1
Pediatrics
HEMOLYTIC ANAEMIA
• Hemolysis is defined as the premature destruction of RBCs (a shortened RBC life
span). Anemia results when the rate of destruction exceeds the capacity of the marrow
to produce RBCs.
• The lifespan of a normal red cell is 120 days. In haemolysis, red cell survival may be
reduced to a few days but bone marrow production can increase about eightfold, so
haemolysis only leads to anaemia when the bone marrow is no longer able to
compensate for the premature destruction of red cells.
• Heightened RBC production, reflected in an increased percentage of reticulocytes in
the blood. Thus, hemolysis should be suspected as a cause of anemia if an elevated
reticulocyte count is present.
• The reticulocyte count may also be elevated
– as a response to acute blood loss.
– for a short period after replacement therapy for iron, vitamin B12, or folate
deficiency.
• The reticulocyte percentage can be corrected to measure the magnitude of marrow
production in response to hemolysis as follows:
Hematology
f
Dr.Athal

2
CLASSIFICATION
1. CELLULAR DEFECTS (Intrinsic Abnormalities)
– Membrane Defects:
• Hereditary spherocytosis
– Enzyme Deficiencies:
• G6PD deficiency
– Hemoglobinopathies: defect in
• HEMOGLOBIN S (Sickle Cell Disease)
• HEMOGLOBIN C
• HEMOGLOBIN E
• HEMOGLOBIN D
• Thalassemia (α and β)
2. EXTRACELLULAR DEFECTS (Extrinsic Abnormalities)
– Antibodies:
• Autoimmune HA
– Mechanical factors:
• DIC, TTP, HUS
• Prosthetic heart valve
• Burns, thermal injury
• Hypersplenism
– Plasma factors:
• Liver disease
• Wilson disease
• Infections
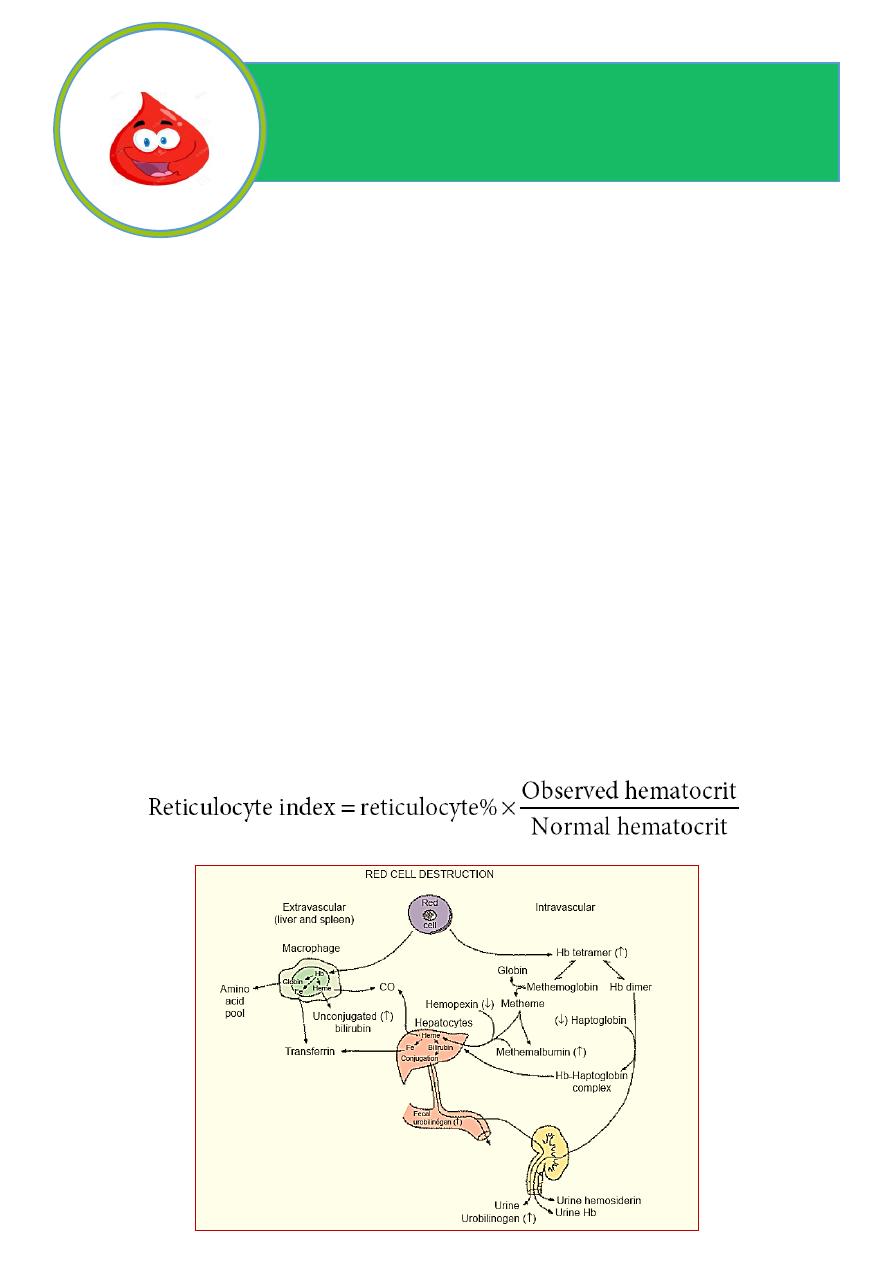
DIAGNOSIS
1. Anaemia(↓ Hb).
2. Raised reticulocyte count (polychromasia on blood film).
3. Raised unconjucated bilirubin.
4. Raised LDH
5. Decreased haptoglobins
6. Positive direct antiglobulin test (only if an immune cause as this test identifies
antibody-coated red blood cells)
3
7. Increased erythropoiesis in the bone marrow.
8. Abnormal appearance of the red cells on a blood film (e.g. spherocytes, sickle shaped)
9. Reticuloendothelial hyperplasia - hepatomegaly and splenomegaly
Heredity Spherocytosis
• HS is transmitted as an autosomal dominant or, less commonly, as an autosomal
recessive disorder.
