
HEMOSTASIS
NORMAL HEMOSTASIS
Haemostasis describes the normal process of blood clotting. It takes place via a series of
complex, tightly regulated interactions involving cellular and plasma factors.
There are five main components:
1.
Vessel wall.
2.
Platelets.
3.
Coagulation proteins.
4.
Anticoagulant proteins.
5.
Fibrinolytic system.
Approach to the diagnosis of bleeding disorders
The diagnostic evaluation of an infant or child for a possible bleeding disorder includes:
1.
Identifying the clinical presentation that suggest the underlying diagnosis.
2.
Initial laboratory screening tests to determine the most likely diagnosis. The most useful
initial screening tests are:
Full blood count and blood film
Prothrombin time (PT) - measures the activity of factors II, V, VII and X
Activated partial thromboplastin time (APTT) - measures the activity of factors II, V, VIII,
IX, X, XI and XII
If PT or APTT is prolonged, a 50: 50 mix with normal plasma will distinguish between
possible factor deficiency or presence of inhibitor
Fifth stage Lec -3
DR. ATHL HUMO
Pediatric
4/2017
Thrombin time - tests for deficiency or dysfunction of fibrinogen
Bleeding time ? Platelet function analyzer.
Quantitative fibrinogen assay
D-dimers
Biochemical screen including renal and liver function tests.
3.
Specialist investigation to characterize a deficiency or exclude important conditions that
can present with normal initial investigations, e.g. mild von Willebrand's disease, factor XIII
deficiency and platelet function disorders.
HEMOPHILIA
The commonest severe inherited coagulation disorders are haemophilia A (FVIII deficiency)
and haemophilia B (FIX deficiency). Both have X-linked recessive inheritance. Identifying
female carriers requires:
1.
A detailed family history.
2.
Analysis of coagulation factors.
3.
DNA analysis.
Prenatal diagnosis is available using DNA analysis.
Clinical Features
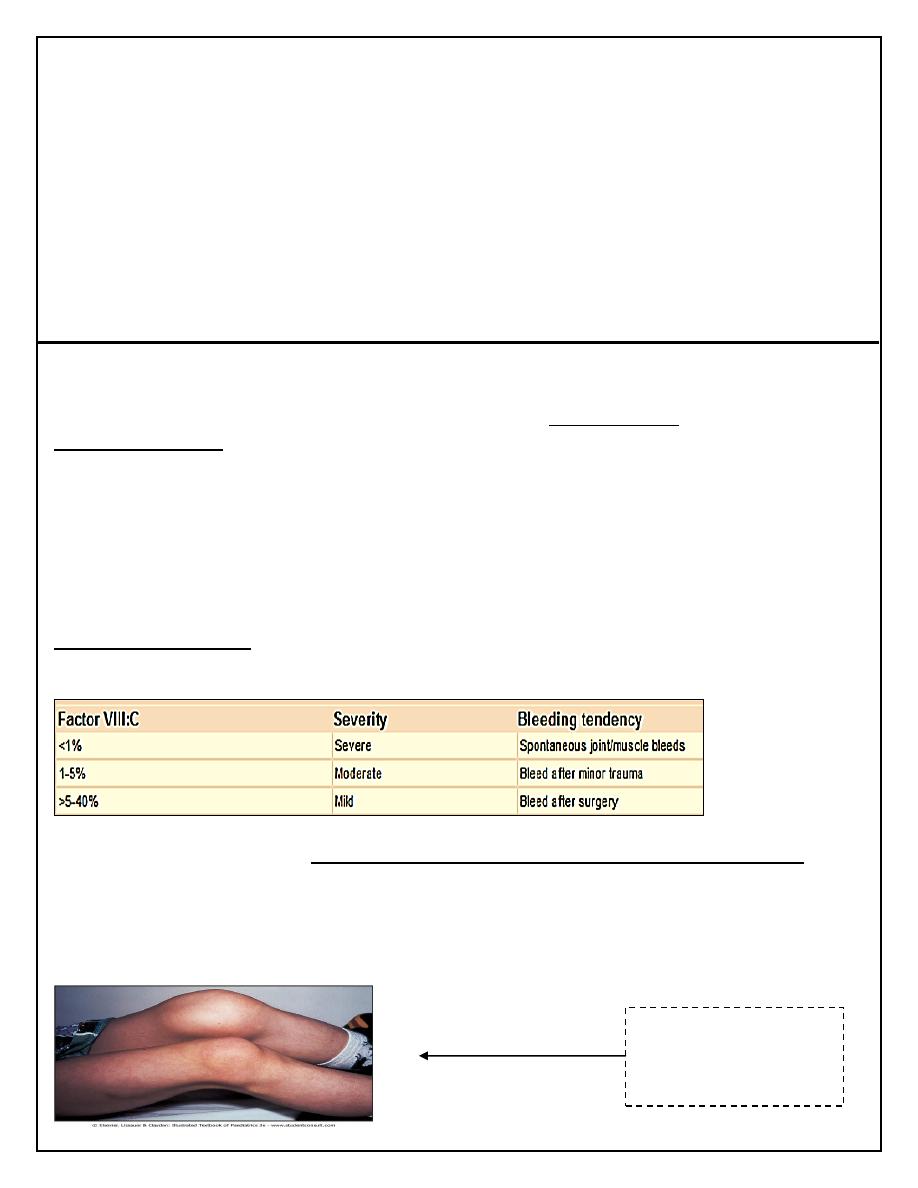
The disorder is graded as severe, moderate or mild depending on the FVIII:C (or IX:C in
haemophilia B) level.
The hallmark of the disease is recurrent spontaneous bleeding into joints and muscles,
which can lead to crippling arthritis if not properly treated. Most children present towards the
end of the first year of life, when they start to crawl then walk (and fall over).
Almost 40% of cases with severe disease present in the neonatal period, particularly with
intracranial hemorrhage, bleeding post-circumcision or prolonged oozing from heel stick and
venepuncture sites. The severity remains constant within a family.
Severe arthropathy
from recurrent joint
bleeds in haemophilia

