Year three: Pathology: Heart Diseases:
Lecture 2: Ischemic heart Disease: February 2018. By Dr. Ehsan.
IHD is a group of pathophysiologically related syndromes resulting from myocardial ischemia—an imbalance between the supply (perfusion) and demand of the heart for oxygenated blood.
Ischemia brings not only an insufficiency of oxygen, but also reduces the availability of nutrients and the removal of metabolites
In more than 90% of cases, the cause of myocardial ischemia is reduced blood flow due to obstructive atherosclerotic lesions in the coronary arteries. Thus, IHD is often termed coronary artery disease (CAD) or coronary heart disease.
In most cases there is a long period (up to decades) of silent, slow progression of coronary lesions before symptoms appear. Thus, the syndromes of IHD are only the late manifestations of coronary atherosclerosis that may have started during childhood or adolescence
Classification
Acute coronary syndromes:
Myocardial infarction (MI)
Sudden cardiac death
Angina pectoris
Chronic IHD with heart failure
Pathogenesis:
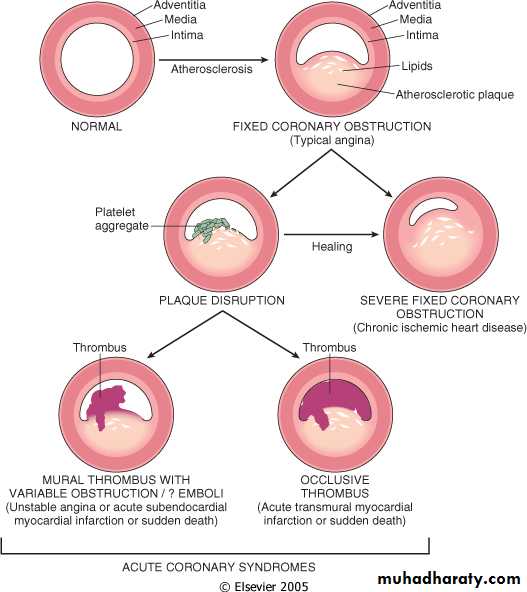
The dominant cause of IHD syndromes is insufficient coronary perfusion relative to myocardial demand, due to chronic, progressive atherosclerotic narrowing of the epicardial coronary arteries, and variable degrees of superimposed acute plaque change, thrombosis and vasospasm. This occurs in 3 steps:
Chronic Atherosclerosis.
More than 90% of patients with IHD have atherosclerosis of one or more of the epicardial coronary arteries. The clinical manifestations of coronary atherosclerosis are generally due to progressive narrowing of the lumen leading to stenosis (“fixed” obstructions) or to acute plaque disruption with thrombosis, both of which compromise blood flow.
A fixed lesion obstructing 75% or greater of the lumen is generally required to cause symptomatic ischemia precipitated by exercise (most often manifested as chest pain, known as angina).
Obstruction of 90% of the lumen can lead to inadequate coronary blood flow even at rest. The progressive myocardial ischemia induced by slowly developing occlusions may stimulate the formation of collateral vessels over time, which can protect against myocardial ischemia and infarction.
Although only a single major coronary epicardial trunk may be affected, two or all three—the left anterior descending (LAD), the left circumflex (LCX), and the right coronary artery (RCA)—are often involved by atherosclerosis. Clinically significant stenosing plaques may be located anywhere within these vessels but tend to predominate within the first several centimeters of the LAD and LCX and along the entire length of the RCA. Sometimes the major secondary epicardial branches are also involved, but atherosclerosis of the intramural (penetrating) branches is rare.
Acute Plaque Change.
The risk of an individual developing clinically important IHD depends in part on the number, distribution, structure, and degree of obstruction of atheromatous plaques.
However, the varied clinical manifestations of IHD cannot be explained by the anatomic disease burden alone. This is particularly true for the so-called acute coronary syndromes, unstable angina, acute MI, and sudden death. The acute coronary syndromes are typically initiated by an unpredictable and abrupt conversion of a stable atherosclerotic plaque to an unstable and potentially life-threatening atherothrombotic lesion through rupture, superficial erosion, ulceration, fissuring, or deep hemorrhage. In most instances, the plaque change causes the formation of a superimposed thrombus that partially or completely occludes the affected artery.
