
Lec.6
HEMATOPAYHOLOGY
Dr. Ali Obeid Ibrahim
Blood Coagulation and Haemostasis:
Haemostasis: the process by which the bleeding or haemorrhage is arrested.
The normal haemostatic response to vascular damage depends on closely linked interaction
between the blood vessel wall, circulating platelets and blood coagulation factors. An
efficient and rapid mechanism for stopping bleeding from sites of blood vessel injury is
clearly essential for survival. Nevertheless, such a response needs to be tightly controlled to
prevent extensive clots developing and to break down such clots once damage is repaired.
The haemostatic system thus represents a delicate balance between procoagulant and
anticoagulant mechanisms allied to a process for fibrinolysis. The five major components
involved are platelets, coagulation factors, coagulation inhibitors, fibrinolysis and blood
vessels.
Platelets:
Platelets are produced within the vascular channels (sinusoids) of the bone marrow by the
fragmentation of the protruding cytoplasm of large bone marrow cells known as
megakaryocytes. Thus they are not cells but rather are fragments of the cytoplasm of cells.
Platelets are considerably smaller than red cells and white cells. They are pale blue with fine
granules which tend to be clustered in the centre of the platelet. When blood films are made,
from anticoagulated blood, the platelets are usually discrete and separate from each other,
but in some circumstances they form clumps or aggregates.
The normal platelet count is approximately 250 x 10
9
/L (range 150-400 x 10
9
/L) and the
normal platelet lifespan is 7-10 days. Up to one-third of the marrow output of platelets may
be trapped at any one time in the normal spleen but this rises to 90% in cases of massive
splenomegaly.
Thrombopoietin is the major regulator of platelet production and is
constitutively produced by the liver and kidneys. Platelets are extremely small and discoid,
3.0 x 0.5
ɱm in diameter, with a mean volume 7-11 fL.
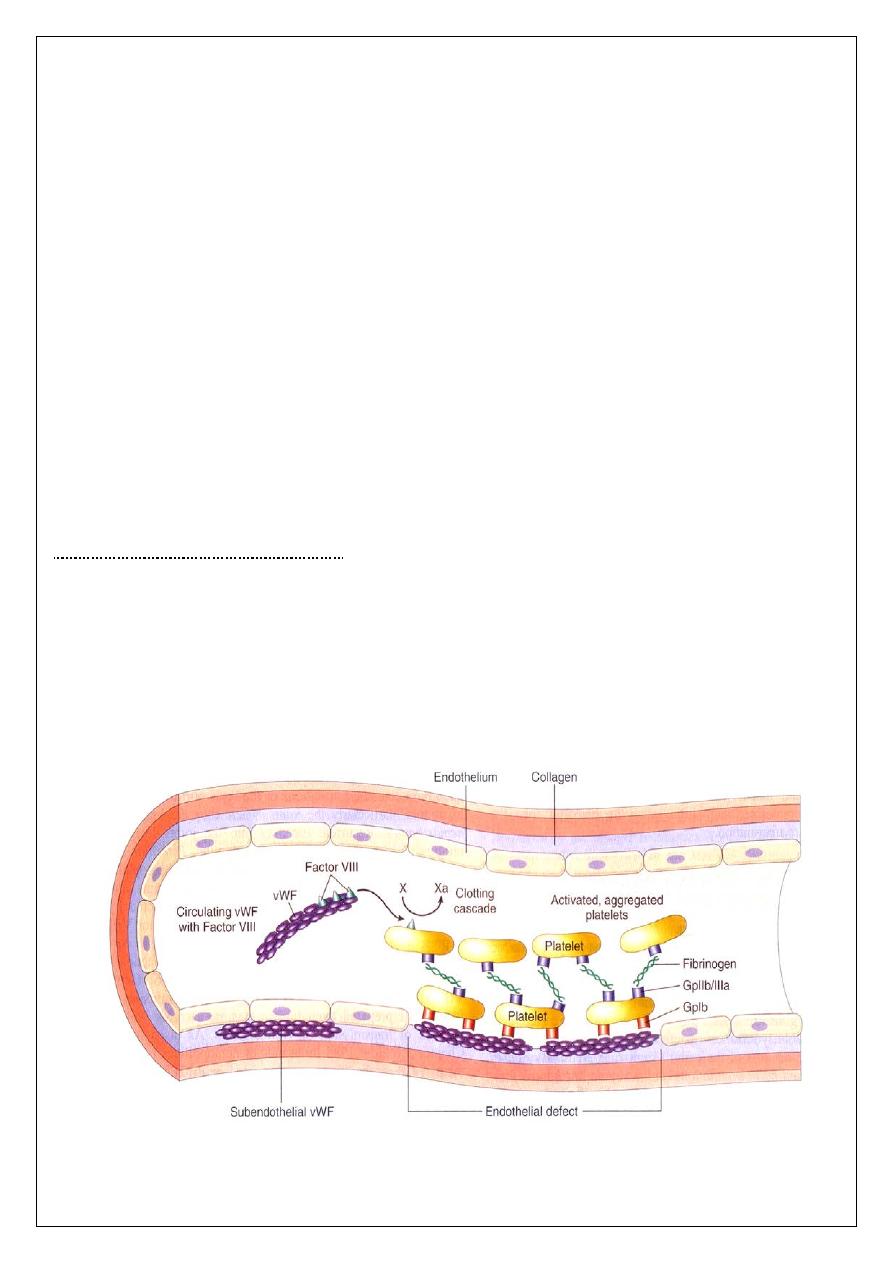
The main function of platelets is the formation of mechanical plugs during the normal
haemostatic response to vascular injury. In the absence of platelets, spontaneous leakage of
blood through small vessels may occur. The immobilization of platelets at the sites of
vascular injury requires specific platelet-vessel wall (adhesion) and platelet-platelet
(aggregation) interactions.
Factors:
Clotting)
Coagulation (
Clotting factors are plasma protein which synthesized in the liver. The term coagulation
cascade refers to the sequential activation of coagulation factors, resulting in the conversion

