
HematopatHology
LEC.5
CHRONIC LEUKEMIA
The chronic leukemias are distinguished from acute leukemias by their slower progression.
Paradoxically, they are also more difficult to cure. Chronic leukemias can be broadly
subdivided into myeloid and lymphoid groups.
CML
Chronic myeloid leukemia
Chronic myeloid leukemia (CML) is a clonal disorder of a pluripotent stem cell. The disease
accounts for around 15% of leukemias and may occur at any age. The diagnosis of CML is
rarely difficult and is assisted by the characteristic presence of the Philadelphia chromosome,
this Ph chromosome results from the t(9; 22) translocation between chromosomes 9 and 22.
The abnormal chromosome 22 is the Ph chromosome.
The Ph translocation is also seen in a
minority of cases of ALL. A great increase in total body myeloid cell mass is responsible for
most of the clinical features.
Clinical features:
This disease occurs in either sex, most frequently between the ages of 40 and 60 years.
In most cases there are no predisposing factors but the incidence was increased in survivors
of the atom bomb exposures in Japan.
Its clinical features include the following:
1 Symptoms related to hypermetabolism (e.g. weight loss, anorexia or night sweats).
2 Splenomegaly is nearly always present and is frequently massive. In some patients, splenic
enlargement is associated with considerable discomfort, pain or indigestion.
3 Features of anemia may include pallor, dyspnea and tachycardia.
4 Bruising, epistaxis, menorrhagia.
5 Gout or renal impairment caused by hyperuricemia.
6 In up to 50% of cases the diagnosis is made incidentally from a routine blood count.
7 Rare symptoms include visual disturbances and Hyperviscosity due to leucostasis.
The disease progresses through three stages:
1 Stable chronic phase: Non-specific symptoms ; tiredness or an awareness of the big
spleen as a vague mass in the abdomen. Chemotherapy causing decrease in the white cell
and platelet counts, and decrease in size of the enlarged spleen.
2 Accelerated phase: Blood counts become difficult to control and the spleen increases to
often a massive size, leading to infarction and pain.
3 Transformation to acute leukemia (Blast crisis): Blast crisis is defined by the presence of
>20% blasts in the blood and/or bone marrow. In most cases, the blasts have a myeloid
phenotype, but approximately 25 to 30% of cases have a lymphoid. Patient develops features
of acute leukemia, lymphadenopathy, high fever and infections.
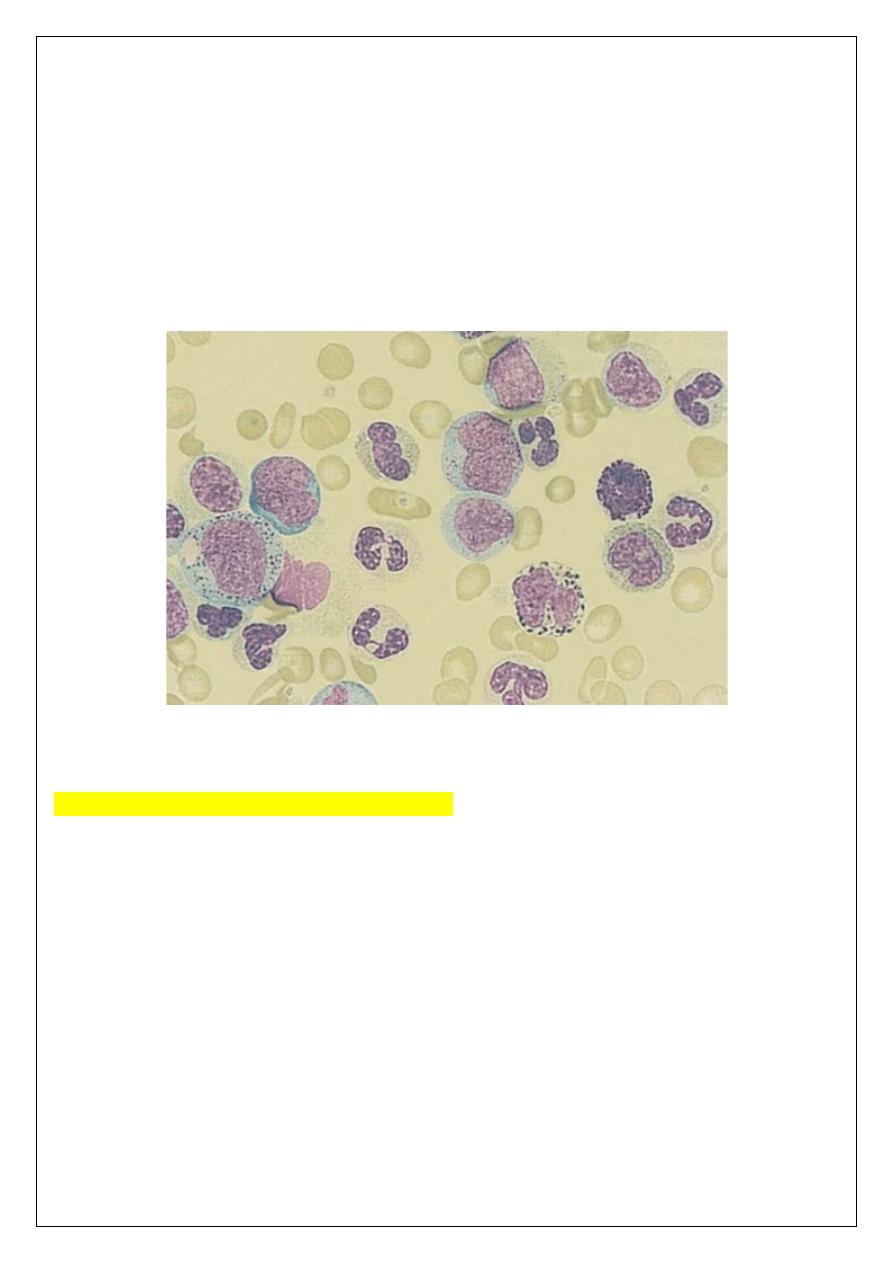
Laboratory findings:
1 Leucocytosis is usually >50 x 10
9
/L and sometimes >500 x 10
9
/L. A complete spectrum
