14 جمادي الاولى 1435 الموافق 16/3/2014
Blood coagulation disordersThe normal haemostasis prevents:
Spontaneous haemorrhage and undue blood loss from injured vessels
Intravascular thrombus formation.
There are three components of blood coagulation system:

Primary haemostasis (it is enough to stop bleeding from small injuries(
Capillaries and larger blood vessels react to injury by an immediate local temporary vasoconstriction (a reflex nervous mechanism) to reduce the amount of blood lost.Platelets:
adhere to the site of injury
aggregation
release substances from their cytoplasms to initiate blood coagulation ===) haemostatic plug is formed.
Secondary haemostasis (it is necessary to stop bleeding definitely)
Blood coagulation factors are necessary to stop bleeding definitely.
I: fibrinogen
II: prothrombin
III: tissue thromboplastin (tissue factor, TF)
IV : Ca++
V : proaccelerin
VI :
VII : proconvertin
VIII : antihemophilic factor (AHF)
IX : Christmas factor (plasma thromboplastin component)
X : Stuart factor
XI : plasma thromboplastin antecedent (PTA)
XII : Hageman factor (contact factor)
XIII : fibrin stabilizing factor (Laki-Lorand factor
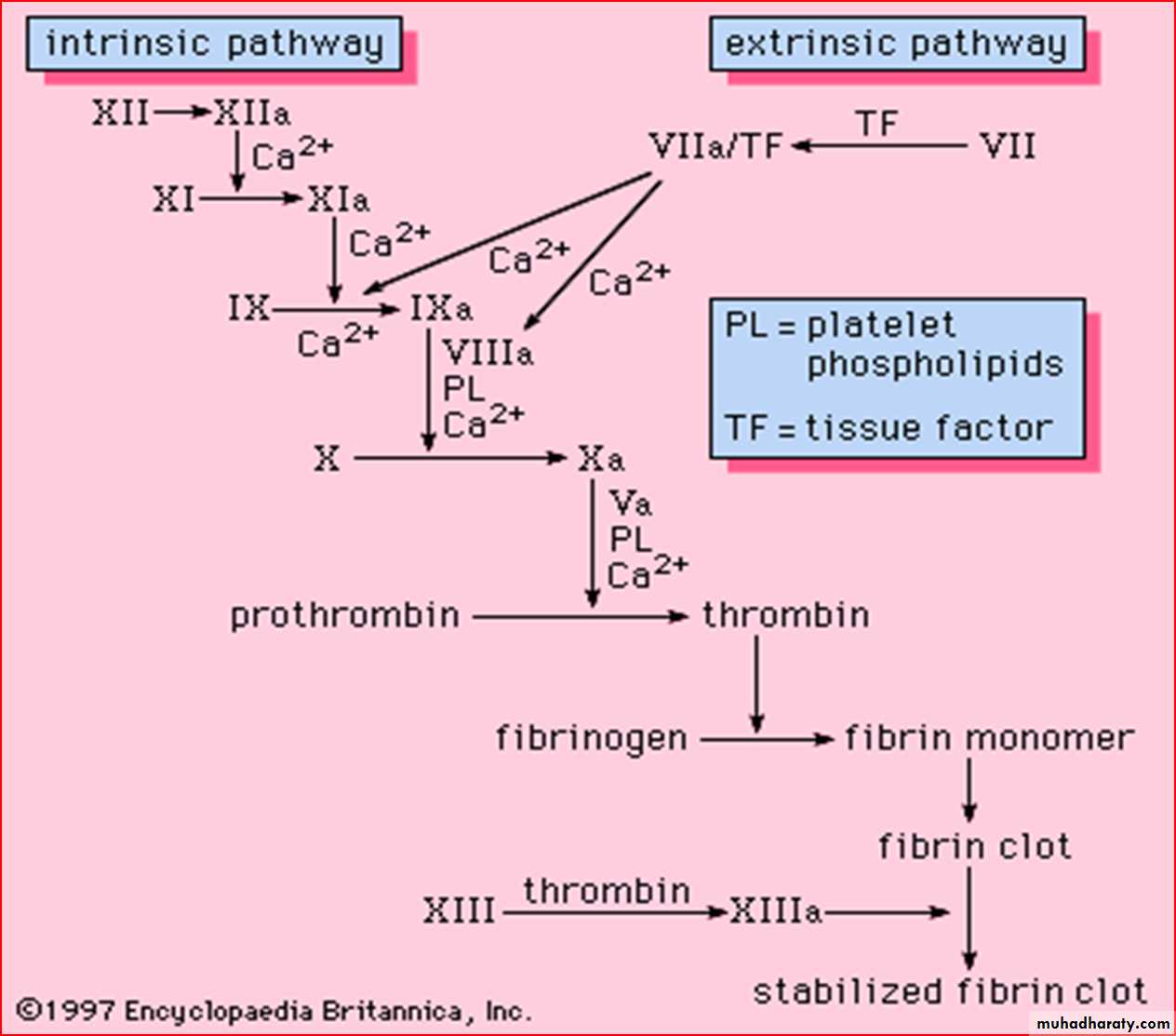
Disorders of the haemostatic mechanism are devided into three main groups:
Purpuric diseases”Disorders of the vessels
Disorders of the platelets
Disorders of the coagulation mechanism (coagulopathies)
The investigation of a patient with a suspected disorder of haemostasis:
case history (personal details, family history)inspection (type of bleeding)
physical examination
other known diseases
drugs and medications
laboratory tests
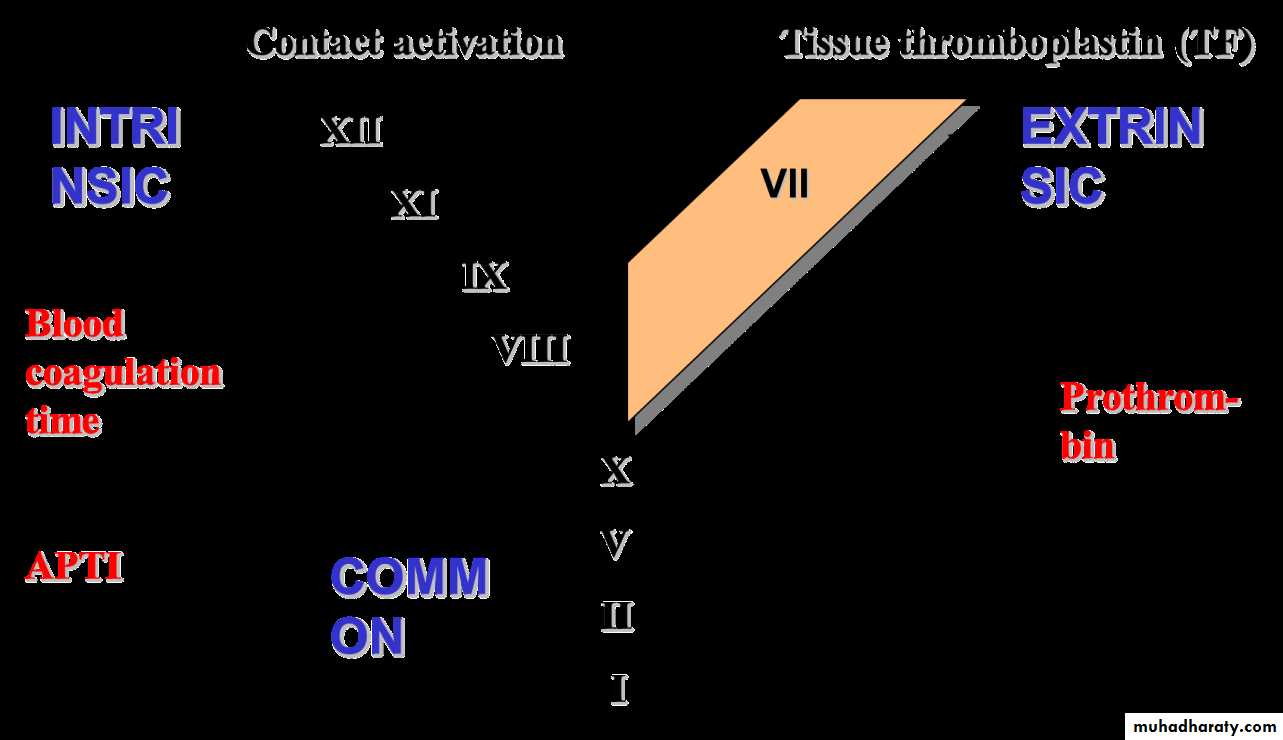
