HEMATOPATHOLOGY
LEC. 3
Enzyme Deficiencies
In order to fulfill its function, the red cell needs a supply of energy in the form of ATP and a
source of reducing power. A mature red cell contains no DNA or RNA and hence is
incapable of protein synthesis, so that the only source of energy as ATP is derived from
anaerobic glycolysis and the linked reducing system of the hexose-monophosphate
shunt (pentose phosphate pathway PPP) and the glutathione cycle. ATP is required to
maintain the membrane in its deformable state. Reducing power is required to counteract the
strong oxidative stresses .
Glucose-6-phosphate dehydrogenase deficiency:
The most common enzyme deficiency worldwide. The main races affected are in
West Africa, the Mediterranean, the Middle East and South-East Asia (approximately 400
million people are affected worldwide).
Genetics
The gene mutations affecting encoding of G6PD are found on the distal long arm of
the X chromosome. This is a typical sex-linked disorder, hence, full expression occurs in
males, while females are carriers (who show approximately half the normal red cell G6PD
values)or may suffer the disease if they are homozygous, which is rarely found. More than
400 mutations have been identified, most being missense mutations.
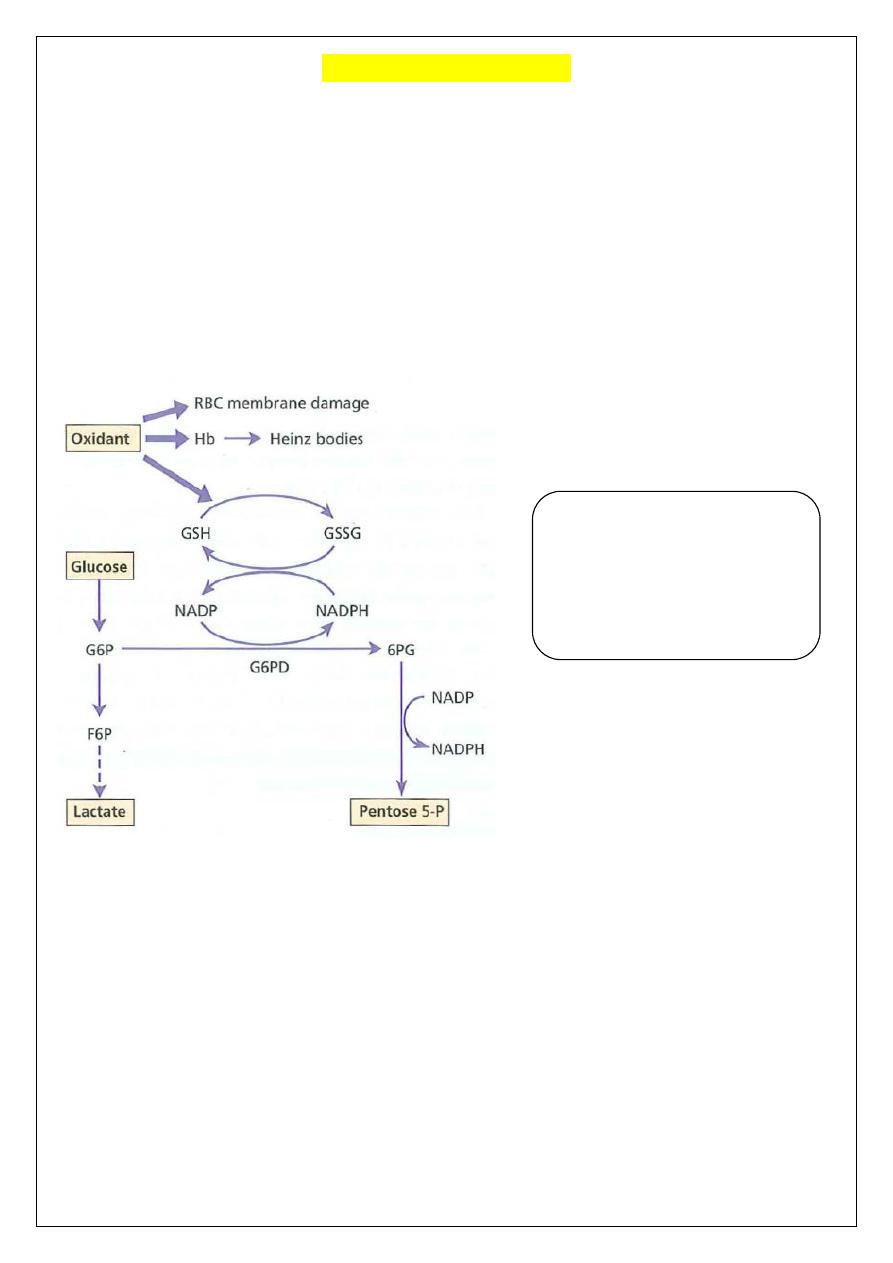
Haemoglobin and red blood cell
(RBC) membranes are usually
protected from oxidant stress
by reduced glutathione (GSH). In
G6PD deficiency, NADPH and
GSH synthesis is impaired.