of myeloid cells is seen in the peripheral blood. The levels of neutrophils and myelocytes
exceed those of blast cells and promyelocytes.
2 Increased circulating Basophils.
3 Normochromic, normocytic anemia is usual.
4 Platelet count may be increased (most frequently), normal or decreased.
5 Bone marrow is hypercellular with granulopoietic predominance.
6 Ph chromosome on cytogenetic analysis of blood or bone marrow.
7 Serum uric acid is usually raised.
LEUKEMIAS:
ONIC LYMPHOID
THE CHR
Several disorders are included in this group characterized by accumulation of mature
lymphocytes of either B- or T-cell type in the blood.
This group is characterized by a chronic persistent lymphocytosis. Subtypes are
distinguished by morphology, immunophenotype and genetic analysis.
There is considerable overlap with the lymphomas. In many cases of NHL non-Hodgkin's
lymphoma, lymphoma cells are found in the blood and the distinction between chronic
leukemia and lymphoma is depending on the relative proportion of the disease in soft tissue
masses compared to blood and bone marrow. In general, these diseases are incurable but tend
to run chronic and fluctuating course.
Chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is by far the most common of the chronic lymphoid
leukemias and has a peak incidence between 60 and 80 years of age. The aetiology is
unknown. There is a seven fold increased risk of CLL in the close relatives of patients. The

tumor cell appears to be a relatively mature B cell. The cells accumulate in the blood, bone
marrow, liver, spleen and lymph nodes as a result of a prolonged lifespan with impaired
apoptosis.
Clinical features
1. The disease occurs in older subjects mainly.
2. Most cases are diagnosed when a routine blood test is performed. With increasing routine
medical check-ups, this proportion is rising.
3. Symmetrical enlargement of cervical, axillary or inguinal lymph nodes is the most
frequent clinical sign.
4. Features of anemia may be present. Patients with thrombocytopenia may show bruising or
purpura.
5. Splenomegaly and, less commonly, hepatomegaly are common in later stages.
6. Immunosuppression is a significant problem resulting from hypogammaglobulinaemia and
cellular immune dysfunction. Early in the disease course bacterial infections predominate but
with advanced disease viral and fungal infections such as herpes zoster are also seen.
Laboratory findings
1. Lymphocytosis. The absolute lymphocyte count is >5 x 10
P
9
P
/L and may be up to
300 X 10
P
9
P
/L or more. Between 70 and 99% of white cells in the blood film appear as small
lymphocytes
usually look monotonous. Smudge or smear cells are also present.