Screening tests of blood coagulation:
Disorders of vessels:Rumpel-Leede test
Disorders of platelets:
Platelet count and morphology
Bleeding time (Ivy)
Coagulopathies:
Coagulation time
Aktivated partial thromboplastin time (APTT)
Prothrombin (INR)
Thrombin time (TT(

HEMOPHILIA
Inherited deficiency of factor VIII (hemophilia A) or factor IX (hemophilia B)Sex-linked inheritance; almost all patients male
Female carriers may have mild symptoms
Most bleeding into joints, muscles; mucosal and CNS bleeding uncommon
Severity inversely proportional to factor level
< 1%: severe, bleeding after minimal injury
1-5%: moderate, bleeding after mild injury
5%: mild, bleeding after significant trauma or surgery
::: Hemophilia A :::
Incidence 1:5,000 - 1:10,000 malesabout as rare as the birth of triplets
~ 1 in 5,000 live male births are affected.
~ 15,000 to 20,000 people with hemophilia in the US
Hemarthroses, post-traumatic and post-surgical bleeding
Severity related to factor VIII level
<1% = severe
1-5% = moderate
5-15% = mild
Inhibitors develop in ~10-20% of severe patients
GENETICS OF HEMOPHILIA:
About half of cases of hemophilia A due to an inversion mutation in intron 1 or 22
Remainder genetically heterogeneous
Nonsense/stop mutations prevent factor production
Missense mutations may affect factor activity rather than production

Deficiency of factor VIII or IX affects the propagation phase of coagulation
Most likely to cause bleeding in situations where tissue factor exposure is relatively lowCOMPLICATIONS OF HEMOPHILIA:
ACUTE
Muscle hematoma (pseudotumor)
Hemarthrosis (joint bleeding)
LONG-TERM
Joint destruction
Nerve damage