Consequences of Myocardial Ischemia.
Stable angina results from increases in myocardial oxygen demand beyond the ability of stenosed coronary arteries to increase oxygen delivery; it is usually not associated with plaque disruption.
Unstable angina is caused by plaque rupture complicated by partially occlusive thrombosis and vasoconstriction, which lead to severe but transient reductions in coronary blood flow. In some cases, microinfarcts can occur distal to disrupted plaques due to thromboemboli.
In MI, acute plaque change induces total thrombotic occlusion and the subsequent death of heart muscle. Finally, sudden cardiac death frequently involves an atherosclerotic lesion in which a disrupted plaque causes regional myocardial ischemia that induces a fatal ventricular arrhythmia. Each of these important syndromes is discussed in detail below, followed by an examination of the important myocardial consequences.
Angina Pectoris:
It is characterized by paroxysmal and usually recurrent attacks of substernal or precordial chest discomfort (variously described as constricting, squeezing, choking, or knifelike) caused by transient (15 seconds to 15 minutes) myocardial ischemia that falls short of inducing myocyte necrosis.Patterns:
Stable angina most common type and called typical angina, occurs due to chronic ischemia due to 75% vessel block, results in transient ( <15 minutes) pain aggravated by exertion, relived by rest & Nitroglycerin (vasodilators).
Prinzmetal variant angina:
is an uncommon from of episodic myocardial ischemia that is caused by coronary artery spasm. Although individuals with Prinzmetal variant angina may well have significant coronary atherosclerosis, the anginal attacks are unrelated to physical activity (not relieved by rest), heart rate, or blood pressure. Prinzmetal angina generally responds promptly to vasodilators, such as nitroglycerin and calcium channel blockers.
Unstable or crescendo angina:
Refers to a pattern of increasingly frequent pain, often of prolonged duration, that is precipitated by progressively lower levels of physical activity or that even occurs at rest. In most patients, unstable angina is caused by the disruption of an atherosclerotic plaque with superimposed partial (mural) thrombosis and possibly embolization or vasospasm (or both). Unstable angina thus serves as a warning that an acute MI may be imminent; indeed, this syndrome is sometimes referred to as preinfarction angina.
Myocardial infarction:
MI, also known as “heart attack,” is the death of cardiac muscle due to prolonged severe ischemia. It is by far the most important form and cause of death of IHD.
Pathogenesis:
Coronary Arterial Occlusion:
•
The initial event is a sudden change in an atheromatous plaque, which may consist of intraplaque hemorrhage, erosion, ulceration, or rupture or fissuring.
•
When exposed to subendothelial collagen and necrotic plaque contents, platelets adhere, become activated, release their granule contents, and aggregate to form microthrombi.
•
Vasospasm is stimulated by mediators released from platelets.
•
Tissue factor activates the coagulation pathway, adding to the bulk of the thrombus.
•
Frequently within minutes, the thrombus evolves to completely occlude the lumen of the vessel.
In approximately 10% of cases, transmural MI occurs in the absence of the typical coronary vascular pathology. In such situations, other mechanisms may be responsible for the reduced coronary blood flow, including:
•
Vasospasm with or without coronary atherosclerosis, perhaps in association with platelet aggregation or due to cocaine abuse
•
Emboli from the left atrium in association with atrial fibrillation, a left-sided mural thrombus, vegetations of infective endocarditis, intracardiac prosthetic material; or paradoxical emboli from the right side of the heart or the peripheral veins, which travel through a patent foramen ovale to the coronary arteries
•
Ischemia without detectable coronary atherosclerosis and thrombosis may be caused by disorders of small intramural coronary vessels, such as vasculitis, hematologic abnormalities such as sickle cell disease, amyloid deposition in vascular walls, and vascular dissection; lowered systemic blood pressure (shock); or inadequate myocardial “protection” during cardiac surgery
Myocardial Response:
Coronary arterial obstruction compromises the blood supply to a region of myocardium, causing ischemia, myocardial dysfunction, and potentially myocyte death. The anatomic region supplied by that artery is referred to as the area at risk. The outcome depends predominantly on the severity and duration of flow deprivation.