Neutrophil
count is decreased as a percentage of white cells, but the absolute neutrophil number may be
normal, increased, or decreased.
2. Immunophenotyping of the lymphocytes shows them to be B cells (surface CD19+),
weakly expressing surface immunoglobulin (IgM or IgD). This is shown to be monoclonal.
3. Normocytic normochromic anemia is present in later stages as a result of marrow
infiltration or hypersplenism. Autoimmune hemolysis may also occur. Thrombocytopenia
occurs in many patients.
4. Bone marrow aspiration shows lymphocytic replacement of normal marrow elements.
Lymphocytes comprise 25-95% of all the cells in BM.
5. Reduced concentrations of serum immunoglobulin are found and this becomes more
marked with advanced disease.
6. Autoimmunity directed against cells of the hemopoietic system is common. Autoimmune
hemolytic anemia is most frequent but immune thrombocytopenia, neutropenia and red cell
aplasia are also seen.
Staging:
It is useful to stage patients at presentation both for prognosis and for deciding on therapy.
The Rai and Binet staging systems
used to classify patients as low, intermediate or high risk.
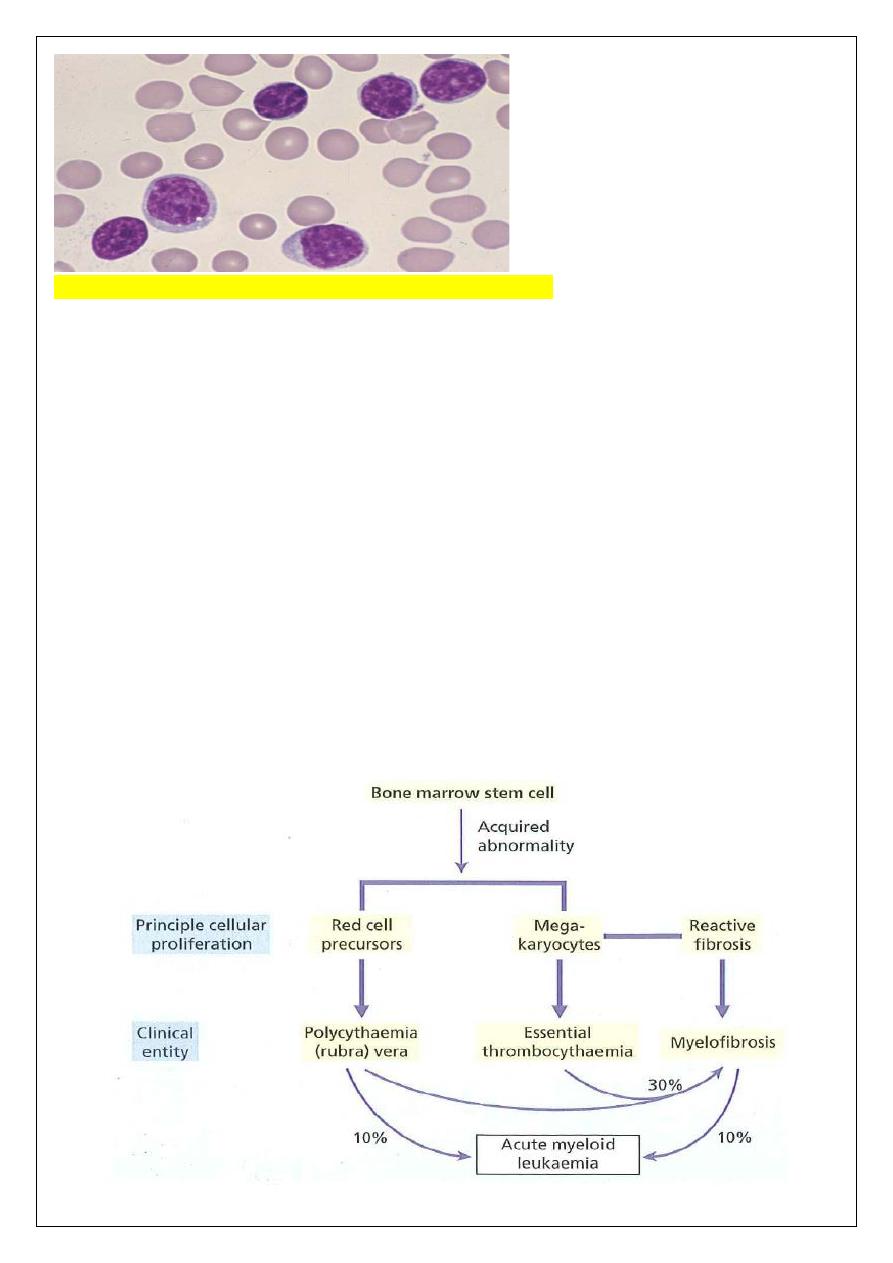
FERATIVE DISORDERS (MPD)
THE MYELOPROLI
The term myeloproliferative disorders describes a group of conditions arising from marrow
stem cells and characterized by clonal proliferation of one or more hemopoietic components
in the bone marrow and, in many cases, the liver and spleen ((regulation of proliferation is
impaired, but differentiation and maturation are generally maintained)).