Hemophilic arthropathy:
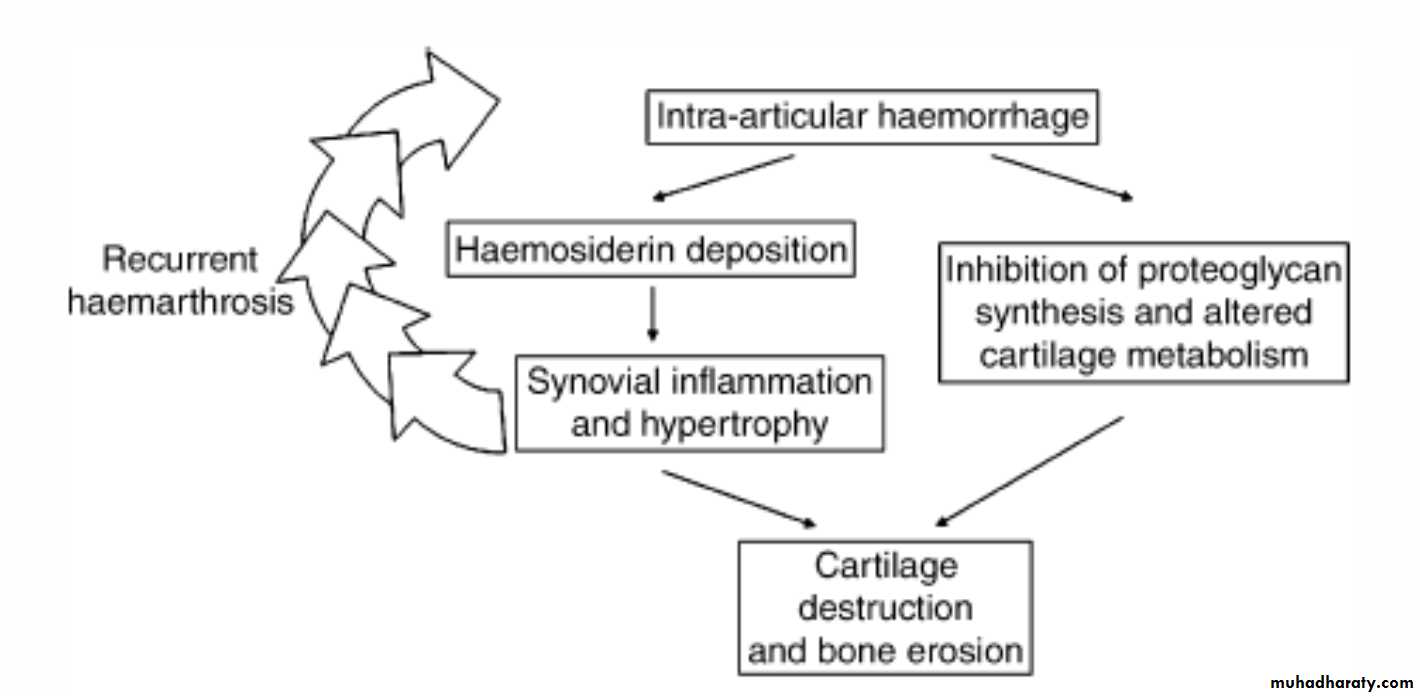
pathogenesis
Management of hemophilic arthropathy
Physical therapyWeight control
COX-2 inhibitors safe and effective
Judicious use of opioids
Surgical or radionuclide synovectomy
Joint replacement
OTHER COMPLICATIONS OF HEMOPHILIA
Pseudotumor: gradually enlarging cyst in soft tissue or bone (requires surgery)
Retroperitoneal hemorrhage
Bowel wall hematoma
Hematuria → renal colic (rule out structural lesion)
Intracranial or intraspinal bleeding (rare but deadly) – usually after trauma
TREATMENT OF BLEEDING EPISODES:
Unexplained pain in a hemophilia should be considered due to bleeding unless proven otherwiseExternal signs of bleeding may be absent
Treatment: factor replacement, pain control, rest or immobilize joint
Test for inhibitor if unexpectedly low response to factor replacement
Dosing clotting factor concentrate
1 U/kg of factor VIII should increase plasma level by about 2% (vs 1% for factor IX)
Half-life of factor VIII 8-12 hours, factor IX 18-24 hours
Volume of distribution of factor IX about twice as high as for factor VIII
Steady state dosing about the same for both factors – initial dose of factor IX should be higher
Give factor q 12 hours for 2-3 days after major surgery, continue with daily infusions for 7-10 days
Trough factor levels with q 12 h dosing after major surgery should be at least 50-75%
Most joint and muscle bleeds can be treated with “minor” (50%) doses for 1-3 days without monitoring
FACTOR VIII CONCENTRATE
Recombinant
Virus-free, most expensive replacement
Treatment of choice for younger/newly diagnosed hemophiliacs
