associated with worse outcomes