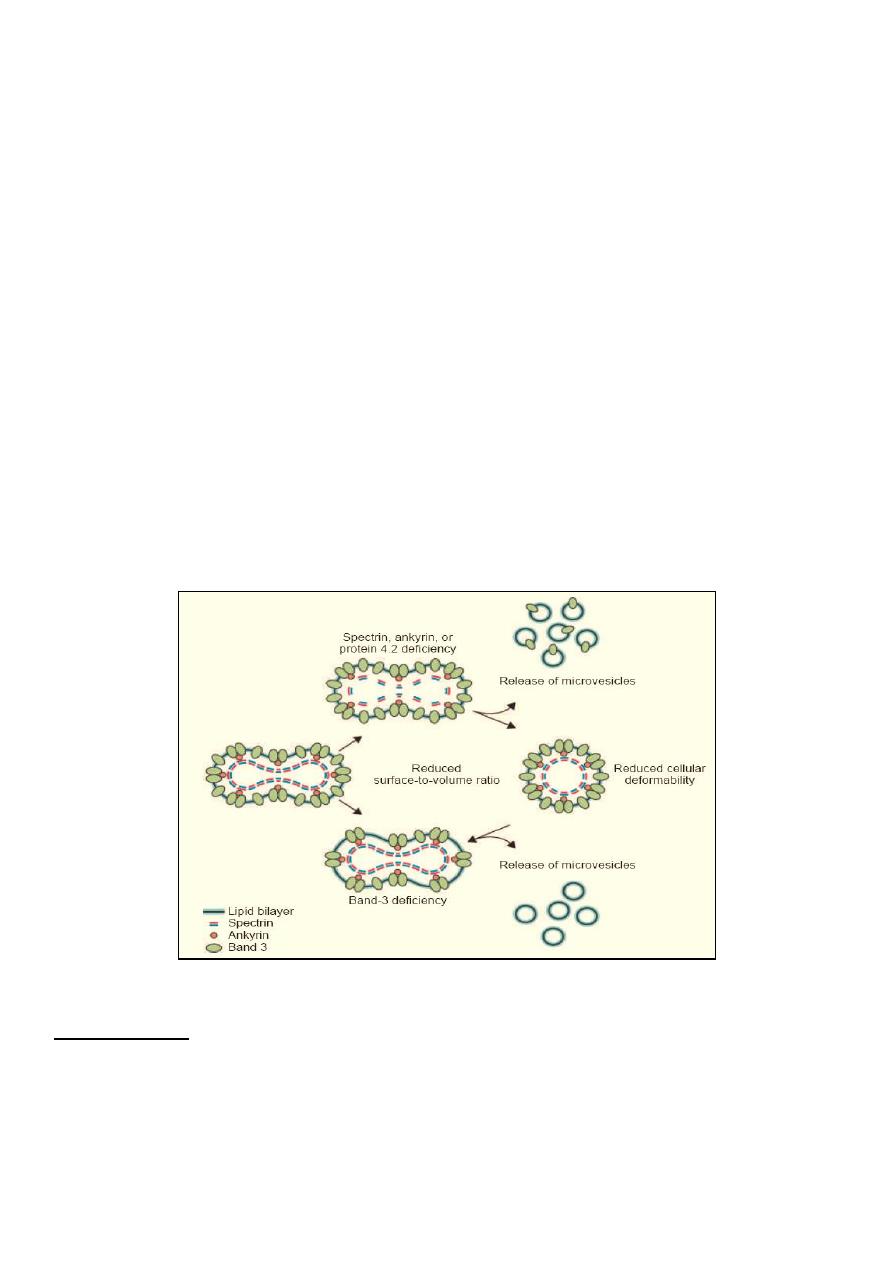
• Molecular defect in protein spectrin, ankyrin or other proteins that are provide stability
of RBC membrane shape. This results in the red cell losing part of its membrane, this
reduction in its surface-to-volume ratio causes the cells to become spheroidal, making
them less deformable than normal red blood cells and leads to their destruction in the
microvasculature of the spleen.
Pathophysiology of Hereditary Spherocytosis
DIAGNOSIS
Diagnosis of HS depend on:
1. Family history.
2. Clinical features.
3. Laboratory study.
4
CLINICAL MANIFESTATIONS
• The clinical spectrum varies widely.
• Affected patients may be asymptomatic, without anemia and with minimal hemolysis.
• Or they may have severe hemolytic anemia associated with growth failure,
splenomegaly, chronic transfusions need splenectomy.
1. Jaundice: usually develops during childhood but may cause severe hemolytic jaundice in
the first few days of life.
2. Anaemia .
3. Mild to moderate splenomegaly - depends on the rate of hemolysis.
4. Aplastic crisis - uncommon, associated with parvovirus B19 infection
5. Gallstones - due to increased bilirubin excretion.
6. Rare complications associated with HS include splenic sequestration crisis, gout,
cardiomyopathy, leg ulcers, and spinocerebellar degeneration.