Pathphysiology
G6PD catalyzes (NADP) to its reduced form, NADPH, in the pentose phosphate
pathway. NADPH protects cells from oxidative damage. Because erythrocytes do not
generate NADPH in any other way, they are more susceptible than other cells to destruction
from oxidative stress, oxidative damage leads to formation of denatured hemoglobin &
Heinz bodies which make the red cells less deformable & liable to splenic destruction.
Clinical features
G6PD deficiency is usually asymptomatic. Although G6PD is present in all cells, the main
syndromes that occur are as follows:
(i) Acute hemolytic anemia in response to oxidant stress, e.g. drugs, fava beans or infections,
The acute hemolytic anemia is caused by rapidly developing intravascular hemolysis IVH
with hemoglobinuria,
The anemia may be self-limiting as new young red cells are made with near normal enzyme
levels.
(ii) Neonatal jaundice.
(iii) Rarely, a congenital non-spherocytic hemolytic anemia. These may result from different
types of enzyme deficiency.
Diagnosis
Between crises the blood count is normal. The enzyme deficiency is detected by one of a
number of screening tests (rapid fluorescent spot test)or by direct enzyme assay on red
cells(Quantitative spectrophotometric analysis). During a crisis the blood film may show
contracted and fragmented cells, 'bite' cells and 'blister' cells which have had Heinz bodies
removed by the spleen. Heinz bodies (oxidized, denatured haemoglobin) may be seen in the
Reticulocyte preparation, particularly if the spleen is absent).
There are also features of intravascular hemolysis. Because of the higher enzyme level in
young red cells, red cell enzyme assay may give a 'false' normal level in the phase of acute