Somewhat lower plasma recovery than with plasma-derived concentrate
Highly purified
Solvent/detergent treated, no reports of HIV or hepatitis transmission
Intermediate purity (Humate-P(
Contains both factor VIII and von Willebrand factor
Solvent/detergent treated, no reports of HIV or hepatitis transmission
Mainly used to treat von Willebrand disease
FACTOR IX CONCENTRATE
Recombinant (slightly lower plasma recovery(Highly purified (solvent/detergent treated, no reports of virus transmission(
Prothrombin complex concentrate
Mixture of IX, X, II, VII
Low risk of virus transmission
Some risk of thrombosis
DDAVP
Releases vWF/fVIII from endothelial cellsFactor VIII levels typically rise 2-4 fold after 30-60 min (IV form) or 60-90 min (intranasal)
Enhanced platelet adhesion due to ↑ vWF
Useful for mild hemophilia (VIII activity > 5%) prior to dental work, minor surgery etc
Trial dose needed to ensure adequate response
Cardiovascular complications possible in older patients
TREATMENT OF HEMOPHILIACS WITH INHIBITORS
Bethesda Assay for Inhibitors
Serial dilutions of patient plasma in normal plasma
Incubate 2 hours
Assay residual factor activity
1 Bethesda Unit neutralizes 50% of factor in an equivalent volume of normal plasma
Example: 1:100 dilution of patient plasma + normal plasma → 50% residual factor activity, so inhibitor titer is 100 BU
Recombinant factor VIIa
FEIBA (Factor Eight Inhibitor Bypassing Activity)
Mixture of partially activated vitamin K-dependent clotting proteases including VIIa
High dose factor VIII (if low titer inhibitor)
Induction of tolerance with daily factor VIII infusions
Optimal dose not established
Role for concomitant immunosuppression?