Three disorders are included in this classification:
1 Polycythaemia rubra vera (PRV).
2 Essential thrombocythemia (ET).
3 Myelofibrosis (MF).
These disorders are closely related to each other. Indeed, transitional forms occur and in
many patients an evolution from one entity into another occurs during the course of the
disease. A single acquired mutation of the cytoplasmic tyrosine kinase Janus-associated
kinase 2 (JAK2) occurs in the marrow and blood of almost all patients with PRV and in
approximately 50% of those with ET and myelofibrosis, showing the common aetiology of
these three diseases.
JAK2 plays a major role in normal myeloid development.
Chronic myeloid leukemia, which used to be classified as a myeloproliferative disorder but
is now recognized as a separate entity.

emia
Polycyth
Polycythaemia (erythrocytosis) is defined as an increase in the hemoglobin concentration
above the upper limit of normal for the patient's age and sex.
Classification of polycythemia
Polycythemia is classified according to its pathophysiology but the major subdivision is into
absolute polycythemia, in which the red cell mass (volume) is raised, and relative or
pseudopolycythaemia in which the red cell volume is normal but the plasma volume is
reduced. Absolute polycythemia can then be subdivided into primary polycythemia (PRV) or
secondary polycythemia.
Polycythaemia (rubra) vera
In PRV, the increase in red cell volume is caused by a clonal malignancy of a marrow stem
cell.
The disease results from somatic mutation of a single
hemopoietic stem cell which gives
its progeny a proliferative advantage. The JAK2 mutation is present in hemopoietic cells in
almost 100% of patients. Although the increase in red cells is the diagnostic finding, in many
patients there is also an overproduction of granulocytes and platelets.
Clinical features
This is a disease of older subjects with an equal sex incidence. Clinical features are the result
of hyperviscosity (Headaches, blurred vision), hypervolemia (dyspnea) or hypermetabolism.

Many patients are found to have an elevated hemoglobin as an incidental finding on
screening blood counts.
laboratory findings
CBC: shows an increase in the hemoglobin ~18 to 24 g/dL, raised hematocrit usually
>60% in men and >55% in women, and raised red cell count. The white cell count is
elevated in over half of patients, predominantly due to mature granulocytes. Basophils are
often slightly increased. Moderate thrombocytosis is present in half of patients.
Bone Marrow is hypercellular, an increase in all cell lines, with a predominant increase in
erythroid precursors, resulting in a decrease in the ratio of myeloid to erythroid cells (M:E
ratio).
The JAK2 mutation is present in the bone marrow and peripheral blood granulocytes in
nearly 100% of patients.
Course and prognosis:
Typically, the prognosis is good with a median survival of 10-16 years. Thrombosis and
haemorrhage are the major clinical problems. Increased viscosity, vascular stasis and high
platelet levels may all contribute to thrombosis, whereas defective platelet function may
promote haemorrhage.
Transition from PRV to myelofibrosis MF occurs in approximately
30% of patients and approximately 5% of patients progress to acute leukaemia.
ESSENTIAL THROMBOCYTHAEMIA
A clonal proliferative disorder of the megakaryocytes in which there is a sustained
increase in platelet count, because of megakaryocyte proliferation and overproduction of
platelets.
A persisting platelet count of >400 x 10
9
/L is the central diagnostic feature but
other causes of a raised platelet count need to be fully excluded before the diagnosis can
be made. Half of patients show the JAK2 mutation.
Clinical and laboratory findings:
The dominant clinical features are thrombosis and haemorrhage (due to abnormal platelet
function). Many cases are symptomless and diagnosed on routine blood counts.