The early biochemical consequence of myocardial ischemia is the cessation of aerobic metabolism within seconds, leading to inadequate production of high-energy phosphates (ATP: adenosine triphosphate) and accumulation of potentially noxious metabolites (such as lactic acid). Because of the dependence of myocardial function on oxygen, severe ischemia induces loss of contractility within 60 seconds. This cessation of function can precipitate acute heart failure long before myocardial cell death. Nevertheless, the early changes are potentially reversible. As demonstrated both experimentally and in clinical studies, only severe ischemia lasting 20 to 30 minutes or longer leads to irreversible damage (necrosis) of cardiac myocytes. Ultrastructural evidence of irreversible myocyte injury (primary structural defects in the sarcolemmal membrane) develops only after prolonged, severe myocardial ischemia (such as occurs when blood flow is 10% or less of normal).
Approximate Time of Onset of Key Events in Ischemic Cardiac Myocytes
Feature
Time
Onset of ATP depletion
Seconds
Loss of contractility
<2 min
ATP reduced
to 50% of normal
10 min
to 10% of normal
40 min
Irreversible cell injury
20–40 min
Microvascular injury
>1 hr
In most cases of MI permanent damage to the heart occurs when the perfusion of the myocardium is severely reduced for an extended interval of 2-4 hours.
This delay provide rationale for rapid diagnosis, rapid intervention by reperfusion and salvage.
Areas of heart affected by vessel obstruction:
Left anterior descending: apex (distal ends of the (ventricles), anterior wall of left ventricle & anterior two thirds of the septum.
LCX: lateral wall of the left ventricle.
RCA: right ventricular free wall, posterobasal wall of the left ventricle, and posterior third of the septum.
Types:
Transmural
Full thickness due to superimposed thrombus in atherosclerosis with focal damage.
Sub-endocardial
A subendocardial infarct (inner 1/3 to half) can occur as a result of a plaque disruption followed by a coronary thrombus that becomes lysed before myocardial necrosis extends across the full thickness of the wall; in this case the infarct will be limited to the distribution of the coronary artery that suffered plaque change.
However, subendocardial infarcts can also result from prolonged, severe reduction in systemic blood pressure, as in shock superimposed on chronic, otherwise noncritical, coronary stenoses. In the subendocardial infarcts that occur as a result of global hypotension, myocardial damage is usually circumferential, rather than being limited to the distribution of a single major coronary artery.
ECG changes resulting from myocardial ischemia/necrosis in various distributions, transmural infarcts are often referred to as “ST elevation infarcts”.
In subendocardial infarcts are known as “non-ST elevation infarcts.”
Morphology: In light microscopy
First 12 hrs. after MI – no change
Up to 3 days = Coagulative necrosis, neutrophils
1-2 weeks = Granulation tissue
≥ 3 weeks = fine scar
≥ 2 months = dense scar
EM – membrane disruption and Mitochondrial densities
Special stain = TTC ( Triphenyl Tetrazolium chloride), (2-3H)
Detects and stains Mahogany brown with Lactate dehydrogenase
Unstained area = infarction
Mahogany brown = viable
White, glistening= scar
Most common and nonspecific change in ischemia = sub-endocardial myocyte vacuolization
Evolution of Morphologic Changes in Myocardial Infarction, in last page.
Reperfusion:
Mechanisms by Intrinsic and Extrinsic mechanisms:
Thrombolytic drugs = < 1hr. After onset of MI
CABG = > 1hr. After onset of MI
The target is clot lysis and restoration of blood flow
Post- reperfusion changes =
Contraction bands = hyper contracting myocytes,
Stunned myocardium = transient, protective dysfunction
Reperfusion damage = mostly apoptosis by free radicals.