Liver disease in hemophilia:
Hepatitis C still a problem, though incidence falling with safer factor concentrates
Treatment for hepatitis C with interferon often causes thrombocytopenia
Liver transplantation done occasionally (cures hemophilia)
All newly diagnosed hemophiliacs should be vaccinated against hepatitis A and B
ACQUIRED FACTOR VIII DEFICIENCY:
Due to antibody to factor VIII (most common autoimmune factor deficiency)
Most patients elderly
Often presents with severe soft tissue or mucosal bleeding (different bleeding pattern than inherited hemophilia)
Laboratory: prolonged aPTT not corrected by mixing, very low factor VIII activity
Normal INR, thrombin time and platelet count
Treatment: rVIIa, FEIBA, immunosuppression
::: VON WILLEBRAND DISEASE :::
Common (most common?) inherited bleeding disorder
Partial lack of VWF causes mild or moderate bleeding tendency
Menorrhagia, bleeding after surgery, bruising
Typically autosomal dominant with variable penetrance
Laboratory:
Defective platelet adherence (PFA-100) or long bleeding time
Subnormal levels of von Willebrand antigen and factor VIII in plasma
Low Ristocetin cofactor activity or VWF activity
Type 1 – decreased production of vWF
Levels 20-50%, antigen ≈ activity
Type 2 – qualitative defect (missense mutation)
Several different types
Usually a disproportionate decrease in vWF activity vs antigen
Type 3 – severe deficiency
Antigen, activity and factor VIII levels < 10%
Hemophilia-like phenotype
Recessively inherited