INVESTIGATIONS
1.
Prolong APTT
2.
Normal PT
3.
Quantitative assessment of factor VIII/IX concentrate
TREATMENT
1.
Recombinant FVIII concentrate for haemophilia A ( or recombinant FIX concentrate for
haemophilia B) is given by intravenous infusion whenever there is any bleeding.
2.
If recombinant products are unavailable, highly purified, virally inactivated
plasma-derived products should be used.
3.
The quantity required depends on the site and nature of the bleeding. In general, raising the
circulating level to 30% of normal is sufficient to treat minor and simple joint bleeding.
Major surgery or life-threatening bleeds require the level to be raised to 100% and then
maintained at 30-50% for up to 2 weeks to prevent secondary hemorrhage. This can only be
achieved by regular infusion of factor concentrate (usually 8- to 12-hourly for FVIII, 12- to
24-hourly for FIX, or by continuous infusion) .
4.
Dose for f VIII : desired level (%) × weight (Kg) × 0.5
Dose for f IX : desired level (%) × weight (Kg) × 1.5
5.
Desmopressin (DDAVP) may allow mild haemophilia A to be managed without the use of
blood products. It is given by infusion and stimulates endogenous release of FVIII:C and
von Willebrand factor. Adequate levels can be achieved to enable minor surgery and dental
extraction to be undertaken. DDAVP is ineffective in haemophila B.
6.
Haemophilia centers should supervise the management of children with bleeding disorders.
They provide a multidisciplinary approach with expert medical, nursing and laboratory
input. Specialised physiotherapy is needed to preserve muscle strength and avoid damage
from immobilization. Psychosocial support is an integral part of maintaining compliance.
7.
NOTE: intramuscular injections, aspirin and NSAI drugs should be avoided in all patients
with haemophilia.
8.
Home treatment is encouraged to avoid delay in treatment which increases the risk of
permanent damage, e.g. progressive arthropathy. Parents are usually taught to give
replacement therapy at home when the child is 2-3 years of age.