A
characteristic symptom is erythromelalgia, a burning sensation felt in the hands or feet
and promptly relieved by aspirin.
Up to 40% of patients will have palpable splenomegaly.
CBC: Platelets count elevated (often above 1000 x10
9
/L), Mild leukocytosis (<20,000/L)
is often present, mild eosinophilia and basophilia are often present.
Blood film: leukocyte differential shows a mild left shift, giant platelets, other bizarre
platelet shapes, and megakaryocyte nuclear fragments are often present.
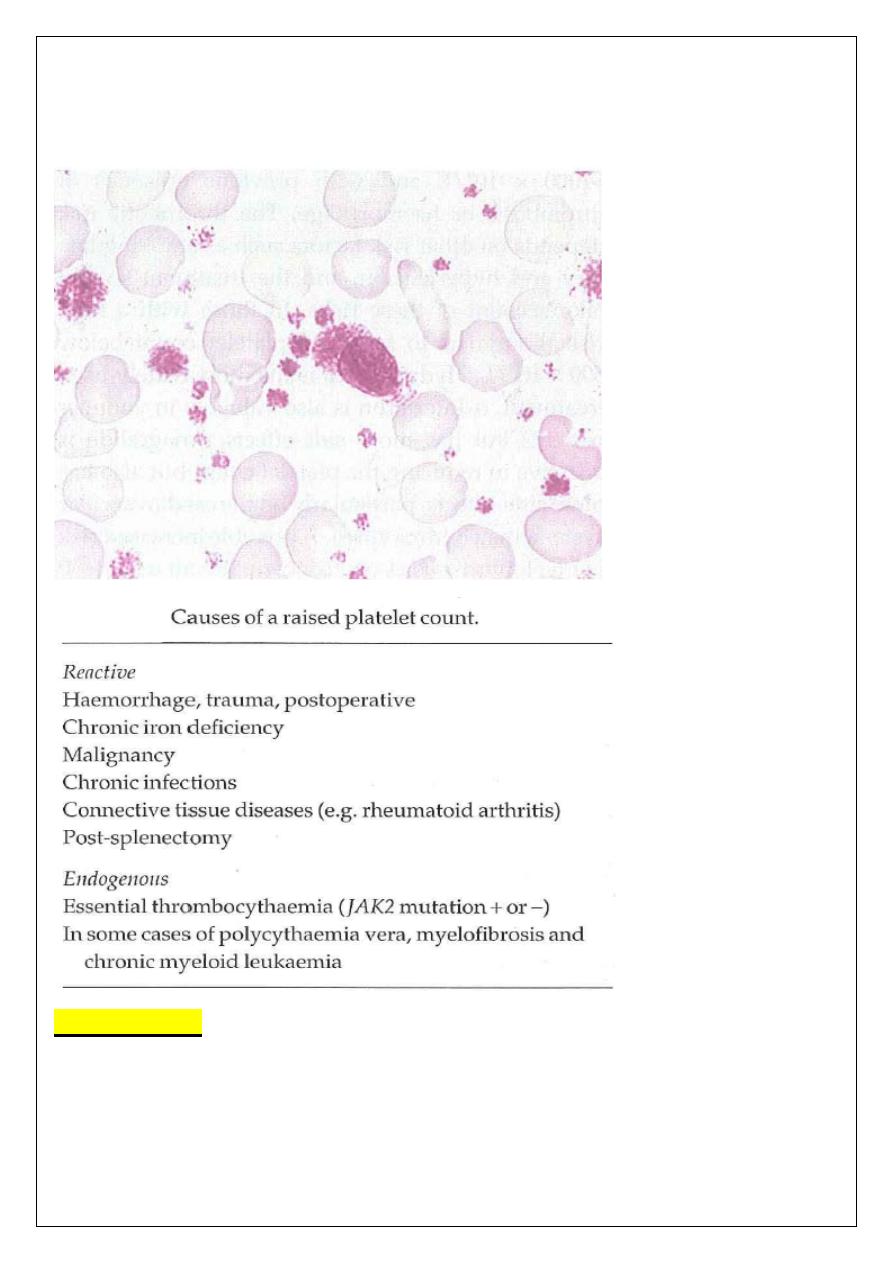
Bone marrow: shows severe megakaryocyte hyperplasia in clusters, often granulocytic
and erythroid hyperplasia. Mild fibrosis may be present.