LABORATORY STUDY
• The hemoglobin level usually is 6-10 g/dL, but it can
be normal.
• MCHC ↑(36-38 g/dl)
• MCV NR.
• Retics ↑(mean 10%).
• Blood film:
– Small spherocyte, hyperchromic with central
pallor is less conspicuous.
– Polychromasia.
• ↓ haptoglobin.
• US may show gallstone.
• If the diagnosis is less certain, the recommended tests that have a high predictive value
for HS are the flow cytometric EMA (eosin-5- maleimide) binding test and the
cryohemolysis test.
• Osmotic fragility test can detect the presence of spherocytes in the blood; however, it
is not specific to HS and may be abnormal in other hemolytic anemias.

5
TREATMENT
1. Folic acid supplementation.
2. Blood transfusion for aplastic crises.
3. Splenectomy is recommended for:
a. patients with severe HS.
b. patients with moderate HS and frequent hypoplastic or aplastic crises, poor
growth, or cardiomegaly.
When splenectomy is indicated, it should be performed after the age of 6 yr, if possible, to
avoid the heightened risk of postsplenectomy sepsis in younger children.Vaccines for
encapsulated organisms, such as pneumococcus, meningococcus, and H. influenzae type b,
should be administered at least 14 days before splenectomy, and prophylactic oral penicillin
prescribed indefinitely.
4. Cholecystectomy for gallstones.
Glucose-6-Phosphate Dehydrogenase Deficiency (G6PD)
• G6PD deficiency, the most frequent disease involving enzymes of the hexose
monophosphate pathway. and is essential for preventing oxidative damage to red cells.
Red cells lacking G6PD are susceptible to oxidant-induced haemolysis.
• G6PD deficency is X-linked inheritance, therefore the symptoms occur more
frequently in males than in females. Rarely, the symptoms appear in heterozygous
females because the random inactivation of the normal X chromosome (Lyon
hypothesis).
• The global distribution of this disorder parallels that of malaria, representing an
example of “balanced polymorphism,” in which there is an evolutionary advantage of
resistance to falciparum malaria in heterozygous females that outweighs the small
negative effect of affected hemizygous males.
• Many different mutations of the gene have been described, leading to different clinical
features in different populations.
• Young red blood cells may have normal enzyme activity whilst older cells are
deficient.

6
Clinical Manifestations
• Neonatal jaundice: onset is usually in the first 3 days of life, even kernicterus may
occur.
• Acute haemolysis: most individuals with G6PD deficiency are asymptomatic, with no
clinical manifestations of illness unless triggered by infection or a substance with
oxidant properties, The degree of hemolysis varies with the inciting agent, amount
ingested, and severity of the enzyme deficiency. The symptoms of intravascular
haemolysis appear 24-48 h after exposure to triggering factors:
1. Anaemia , may be severe lead to shock & acidosis.
2. Jaundice
3. Dark urine, as it contains haemoglobin as well as urobilinogen.
4. Malaise.
Agents Precipitating Hemolysis in G6PD Deficiency
• FOOD: fava beans known as favism. Fava beans contain divicine, convicine &
isouramil which ultimately lead to production of hydrogen peroxide and other reactive
oxygen products.
• MEDICATIONS:
– Antibacterials: sulfonamides, trimethoprim-sulfamethoxazole, nalidixic acid,
chloramphenicol, nitrofurantoin.
– Antimalarials: primaquine, chloroquine, quinacrine
– Others: vitamin K, aspirin
• CHEMICALS:
– Benzene,
– Naphthalene (moth balls)
• ILLNESS:
– Diabetic acidosis
– Hepatitis
– Sepsis
Laboratory Findings
1. Fall in hemoglobin and hematocrit.
2. Retics ↑
3. Bilirubin ↑
4. Haptoglobin ↓.

7
5. hemoglobinuria.
6. Bile pigment in the urine.
7. Blood film: The RBC morphology during episodes of acute hemolysis is striking,
appearing to have “bites” taken out of them (cookie cells). These are areas of absent
hemoglobin that are produced by phagocytosis of Heinz bodies by splenic
macrophages. When a patient with G6PD is exposed to significant oxidant stress,
hemoglobin is oxidized, forming precipitates of hemoglobin called (Heinz bodies)
8. Confirmation of diagnosis: by measuring G6PD activity in RBCs. During a
haemolytic crisis, G6PD levels may be misleadingly elevated due to the higher
enzyme concentration in reticulocytes, which are produced in increased numbers in
response to the destruction of mature red cells. A repeat assay after few weeks is then
required in the steady state to confirm the diagnosis.
Morphologic erythrocyte changes (anisopoikilocytosis,
bite cells) during acute hemolysis in a G6PD-deficient patient. Arrows show bite cells
Treatment and Prevention
The treatment of G6PD deficiency is supportive.
• Transfusions are indicated when significant cardiovascular compromise is present.
• Maintaining hydration and urine alkalization protects the kidneys against damage from
precipitated free hemoglobin.
• Prevention of hemolysis is by:
– Avoidance of known oxidants.
– Serious infection also is a potential precipitant of hemolysis in G6PD-deficient
young children.
8
Sickle Cell Disease
• Sickle cell disease is the collective name given to haemoglobinopathies in which HbS
is inherited.
PATHOPHYSIOLOGY
• HbS forms as a result of a point mutation in codon 6 of the β-globin gene which
causes a change in the amino acid encoded from glutamine to valine.