PROPHYLAXIS
Prophylactic FVIII/IX is given to all children with severe haemophilia to further reduce the
risk of chronic joint damage by raising the baseline level above 2%.
Primary prophylaxis usually begins at age 2-3 years, and is given two to three times per
week. If peripheral venous access is poor, a central venous access device may be required.
Prophylaxis has been shown to result in better joint function in adult life.
Von Willebrand Disease
Von Willebrand disease is a common AD inheritance disorder (found in 1% of the population)
caused by a deficiency of von Willebrand factor
Von Willebrand factor (vWF) has two major roles:
1.
It facilitates platelet adhesion to damaged endothelium.
2.
It acts as the carrier protein for FVIII, protecting it from inactivation and clearance.
vWD results from either a quantitative or qualitative deficiency of von Willebrand factor
(vWF). This causes defective platelet plug formation and, since vWF is a carrier protein for
FVIII, patients with vWD also are deficient in FVIII.
CLINICAL FEATURES
Bruising
Excessive, prolonged bleeding after surgery
Mucosal bleeding such as epistaxis and menorrhagia.
In contrast to haemophilia, spontaneous soft tissue bleeding such as large haematomas and
haemarthroses are rare.
INVESTIGATIONS
1.
VWF antigen (VWF:Ag), measures total amount of VWF protein present.
2.
VWF activity (VWF : Ristocetin Cofactor [Rco]), assess interaction of VWF and platelets
as mediated by ristocetin antibiotic. The rate of Ristocetin induced agglutination is related
to the concentration and functional activity of the plasma von Willebrand factor).
3.
APTT mildly prolonged.
4.
Bleeding time is prolonged.
TREATMENT
1.
Mild vWD can usually be treated with DDAVP, which causes secretion of both FVIII and
vWF into plasma.
2.
More severe types of vWD have to be treated with vWF- FVIII concentrate.
3.
Cryoprecipitate is no longer used to treat vWD as it has not undergone viral inactivation.
4.
Intramuscular injections, aspirin and NSAI drugs should be avoided in all patients with
vWD.

Investigations in haemophilia A & vWD
Lab
Haemophilia A
von Willebrand's disease
PT
Normal
Normal
APTT
↑↑
Normal or ↑
Factor VIII:C
↓↓
Normal or ↓
vWF Ag
Normal
↓
RiCoF (activity)
Normal
↓
vWF multimers
Normal
Variable
Acquired Disorders of Coagulation
The main acquired disorders of coagulation affecting children are those secondary to:
1.
Haemorrhagic disease of the newborn due to vitamin K deficiency.
2.
liver disease.
3.
ITP (immune thrombocytopenia).
4.
DIC (disseminated intravascular coagulation).
THROMBOCYTOPENIA
Thrombocytopenia is a platelet count <150 × 10
9
/L. The risk of bleeding depends on the
level of the platelet count:
1.
Severe thrombocytopenia (platelets <20 × 10
9
/L) - risk of spontaneous bleeding.
2.
Moderate thrombocytopenia (platelets 20-50 × 10
9
/L) - at risk of excess bleeding during
operations or trauma but low risk of spontaneous bleeding.
3.
Mild thrombocytopenia (platelets 50-150 × 10
9
/L) - low risk of bleeding during operations
or trauma.
Thrombocytopenia may result in bruising, petechiae, purpura and mucosal bleeding (e.g.
epistaxis, bleeding from gums when brushing teeth). Major haemorrhage in the form of
severe gastrointestinal haemorrhage, haematuria and intracranial bleeding is much less
common.
While purpura may signify thrombocytopenia, it also occurs with a normal platelet count
from platelet dysfunction and vascular disorders.

Immune Thrombocytopenia
Idiopathic Thrombocytopenic Purpura(ITP)
Childhood ITP is a common disorder that usually follows an acute viral infection. Childhood
ITP is caused by an antibody (IgG or IgM) that binds to the platelet membrane. The
condition results in Fc receptor–mediated splenic destruction of antibody-coated platelets.
Rarely, ITP may be the presenting symptom of an autoimmune disease, such as systemic
lupus erythematosus (SLE).
The reduced platelet count is accompanied by a compensatory increase of megakaryocytes in
the bone marrow.
CLINICAL MANIFESTATIONS
Most children present between the ages of 2 and 10 years, with onset often 1-2 weeks after a
viral infection. Affected children develop petechiae and purpura and superficial bruising . It
can cause epistaxis and other mucosal bleeding but profuse bleeding is uncommon.
Intracranial bleeding is a serious but rare complication, occurring in 0.1-0.5%, mainly in
those with a long period of severe thrombocytopenia.
Significant adenopathy or hepatosplenomegaly is unusual, and the RBC & WBC counts are
normal.
The presence of abnormal findings such as HSM, bone or joint pain, remarkable
lymphadenopathy other cytopenias, or congenital anomalies suggests other diagnoses
(leukemia, syndromes).
When the onset is insidious, especially in an adolescent, chronic ITP or the possibility of a
systemic illness, such as SLE, is more likely.
DIAGNOSIS
ITP is a diagnosis of exclusion, so careful attention must be paid to the:
1.
History.
2.
Clinical features.
3.
Lab finding:
CBC & blood film: ↓platelet count (normal or ↑size platelet), normal RBC & WBC count.
Bone marrow examination [shows normal granulocytic and erythrocytic series, with
characteristically normal or increased numbers of megakaryocytes], usually not done unless
there is atypical finding as :
an abnormal WBC count or differential
unexplained anemia
as well as findings on history and physical examination suggestive of a bone
marrow failure syndrome or malignancy as HSM & LAP.
A. Blood smear (large platelets)
B. Bone marrow aspirate
(Increased numbers of
megakaryocytes, many of
which appear immature)