of fibrinogen to fibrin (by the action of Thrombin IIa ) and the subsequent polymerization of
fibrin into a fibrin clot. Most of the coagulation factors serine proteases. They circulate in the
plasma as inactive precursors, which are converted to the active enzyme by protease
cleavage. One coagulation factor cleaves and activates the next factor along the line and so
on. Since each active enzyme can activate many molecules of the subsequent factor, there is
a geometric increase in the number of molecules activated.
Thrombin IIa

Coagulation factor inhibitors:
It is important that the effect of thrombin is limited to the site of injury. It includes tissue
factor pathway inhibitor (TFPI) , Anti-Thrombin (AT) ,protein C , protein S & others .
:
Fibrinolysis
Fibrinolysis (like coagulation) is a normal haemostatic response to vascular injury.
The
principal functions of the fibrinolytic system are to ensure that excess fibrin deposition is
either prevented or rapidly removed. These include Plasminogen (PLG) and plasmin, PLG
activators and inhibitors of plasmin or inhibitors of the PLG activators.
:
mostatic function
Tests of hae
A number of simple tests are employed to assess the platelet, vessel wall and coagulation
components of haemostasis.
Blood count and blood film examination:
As thrombocytopenia (decrease platelet count) is a common cause of abnormal bleeding,
patients with suspected bleeding disorders should initially have a complete blood count CBC
including platelet count and blood film examination. In addition to establishing the presence
of thrombocytopenia, the cause may be obvious (e.g. acute leukaemia).
Screening tests of blood coagulation:
Screening tests provide an assessment of the 'extrinsic' and 'intrinsic' systems of blood
coagulation and also the central conversion of fibrinogen to fibrin.
The prothrombin time (PT):
measures factors VII, X, V, prothrombin and fibrinogen.
The
normal time for clotting is 10-14 s. It may be expressed as the international normalized ratio
(INR).
The activated partial thromboplastin time (APTT):
measures factors VIII, IX, XI and XII
in addition to factors X, V, prothrombin and fibrinogen.
The normal time for clotting is
approximately 30-40s.
Bleeding time:
The bleeding time is a useful test for abnormal
platelet function including
the diagnosis of VWF
deficiency. It has largely been replaced by the
platelet function
analysis-100 (PFA-100) test.
Bleeding stops normally in
3-8 min.
It will be prolonged in
thrombocytopenia but is normal in vascular causes of abnormal bleeding.
Tests of platelet function:
The most valuable investigation is platelet aggregometry.