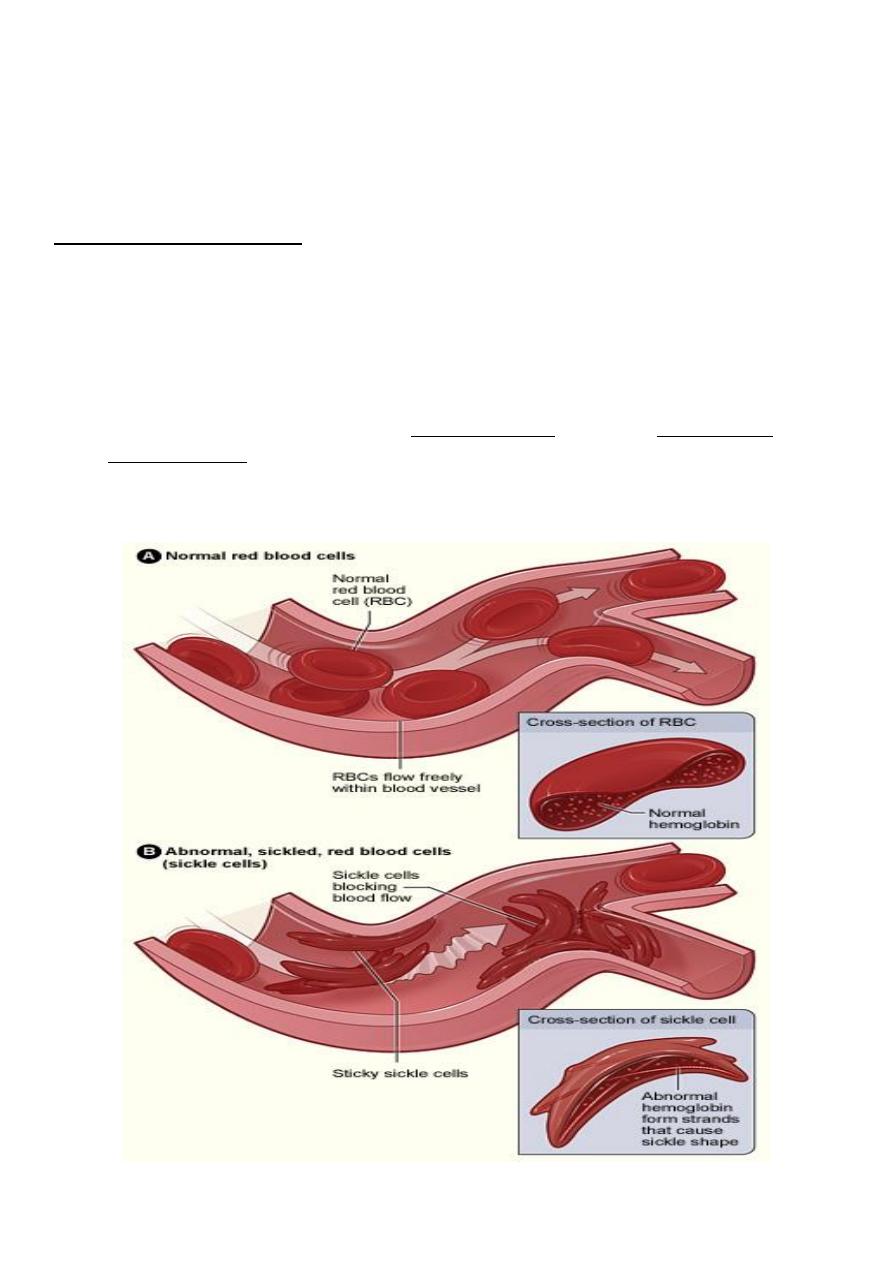
• In HbSS, the haemoglobin molecule becomes deformed (insoluble) in the
deoxygenated state. HbS polymerises within red blood cells forming rigid tubular
spiral bodies which deform the red cells into a sickle shape.
• Irreversibly sickled red cells have a reduced lifespan and may be trapped in the
microcirculation, resulting in thrombosis and therefore ischaemia in an organ or bone.
This is exacerbated by hypoxia, acidosis, fever, hypothermia, and dehydration.

9
• Sickle cell disease refers to not only patients with sickle cell anemia, but also to
compound heterozygotes where one β-globin allele includes the sickle cell mutation
and the second β-globin allele includes a gene mutation other than the sickle cell
mutation, such as HbC & β-thalassemia.
• In sickle cell anemia, HbS is commonly as high as 90% of the total hemoglobin;
whereas as in sickle cell disease, HbS is >50% of all hemoglobin.
There are four main forms of SCD:
1-Sickle cell anaemia (HbSS) - patients are homozygous for HbS, i.e. virtually all their Hb
is HbS; they have no HbA because they have no normal β-globin genes.
2-SC disease (HbSC) - affected children inherit HbS from one parent and HbC from the
other parent (HbC is formed as a result of a different point mutation in β-globin), so they
also have no HbA because they have no normal β-globin genes.
3-Sickle β-thalassaemia - affected children inherit HbS from one parent and β-thalassaemia
trait from the other. They have no normal β-globin genes and most patients can make no
HbA and therefore have similar symptoms to those with sickle cell anaemia.
4-Sickle trait - inheritance of HbS from one parent and a normal β-globin gene from the
other parent, so approximately 40% of the haemoglobin is HbS. They do not have sickle cell
disease but are carriers of HbS, so can transmit HbS to their offspring. They are
asymptomatic and are only identified as a result of blood tests. Rarely causes problems
except under conditions of low oxygen tension.