hemolysis with a Reticulocyte response. Subsequent assay after the acute phase reveals the
low G6PD level when the red cell population is of normal age distribution.
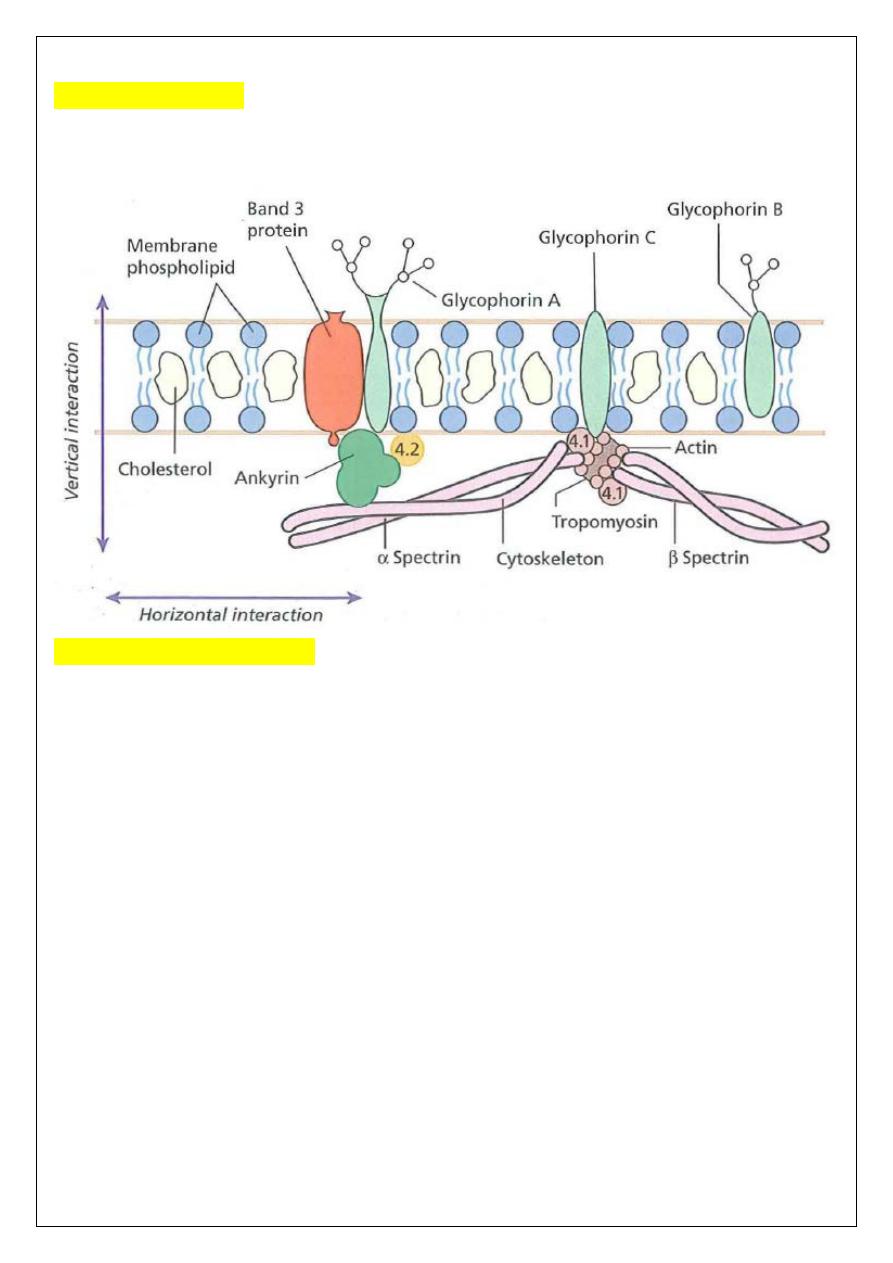
Membrane defects
The red cell membrane, like all other cell membranes, consists of a lipid bilayer that is
stabilized and given specific properties by the proteins, glycolipids and other specialized
molecules and structures with which it is associated.
Hereditary Spherocytosis
Hereditary Spherocytosis (HS) is one of the most common hereditary hemolytic anemia in
white populations, it's a genetically determined hemolytic anemia characterized by the
spherical shape of the affected red cells.
Pathogenesis:
HS is usually caused by defects in the proteins involved in the vertical interactions between
the membrane skeleton and the lipid bilayer of the red cell. The loss of membrane may be
caused by the release of parts of the lipid bilayer that are not supported by the skeleton. The
marrow produces red cells of normal biconcave shape but these lose membrane and become
increasingly spherical (loss of surface area relative to volume) as they circulate through the
spleen and the rest of the RE system. Finally, the spherocytes are unable to pass through the
splenic microcirculation where they die prematurely.
Clinical features:
The inheritance is autosomal dominant (AD) with variable expression; rarely it may be
autosomal recessive (AR).
The anemia can present at any age from infancy to old age. Jaundice is typically fluctuating;
splenomegaly occurs in most patients (splenectomy reverses the mechanisms of cell
destruction and alleviates the anemia). Pigment gallstones are frequent; aplastic crises,
usually precipitated by parvovirus infection, may cause a sudden increase in severity of
anemia.