Conditions associated with secondary reactive thrombocytosis must be excluded.
Myelofibrosis:
The predominant feature of myelofibrosis is a progressive generalized reactive fibrosis of
the bone
marrow in association with the development of hemopoiesis in the spleen and
liver.
Clinically this leads to anaemia and massive hepatosplenomegaly.
Myelofibrosis is a
clonal stem cell disease. The fibrosis of the bone marrow is secondary to hyperplasia of
abnormal megakaryocytes. It is thought that fibroblasts are stimulated by platelet-derived

growth factor and other cytokines secreted by megakaryocytes and platelets. The JAK2
mutation occurs in approximately 50% of patients.
One-third or more of the patients have
a previous history of PRV and some patients present with clinical and laboratory features
of both disorders.
Clinical features
1 An insidious (gradual) onset in older people is usual with symptoms of anaemia.
2 Symptoms resulting from massive splenomegaly (e.g. abdominal discomfort, pain or
indigestion) are frequent; splenomegaly is the main physical finding.
3 Hypermetabolic symptoms such as loss of weight, anorexia, fever and night sweats are
common.
4 Bleeding problems, bone pain or gout occur in a minority of patients.
Myelofibrosis and chronic myeloid leukaemia are responsible for most cases of massive
(>20 cm) splenic enlargement in the UK and North America.
Laboratory findings
1 Anaemia is usual but a normal or increased hemoglobin level may be found in some
patients.
2 The white cell and platelet counts are frequently high at the time of presentation. Later
in the disease leucopenia and thrombocytopenia are common.
3 A leucoerythroblastic blood picture is found. The red cells show characteristic 'tear-drop'
poikilocytes.
4 Bone marrow is usually unobtainable by aspiration (dry tap). Trephine biopsy shows a
fibrotic hypercellular marrow. Increased megakaryocytes are frequently seen. In 10% of
cases there is increased bone formation with increased bone density on X-ray.
5 JAK2 kinase is mutated in approximately 50% of cases.
6 Transformation to acute myeloid leukaemia occurs in 10-20% of patients.

MDS
Myelodysplasia (myelodysplastic syndromes)
This is a group of clonal disorders of multipotent hemopoietic stem cells which are
characterized by increasing bone marrow failure with quantitative and qualitative
abnormalities of all three myeloid cell lines. A hallmark of the disease is ineffective
haemopoiesis so that cytopenias often accompany a marrow of normal or increased
cellularity ((increased stem cell proliferation but ineffective differentiation and
maturation, leading to the paradox of a hypercellular bone marrow but a pancytopenia in
peripheral blood)). There is a tendency to progress to (AML), although death often occurs
before this develops. In most cases, the disease is primary but in a significant proportion
of patients the disease is secondary to chemotherapy and/or radiotherapy which has been
given for treatment of another malignancy.
Clinical features:
The disease has low incidence and a slight male predominance. Over half of patients are
over 70 years and fewer than 25% are less than 50 years old. The evolution is often slow
and the disease may be found by chance when a patient has a blood count for some
unrelated reason. The symptoms, if present, are those of anaemia, infections or of easy
bruising or bleeding.
laboratory findings
Peripheral blood: Pancytopenia is a frequent finding. The red cells are usually
macrocytic or dimorphic but occasionally hypochromic; normoblasts may be present. The
reticulocyte count is low. Granulocytes
are often reduced in number and frequently show
lack of granulation. Their chemotactic, phagocytic and adhesive functions are impaired.
The Pelger abnormality (single or bilobed nucleus) is often present. The platelets may be
large or small and are usually decreased in number but in 10% of cases are elevated. In
poor prognosis cases variable numbers of myeloblasts are present in the blood
Bone marrow The cellularity is usually increased but is decreased in approximately 20%
of cases. Multinucleate normoblasts and other dyserythropoietic features are seen.
ﺍﻟﺪﻛﺘﻮﺭﺍﻻﺧﺘﺼﺎﺹ
ﻋﻠﻲ ﻋﺒﻴﺪ ﺍﺑﺮﺍﻫﻴﻢ ﺍﻟﺨﻔﺎﺟﻲ
ﺩﻛﺘﻮﺭﺍﺓ ) ﺑﻮﺭﺩ ( ﺍﻣﺮﺍ
ﺽ ﺍﻟﺪﻡ
M.B.Ch.B. F.I.C.M.S