CLINICAL MANIFESTATIONS
1. ANAEMIA & JAUNDICE: Moderate anaemia (Hb usually 6-8g/dl) and jaundice due
to chronic hemolysis.
2. INFECTON :
- Increase susceptibility to infection from encapsulated organism, also ↑incidence
of osteomyelitis caused by Salmonella & other organisms. This due to
hyposplenism 2ry to chronic sickling & microinfarction in the spleen .
- Acute chest syndrome (most common pathogens are S. pneumoniae,
Mycoplasma pneumoniae, and Chlamydia sp.)
3. PAINFUL CRISES:
- Vaso-occlusive crises causing pain may affect all organs of the body with
varying frequency & severity.
- Hand-foot syndrome (dactylitis with swelling & pain of fingers & feet from vaso-
occlusion). The bones pain of the limbs & spine are common . Also avascular
necrosis of femoral head.
10
- Cerebral infarction presented as acute stroke & hemiparesis
- Acute chest syndrome (severe and may be fatal due to vasooclusive crises leading
to hypoxia )
4. ACUTE ANAEMIA: Sudden ↓in Hb from:
- Haemolytic crises associated with infection
- Aplastic crises due to parvovirus infection causes complete though temporary
cessation of RBC production.
- Sequestration cises: sudden splenic enlargement, abdominal pain & circulatory
collapse from accumilation of sickled cell in spleen.
5. PRIAPISM: Due to occlusion of corpora cavernosa; needs treated urgently by
exchange blood transfusion otherwise may lead to damage and fibrosis.
6. LONG TERM COMPLICATIONS:
- Short stature & delay puberty
- Cognitive problem & poor school performance.
- HF from chronic uncorrected anaemia
- Renal dysfunction
- Gall stone
- Leg ulcers
- Psychological proplem
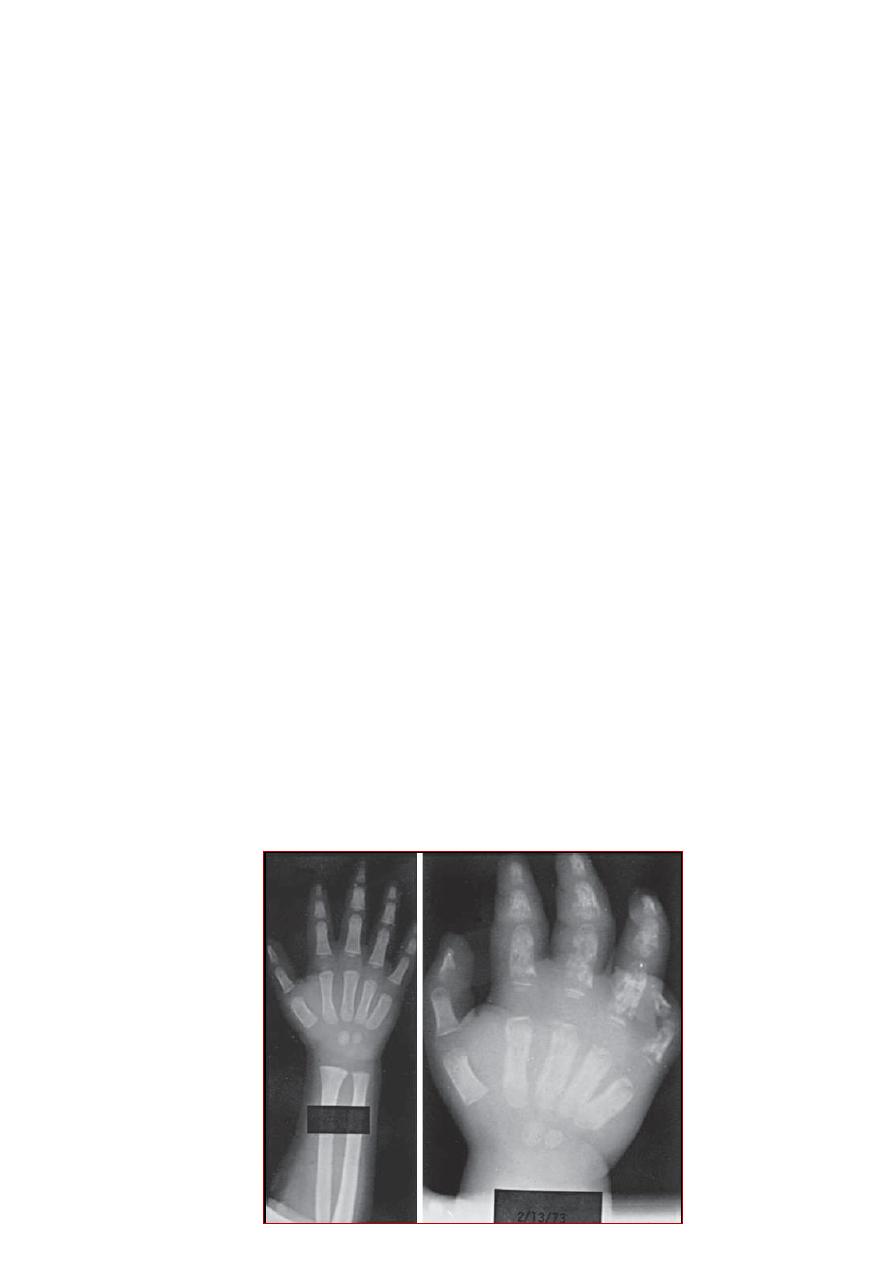
XR of an infant with SCA and acute dactylitis.