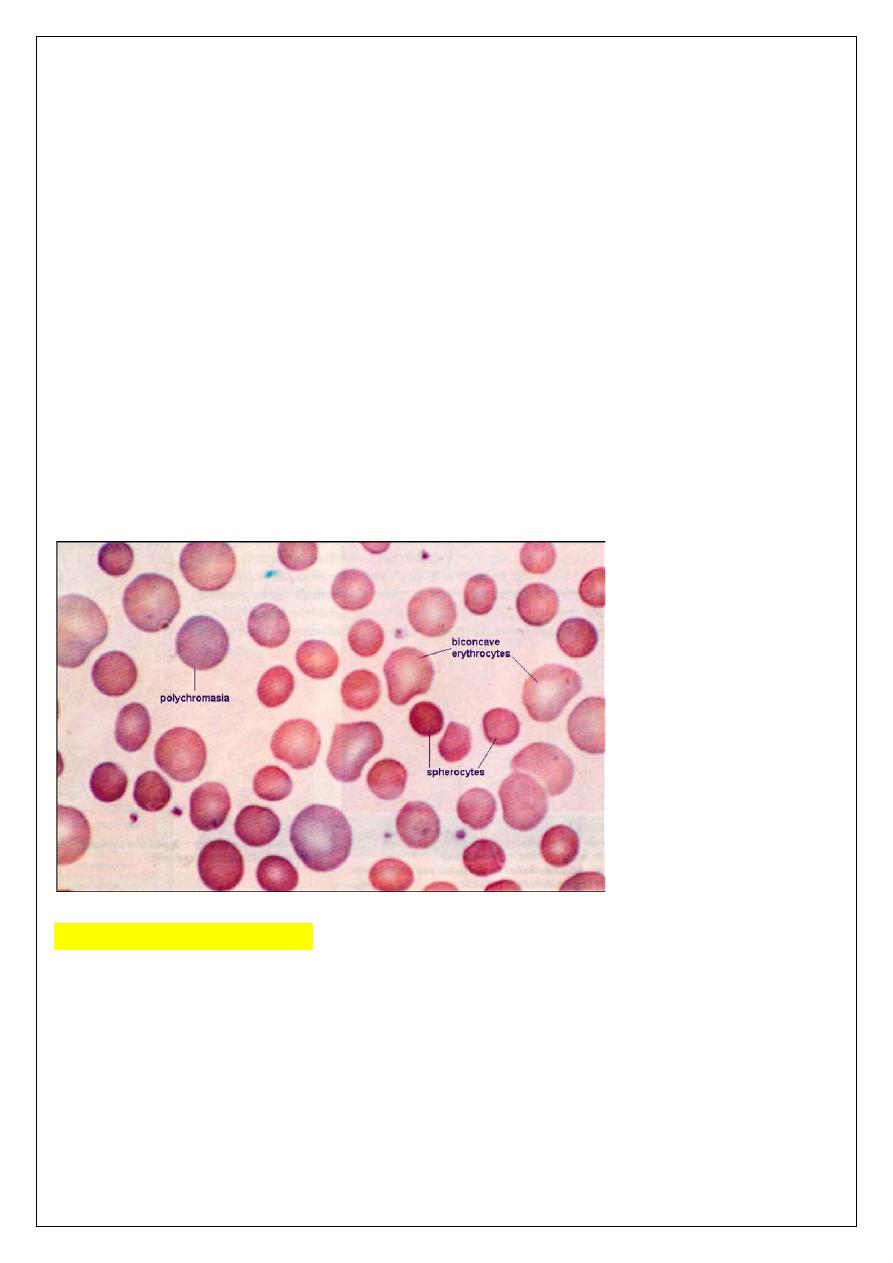
Laboratory Findings: The diagnosis is usually made on the basis of morphology of the
blood, backed up where possible with a family history.
Hematological findings:
Anemia is usual; its severity tends to be similar in members of the same family.
Reticulocytes are usually 5-20%. The blood film shows microspherocytes which are densely
staining with smaller diameters than normal red cells(which lack the area of central pallor of
the normal biconcave discs).
The MCHC is often increased above 35 g/dL in HS, but the presence of macrocytic
Reticulocytes usually results in a low normal MCV rather than true microcytosis.
The diagnosis is made after incubation in reducing concentrations of sodium chloride - called
the osmotic fragility test, to demonstrate the poor tolerance of the cells to a hypotonic
environment →swelling and rupture of the red cells.
The direct Antiglobulin (Coombs') test is normal, excluding an autoimmune cause of
spherocytosis and hemolysis.
Hereditary elliptocytosis:
This has similar clinical and laboratory features to HS except for the appearance of the blood
film (shows large numbers of elliptocytes and/or ovalocytes), but it is usually a clinically
milder disorder.
It is usually discovered by chance on a blood film and there may be no evidence of
hemolysis or only mild hemolysis.
Occasional patients require splenectomy.
A number of genetic mutations affecting horizontal interactions have been detected
(inherited as an autosomal dominant trait). Patients with homozygous or doubly

heterozygous elliptocytosis present with a severe hemolytic anemia with microspherocytes,
poikilocytes and splenomegaly (hereditary pyropoikilocytosis).