TREATMENT
In about 80% of children, the disease is acute, benign and self-limiting, usually remitting
spontaneously within 6-8 weeks. Most children can be managed at home and do not require
hospital admission.
The American Society of Hematology Guidelines:
1.
No therapy, except to increase platelet count > 20.
2.
Prednisone 1-4mg/kg/day, short course until plat >20.
3.
IVIG: 1g/kg single dose for 2 days (downregulating Fc-mediated phagocytosis of
antibody-coated platelets.
4.
Intravenous anti-D therapy: for Rh-positive patients at a dose of 50-75 μg/kg for 2-3 days
(induces mild hemolytic anemia. RBC-antibody complexes bind to macrophage Fc receptors
and interfere with platelet destruction)
5.
Splenectomy:
Acute ITP with ICH & life-threatening bleeding not respond to platelet
transfusion & other therapy.
Child ≥ 4 years with severe chronic ITP, not respond to other therapy.
Chronic ITP
In 20% of children the platelet count remains low for 6-12 months after diagnosis; this is
known as chronic ITP.
No treatment is given unless there is major bleeding, treatment is mainly supportive, the
child should avoid contact sports but be encouraged to continue normal activities, including
schooling.
Splenectomy may benefit patient with significant bleeding, but has significant morbidity and
may be unsuccessful in up to 25% of cases.
Rituximab, has been used to treat chronic ITP.
Thrombopoiesis stimulator, romiplastin and eltrombopag are encouraging for treating
chronic ITP.
If ITP in a child becomes chronic, regular screening for SLE should be performed, as the
thrombocytopenia may predate the development of autoantibodies.
Disseminated Intravascular Coagulation
DIC describes a disorder characterised by coagulation pathway activation leading to diffuse
fibrin deposition in the microvasculature and consumption of coagulation factors and platelets.
This altered balance of hemostasis usually caused by life-threatening severe systemic disease
associated with hypoxia, acidosis, tissue necrosis, shock, and/or endothelial damage.

CLINICAL MANIFESTATIONS
Bleeding frequently first occurs from sites of venipuncture or surgical incision.
The skin may show petechiae and ecchymoses.
Tissue necrosis as infarction of large areas of skin, subcutaneous tissue, or kidneys.
Microangiopathic hemolytic anemia.
LABORATORY FINDINGS
No single test reliably diagnoses DIC. However, DIC should be suspected when the following
abnormalities coexist:
1.
Thrombocytopenia.
2.
Prolonged PT (prothrombin time).
3.
Prolonged APTT.
4.
Low fibrinogen.
5.
Raised fibrinogen degradation products and D-dimers.
6.
Microangiopathic haemolytic anaemia.
7.
There is also usually a marked reduction in the naturally occurring anticoagulants, protein C,
S and antithrombin.
TREATMENT
1.
Treat underlying cause.
2.
Supportive:
correcting hypoxia, acidosis and poor perfusion.
replace depleted blood-clotting factors, platelets, and anticoagulant proteins by transfusion.
3.
Heparin may be used to treat significant arterial or venous thrombotic disease unless sites of
life-threatening bleeding coexist.

THROMBOSIS
Thrombosis is uncommon in children. It may be due to an:
1.
Inherited disorder:
protein C deficiency
protein S deficiency
antithrombin deficiency.
2.
Acquired secondary to another underlying illness or its treatment:
catheter-related thrombosis.
DIC.
polycythaemia (e.g. due to congenital heart disease).
malignancy .
SLE.
All children with thrombosis should be screened for inherited or acquired predisposing
disorders.