A. The bones appear normal at the onset of the episode.
B. Destructive changes and periosteal reaction are evident 2 wk later.

11
MANAGEMENT
1. Prophylactic Penicillin: children with sickle cell anemia should receive prophylactic oral
penicillin until at least 5 yr of age to prevent pneumococcus infection. An alternative for
children who are allergic to penicillin is erythromycin.
2. Immunizations: routine childhood immunizations, especially against pneumococcus &
Haemophilus influenzae type b, as well as the annual administration of influenza
vaccine, are highly recommended.
3. Folic acid: oral once daily .
4. Minimized vaso-occlusive crises, by avoiding exposure to cold, dehydration, excessive
exercise, undue stress or hypoxia.
5. Treatment of acute painful crises should be with oral or intravenous analgesia according
to need (may require opiates) and good hydration (oral or intravenous as required);
infection should be treated with antibiotics; oxygen should be given if the oxygen
saturation is reduced.
6. RBC transfusions or exchange transfusion is indicated for acute chest syndrome, stroke
and priapism.
7. Treatment of chronic problems, children who have recurrent hospital admissions for
painful vaso-occlusive crises or acute chest syndrome may benefit from hydroxyurea, a
drug which increases their HbF production and helps protect against further crises.
8. BM transplantation, this is the only cure for sickle cell disease.
THALASSEMIA SYNDROME
• Thalassemia is hereditary hemolytic anemia characterized by decreased or absent
synthesis of one or more globin subunits of the hemoglobin molecule.
• α-Thalassemia results from reduced synthesis of α-globin chains.
• β-thalassemia results from reduced synthesis of β-globin chains.
PATHOPHYSIOLOGY
An imbalance in globin chain production is a hazard to the RBC, because it results in
precipitation of excess unpaired globin chains within the red cell membrane, leading to:
• Ineffective erythropoiesis, cell death within the bone marrow.
12
• Hemolytic anaemia, premature removal of circulating red cells by the spleen.
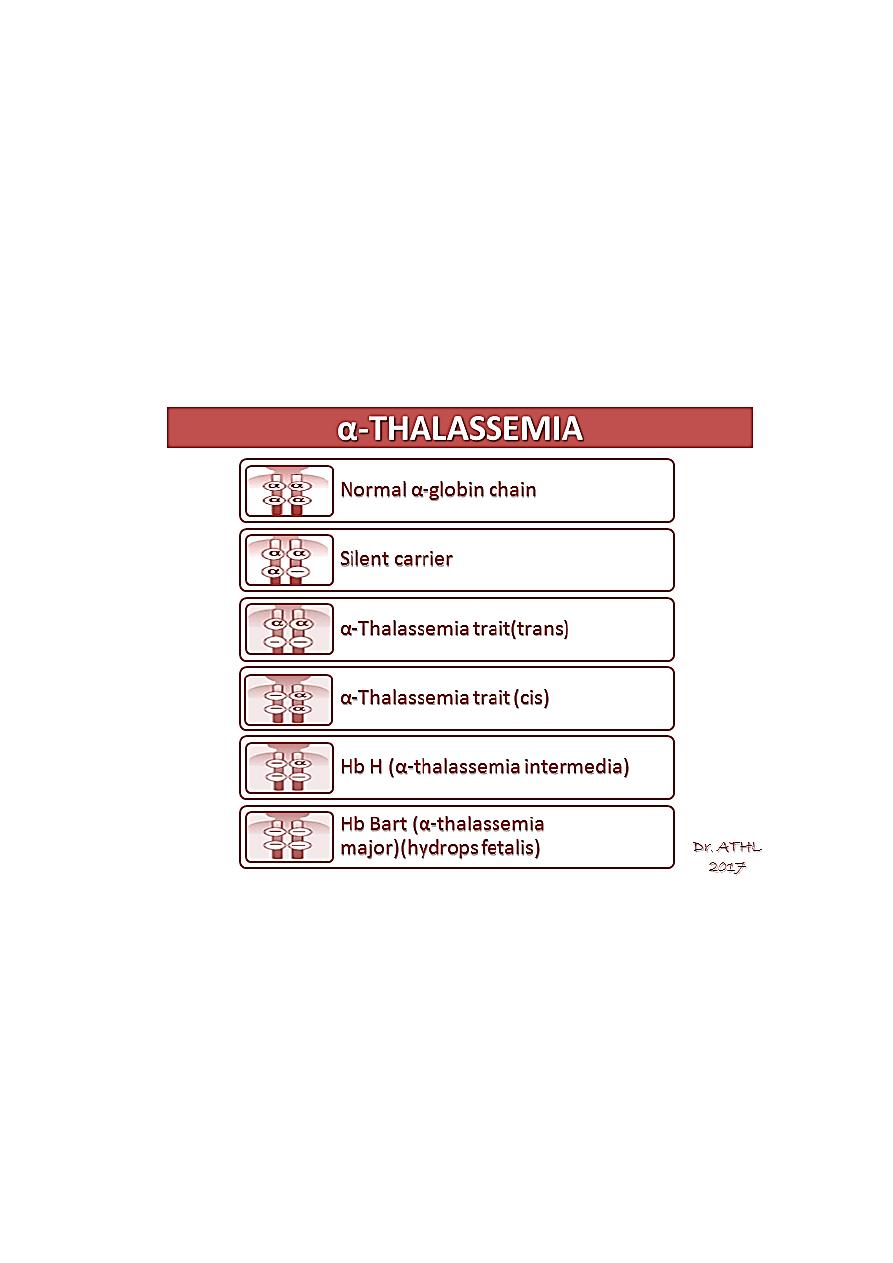
α-THALASSEMIA
– α-Thalassemias are usually the result of α gene deletion.