Bleeding disorders
Abnormal bleeding may result from:
1 Vascular disorders;
2 Thrombocytopenia;
3 Defective platelet function; or
4 Defective coagulation.
Vascular bleeding disorders:
The vascular disorders are a heterogeneous group of conditions characterized by easy
bruising and spontaneous bleeding from the small vessels. The underlying abnormality is
either in the vessels themselves or in the perivascular connective tissues. Most cases of
bleeding caused by vascular defects alone are not severe. Frequently, the bleeding is mainly
in the skin causing petechiae, ecchymosis or both. In some disorders there is also bleeding
from mucous membranes. In these conditions the standard screening tests are normal. The
bleeding time is normal and the other tests of haemostasis are also normal. Vascular defects
may be inherited (Hereditary haemorrhagic telangiectasia) or acquired (senile purpura and
Henoch-Schonlein syndrome).
Thrombocytopenia:
a subnormal number of platelets in the circulating blood, usually below 100 × 10
9
/L. Acute
thrombocytopenia is the most frequent cause of severe bleeding and the risk of hemorrhage
is inversely proportional to the platelet count, with spontaneous bleeding occurring
frequently at a platelet count below 20 × 10
9
/L.
Causes of Thrombocytopenia:
1) Failure of platelet production as: in cases of aplastic anemia , invasion of bone marrow by
leukemia or solid malignancy, drugs or viral infections .
2) Increased consumption of platelets: as in Immune thrombocytopenia purpura (ITP) as
immune cause and DIC as non-immune.
3) Abnormal distribution of platelets: Splenomegaly
4) Dilutional loss: Massive transfusion of stored blood to bleeding patients.

Disorders of platelet function:
Are suspected in patients who show skin and mucosal haemorrhage and in whom the
bleeding time is prolonged despite a normal platelet count. These disorders may be
hereditary (like Bernard-Soulier syndrome & Glanzmann's disease) or acquired (use
Antiplatelet drugs like Aspirin, uremia).
Coagulation disorders:
Hereditary coagulation disorders:
Hereditary deficiencies of the coagulation factors. Haemophilia A (factor VIII deficiency),
haemophilia B (Christmas disease, factor -IX deficiency) and von Willebrand disease
(VWD) are the most common; the others are rare.
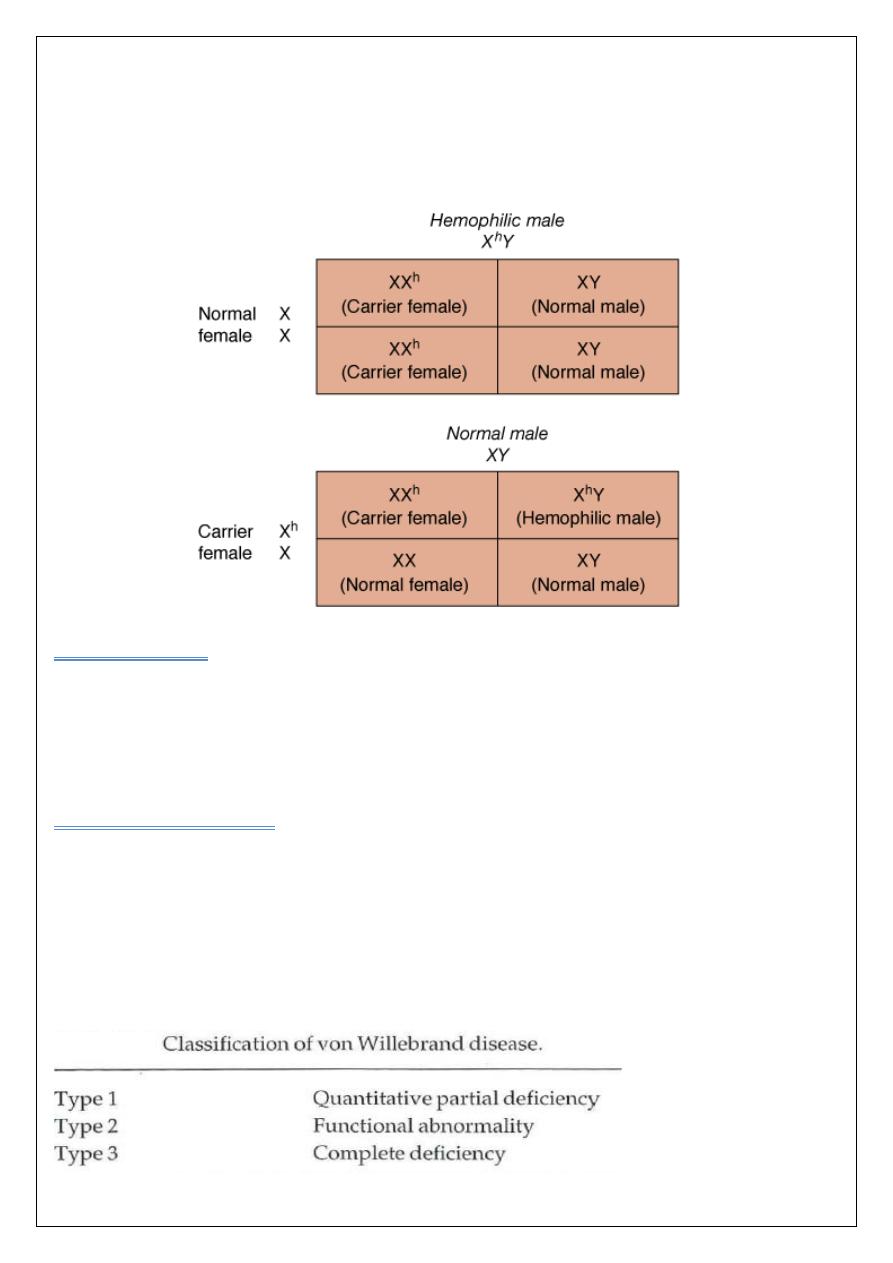
Haemophilia A:
Is the most common of the hereditary clotting factor deficiencies. The prevalence of
Haemophilia A affects approximately 1 in 10 000 live births and is equally common in all
ethnic groups population. The inheritance is sex-linked (X-linked) it occurs in males or in
homozygous females.
The defect is an absence or low level of plasma factor VIII.