Osmotic fragility is usually normal. Reticulocytes are mildly increased (<5%). RBC
hemolysis occurs in the spleen, thus splenectomy corrects the hemolysis, but not the RBC
membrane defect.
Acquired hemolytic anemias:
Acquired hemolytic anemias are usually divided into two main categories depending
on the mechanism by which the premature destruction of red blood cells is produced.
In the immune hemolytic anemias, antibodies are the main agents of destruction. The non -
immune acquired hemolytic anemias include diverse causes and mechanisms of hemolysis.
In most anemias, there is some shortening of red cell survival, but in the haemolytic
anaemias this shortening of red cell lifespan is the major cause of the anaemia and produces
the classical features of hemolysis.
Unlike the inherited hemolytic anemias, which predominantly involve abnormalities intrinsic
to the erythrocyte, the acquired hemolytic anemias (with exception of paroxysmal nocturnal
hemoglobinuria PNH) involve abnormalities that are extrinsic to the erythrocyte.
IMMUNE HEMOLYTIC ANEMIA:
Immune hemolytic anemia is the most common form of acquired hemolytic anemia
and it can be classified as: 1) auto-immune, 2) allo-immune or 3) drug-induced. Irrespective
of the etiology, the typical feature in the peripheral blood smear is microspherocytosis.
1.
Autoimmune hemolytic anemia (AIHA):
Are caused by antibody production by the body against its own red cells. They are
characterized by a positive direct antiglobulin test (DAT) also known as the Coombs'
test and divided into 'warm' and 'cold' types according to whether the antibody reacts
more strongly with red cells at 37°C or 4°C. AIHA may be idiopathic (no underlying
cause identified) or secondary.
Warm Antibody Hemolytic Anemia:
This is the most frequent immune hemolytic anemia, accounts for more than 70% of AIHA.
The red cells are coated with immunoglobulin (Ig), usually immunoglobulin G (IgG) alone
or with complement, and are therefore taken up by RE macrophages which have receptors
for the Ig Fc fragment. Part of the coated membrane is lost so the cell becomes progressively
more spherical to maintain the same volume and is ultimately prematurely destroyed,
predominantly in the spleen.

Clinical features:
The disease may occur at any age, in both sex, and presents as a haemolytic anaemia of
varying severity (mild chronic anemia to a life threatening condition). The spleen is often
enlarged. The disease tends to remit and relapse. It may occur alone or in association with
other diseases. Certain common events are known to precipitate episodes of increased RBC
destruction: surgery, pregnancy, acute and chronic infections, etc.
Most warm autoantibodies do not cause autoagglutination or intravascular hemolysis.
Laboratory findings:
• CBC & blood film: anemia, spherocytosis, polychromasia (reticulocytosis),
normoblastaemia, neutrophilia common, lymphocytosis in lymphoproliferative
disorders.
• Coombs test positive: RBC coated with IgG, complement or both.
• Unconjugated hyperbilirubinemia with increased urobilinogen in the urine
and raised LDH.
The antibodies both on the cell surface and free in serum are best detected at 37°C.
Cold Antibody Hemolytic Anemia:
Is much less common than Warm AIHA (15 %).
These autoantibodies attaches to red cells
mainly in the peripheral circulation where the blood temperature is cooled, In this condition
the IgM antibody and complement bind to red cells, agglutinating them when exposed to
temperatures below the body temperature, best at 4°C. Upon rewarming, IgM dissociates but
the complement is activated, leading to intravascular hemolysis. It affects both sexes
equally, with a peak incidence in the 5th decade.
Clinical features
The patient may have a chronic hemolytic anemia aggravated by the cold and often
associated with intravascular hemolysis (hemoglobinuria). Mild jaundice and splenomegaly
may be present. The patient may develop acrocyanosis (purplish skin discoloration) at the tip
of the nose, ears, fingers and toes caused by the agglutination of red cells in small vessels.
Laboratory findings:
are similar to those of warm AIHA except that spherocytosis is less marked, agglutination of
red cells on the slide (RBC clumping), is prominent and diagnostic. sometimes a high MCV
(due to agglutination).

2.
Alloimmune hemolytic anemia:
In these anaemias, antibody produced by one individual reacts with red cells of another.
Typically, this occurs following a blood transfusion, during or after pregnancy, or following
bone marrow or stem cell transplantation.