Most widely used is Streptokinase and plasminogen activator.
Balloon angioplasty with or without stenting can be done urgently.
Coronary artery bypass graft also done f delayed more than 1 hour.
Clinical features:
Silent MI = DM, Elderly, Cardiac transplantation recipients,
Typical features = Rapid, weak pulse and sweating profusely (diaphoretic), Dyspnea, chest pain
Lab: A- Diagnostic
Best markers = Troponins ( T & I), both sensitive and cardio – specific
Next best – CK-MB
Predictive: CRP- >3mg/l – highest risk
MI –Complications
Poor prognosis in = elderly, females, DM, old case of MI, Anterior wall infarct – worst, posterior –worse, Inferior wall – best
1. Arrhythmia = Ventr. Fibrillation – lead to sudden death in MI patients, before they reach hospital.
2. Pump failure – LVF, cardiogenic shock, if >LV wall infarcts, lead to death ( 70% of hospitalized MI patients)
3. Ventricular rupture = Free or lateral LV wall – septum, or papillary muscle cause severe Mitral regurg. (3-7 days)
4. True aneurysm = rupture is very rare
5. Pericarditis, 2-3 days after transmural MI.
6. Mural thrombus.
7. Papillary muscle dysfunction.
8. Progressive late heart failure.
9. Infarct expansion.
10. Recurrence
Sudden cardiac death:
Unexpected death from cardiac causes either without clinical symptoms or within 1-24 hours of symptoms.
Cause – Atherosclerosis ( 90%), others (10%)
Mechanism- Most likely due to arrhythmias ( VF)
Patients – young athletes, with Pul. HTN, IHD
Morphology:
Prominent finding – increased heart mass
Vacuolations in Subendocardial myocardium
Non atherogenic causes of SCD:
Congenital coronary artery D.
Aortic valve stenosis.
Mitral valve prolapse.
Myocarditis or sarcoidosis.
Dilated or hypertrophic cardiomyopathy.
Pulmonary hypertension.
Hereditary or acquired causes of cardiac conduction defects. As Romano- Ward syndrome – Long Q-T syndrome with defect in cardiac ion channels as K+, Na+ channel defects.
Chronic ischemic heart disease:
also called ischemic cardiomyopathy
Patients = post heart transplant receipts, previous MI or CABG pts
Cause =compromised ventricular function
Morphology = enlarged heart with left ventricular dilation and hypertrophy. Microscopically there is vacuoles in Myocyte with myocyte hypertrophy
Evolution of Morphologic Changes in Myocardial Infarction, related to morphology:
Time
Gross Features
Light Microscope
Electron Microscope
REVERSIBLE INJURY
0–½ hr
None
None
Ralaxation of myofibrils; glycogen loss; mitochondrial swelling
IRREVERSIBLE INJURY
½–4 hr
None
Usually none; variable waviness of fibers at border
Sarcolemmal disruption; mitochondrial amorphous densities
4–12 hr
Dark mottling (occasional)
Early coagulation necrosis; edema; hemorrhage
12–24 hr
Dark mottlingOngoing coagulation necrosis; pyknosis of nuclei; myocyte hypereosinophilia; marginal contraction band necrosis; early neutrophilic infiltrate
1–3 days
Mottling with yellow-tan infarct center
Coagulation necrosis, with loss of nuclei and striations; brisk interstitial infiltrate of neutrophils
3–7 days
Hyperemic border; central yellow-tan softeningBeginning disintegration of dead myofibers, with dying neutrophils; early phagocytosis of dead cells by macrophages at infarct border
7–10 days
Maximally yellow-tan and soft, with depressed red-tan marginsWell-developed phagocytosis of dead cells; early formation of fibrovascular granulation tissue at margins
10–14 days
Red-gray depressed infarct bordersWell-established granulation tissue with new blood vessels and collagen deposition
2–8 wk
Gray-white scar, progressive from border toward core of infarctIncreased collagen deposition, with decreased cellularity
>2 mo
Scarring completeDense collagenous scar