Approximately half of the patients have missense or frameshift mutations or deletions in the
factor VIII gene.
Clinical features
Infants may suffer from profuse post-circumcision haemorrhage or develop joint and soft
tissue bleeds
and excessive bruising when they start to be active. Recurrent painful
haemarthroses and muscle haematomas dominate the clinical course of severely affected
patients and if poorly treated may lead to progressive joint deformity and disability.
Laboratory findings:
The following tests are abnormal: Activated partial thromboplastin time (APTT) & Factor
VIII clotting assay. While the bleeding time and prothrombin time (PT) tests are normal.
Treatment: of hemophilia A involve infusion of factor VIII.
:
aemophilia B
H
The inheritance and clinical features of factor IX deficiency (Christmas disease, haemophilia
B) are identical to those of haemophilia A. Indeed, the two disorders can only be
distinguished by specific coagulation factor assays. The incidence is one-fifth that of
haemophilia A.
:
Von Willebrand disease
The most common inherited bleeding disorder ,in this disorder there is either a reduced level
or
abnormal function of VWF resulting from a point mutation or major deletion in VWF
gene . VWF is produced in endothelial cells and megakaryocytes. It has two roles. It
promotes platelet adhesion to damaged endothelium and it is the carrier molecule for factor
VIII, protecting it from premature destruction ((explains the occasional reduced factor VIII
levels found in VWD)).Three types of VWD have been described.
Type 2 is divided into four subtypes depending on the type of functional defect.
Usually, the
inheritance is autosomal dominant with varying expression. The severity of the bleeding is
variable. Typically, there is mucous membrane bleeding (e.g. epistaxis, menorrhagia),
excessive blood loss from superficial cuts and abrasions, and operative and post-traumatic
haemorrhage. The severity is variable in the different types. Haemarthroses and muscle
haematomas are rare, except in type 3 disease.
Laboratory findings:
1 The bleeding time can be prolonged. This test is usually replaced by the PFA-100 test.
2 VWF levels are usually low.
3 The APTT may be prolonged.
4 Factor VIII levels are often low. If low, a factor VIII-VWF binding assay is performed.
5 Multimers analysis is useful for diagnosing different subtypes.
6 The platelet count is normal.
Acquired coagulation disorders:
The acquired coagulation disorders are more common than the inherited disorders. Unlike
the inherited disorders, multiple clotting factor deficiencies are usual.
1 Vitamin K deficiency.
2 Liver disease .
3 Disseminated intravascular coagulation DIC.
ﺍﻟﺩﻛﺗﻭﺭ ﺍﻻﺧﺗﺻﺎﺹ
ﻋﻠﻲ ﻋﺑﻳﺩ ﺍﺑﺭﺍﻫﻳﻡ ﺍﻟﺧﻔﺎﺟﻲ
ﺑﻭﺭﺩ ) ﺩﻛﺗﻭﺭﺍﺓ ( ﺍﻣﺭﺍﺽ ﺍﻟﺩﻡ
M.B.Ch.B. F.I.C.M.S