Type 2 vWD :
2A: Selective deficiency of large multimers
Defective assembly
Increased susceptibility to proteolysis
2B: Increased affinity for platelet Gp Ib
Large multimers bind spontaneously to platelets and cleared from blood
Rarely, a mutation in Gp Ib may have the same effect (“platelet-type” vWD)
2M: Decreased vWF function but no loss of large multimers
2N: Decreased binding of factor VIII to vWF (recessive)
Desmopressin (DDAVP) in vWD:
DDAVP releases vWF from endothelial cells
Can be given IV or intranasally
0.3 mcg/kg IV, or 150 mcg per nostril
Typically causes 2-4 fold increase in blood levels of vWF (in type 1 vWD), with half-life of 8+ hours
Response to DDAVP varies considerably
Administration of a trial dose necessary to ensure a given patient responds adequately
Peak response
Duration of response
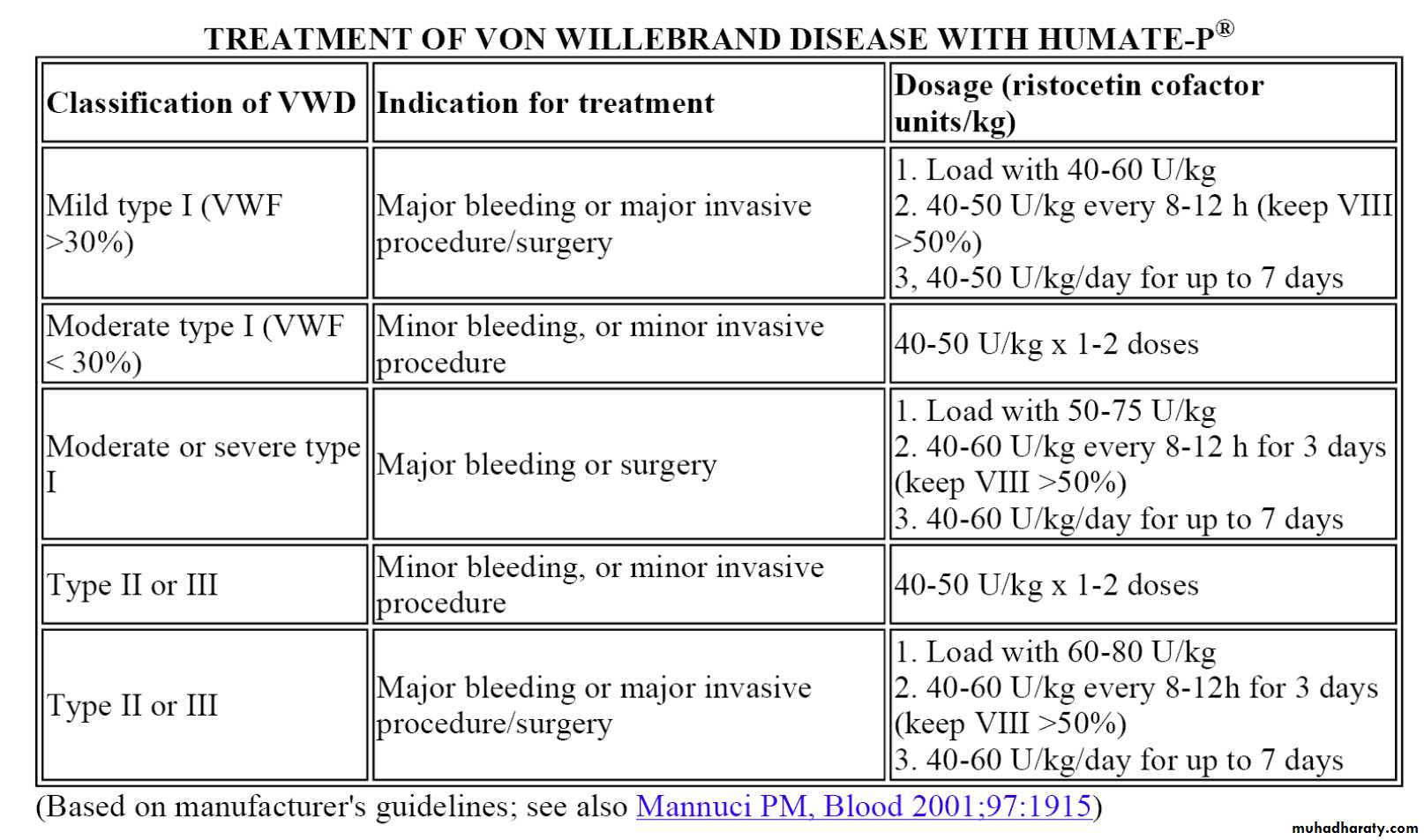
Indications for clotting factor concentrate administration in vWD:
Type 2 or 3 vWD
Active bleeding
Surgery or other invasive procedure
Type 1 vWD with inadequate response to DDAVP

Acquired von Willebrand disease:
Monoclonal gammopathy: vWF neutralized by paraprotein (?)Autoimmune disorders: Autoantibody to vWF
Myeloproliferative disorder: large multimers absorbed onto neoplastic cells (platelets?)
Cardiovascular diseases (AS, VSD, etc): High shear stress causes unfolding/proteolysis of large multimers
Hypothyroidism: Decreased release of vWF from endothelial cells
Treatment varies depending on cause/mechanism