Two important situations are transfusion of ABO-incompatible blood and hemolytic disease
of the newborn.
Transfusion reactions due to immune mediated Hemolysis HTR
There are two major types of immune-mediated hemolysis following blood transfusion:
acute and delayed. More than 80% of acute hemolytic transfusion reactions are due to ABO
incompatibility. Transfusion reactions due to intravascular hemolysis are generally more
severe than with extravascular hemolysis. Patients may experience fever, chills, joint pain,
shock, renal failure and/or disseminated intravascular coagulation DIC. Delayed hemolytic
transfusion reactions may be caused by primary alloimmunization but, more frequently, the
transfused red cells trigger a delayed (anamnestic) IgG mediated transfusion reaction 7–14
days after the blood transfusion.
Hemolytic disease of the newborn HDN
Is hemolysis in the fetus caused by transplacental transfer of maternal IgG.
During pregnancy, alloimmune hemolytic anemia may occur in the fetus if there is a blood
group incompatibility between the mother and the fetus. The antibodies against the Rh and
the ABO antigen systems are the two major causes of hemolytic disease of the newborn,
with anti-D causing the most severe cases.
3. Drugs-induced immune hemolytic anemias:
Drugs may cause immune haemolytic anaemias via several mechanisms
.

NON-IMMUNE HEMOLYTIC ANEMIA:
1. Red cell fragmentation syndromes:
These arise through physical damage to red cells either on abnormal surfaces (e.g. artificial
heart valves or arterial grafts), arteriovenous malformations or as a microangiopathic
haemolytic anaemia MAHA . This is caused by red cells passing through abnormal small
vessels. The MAHA may be caused by deposition of fibrin strands often associated with
disseminated intravascular coagulation (DIC) or platelet adherence as in thrombotic
thrombocytopenic purpura (TTP) or vasculitis (e.g. polyarteritis nodosa).
The peripheral blood contains many deeply staining red cell fragments . Clotting
abnormalities typical of DIC with a low platelet count are also present when DIC underlies
the hemolysis.
2. March haemoglobinuria:
This is caused by damage to red cells between the small bones of the feet, usually during
prolonged marching or running. The blood film does not show fragments.
3. Infections:
Infections can cause hemolysis in a variety of ways. They may precipitate an acute hemolytic
crisis
in G6PD deficiency or cause microangiopathic hemolytic anemia (e.g., with
meningococcal or pneumococcal septicemia). Malaria causes hemolysis by extravascular
destruction of parasitized red cells as well as by direct intravascular lysis.
Blackwater fever is an acute intravascular hemolysis accompanied by acute renal failure
caused by Falciparum malaria. Clostridium perfringens septicemia
can cause intravascular
hemolysis with marked microspherocytosis.
4. Chemical and physical agents:
Certain drugs in high doses cause oxidative intravascular hemolysis with Heinz body
formation in normal subjects. In Wilson's disease an acute haemolytic anaemia can occur as
a result of high levels of copper in the blood. Chemical poisoning (e.g. with lead, chlorate or
arsine) can cause severe hemolysis. Severe burns damage red cells causing acanthocytosis or
spherocytosis.

5. Secondary haemolytic anaemias:
In many systemic disorders red cell survival is shortened. This may contribute to anemia.
6. Paroxysmal Nocturnal Hemoglobinuria (PNH):
PNH is a rare, acquired, clonal disorder of marrow stem cells in which
the patient’s
red cells are abnormally sensitive to lysis by the complement of normal plasma,
characterized by hemolysis, particularly during sleep, leading to the passage of dark
urine (due to haemoglobinuria),
PNH is almost invariably associated with some form
of bone marrow hypoplasia, often frank aplastic anaemia.
ﺍﻟﺪﻛﺘﻮﺭﺍﻻﺧﺘﺼﺎﺹ
ﻋﻠﻲ
ﻋﺒﻴﺪ
ﺍﺑﺮﺍﻫﻴﻢ
ﺍﻟﺨﻔﺎﺟﻲ
ﺩﻛﺘﻮﺭﺍﺓ
)
ﺑﻮﺭﺩ
(
ﺍﻣﺮﺍﺽ
ﺍﻟﺪﻡ
M.B.Ch.B. F.I.C.M.S