– α-Thalassemia variants are found most often in populations of African or East
Asian ancestry.
– Normally there are four α-globin genes; clinical manifestations of α-thalassemia
variants reflect the number of genes affected.
– An α
0
-mutation indicates no α-chains produced from that gene. An α
+
mutation
produces a decreased amount of α-globin chain.
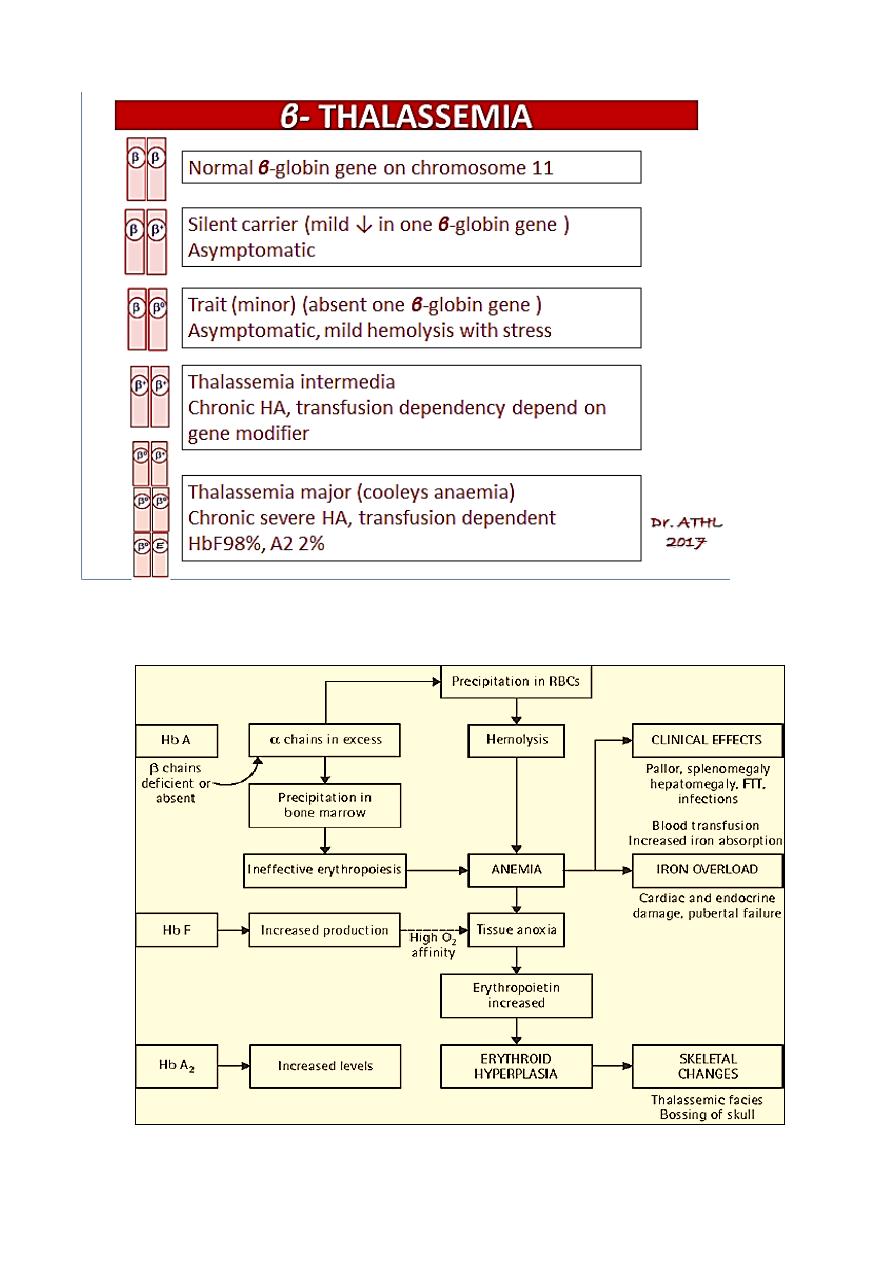
β- THALASSEMIA
• The clinical phenotype of β-thalassemia is related to the degree of globin chain
imbalance.
• Normally there are two β-globin genes; clinical manifestations of β-thalassemia
variants reflect the number of genes affected.
Heterozygous β-thalassemia(β-thalassemia minor).
Homozygous β-thalassemia (β-thalassemia major ‘Cooley anemia’ and
intermedia).
13
β- THALASSEMIA PATHOLOGY

14
Heterozygous β-thalassemia(β-thalassemia minor, trait)
Clinical Features:
1. Mild anemia (Hb about 10 gm/dl)
2. Normal growth and development.
3. Blood film: Hypochromia, microcytosis, and anisocytosis.
4. Hemoglobin electrophoresis shows elevation of the Hb A2 level and, sometimes,
elevation of the Hb F level.
5. RBC count is elevated.
Therapy: No treatment is necessary.
• It is important, however, that thalassemia minor is distinguished from iron deficiency
to prevent inappropriate therapy with iron.
• Folic acid may be given.
• Genetic counseling is also important.
Homozygous β-thalassemia
Homozygous β-thalassemia (β-thalassemia major’ Cooley anemia’ and intermedia). Its
autosomal recessive inheritance of mutation in B globin chain genes.
Defect: Molecular defects range from complete absence of β-globin synthesis (genotype
β0/β0) to partial reduction in the gene product from the affected locus (genotype β+/β+).
Clinical Features: begin in the middle of the first year of life.
1. The infant manifests a progressively severe hemolytic anemia and jaundice.
2. Marked HSM.
3. FTT.
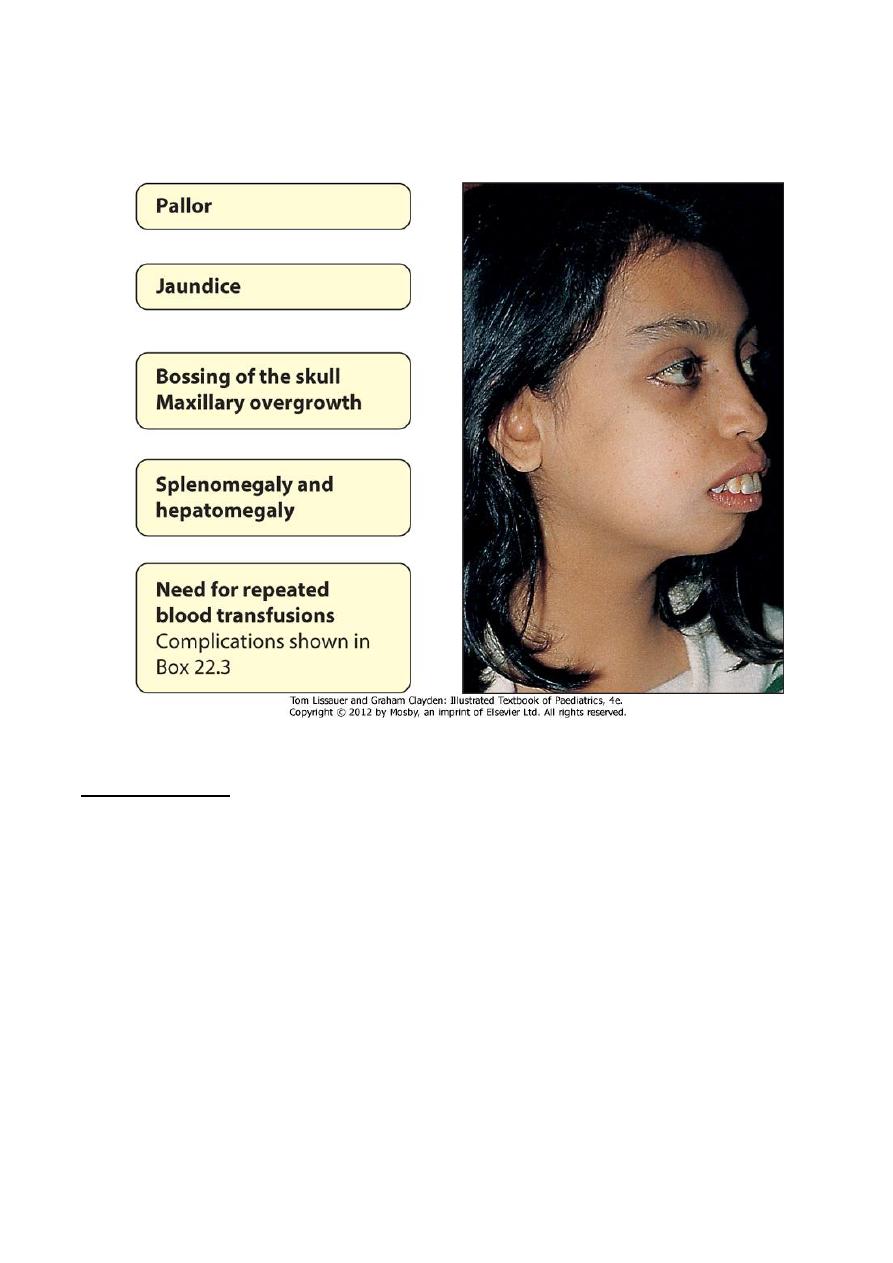
4. The BM hyperplasia produces characteristic features such as tower skull, frontal
bossing, maxillary hypertrophy with prominent cheekbones, and overbite.
5. Hemochromatosis: Even in the untransfused state, iron overload develops in
thalassemic patients because of hyper absorption of dietary iron. The iron load
becomes even greater with chronic transfusion therapy. When the bone marrow
storage capacity for iron is exceeded, iron accumulates in parenchymal organs such
15
as the liver, heart, pancreas, gonads, and skin, producing the complications of
hemochromatosis. Many patients develop congestive heart failure, hypogonadism,
DM, hypothyroidism, liver cirrhosis and short stature.
Investigations
:
1. CBC & smear: hypochromic microcytic anemia, with nucleated RBC &
reticulocytopenia.
2. Elevated unconjucated bilirubin.
3. Hemoglobin electrophoresis, Hb A is either markedly decreased or totally absent. Of
the total hemoglobin concentration, 30% to 90% is Hb F.
4. BM: hyperplasia is seen in bone XR.
5. Iron study: elevated S. ferritin & transferrin saturation.

16
Treatment
:
1. The mainstay of treatment is transfusion with packed RBCs using irradiated CMV –
ve blood, a post transfusion Hb level of 10 gm/dl is the goal.
2. In an effort to prevent hemochromatosis, patients who receive chronic transfusion
regimens are treated with chelating agents (e.g.deferoxamine, deferasirox) that
promote iron removal from the body through excretion in the urine and the stool.
3. Splenectomy is usually considered when transfusion requirements exceed 250 ml
/kg/year.
4. Stem cell transplantation can cure the patients.
5. Genetic counseling.
:D