
1
CHOREA
Definition
excessive spontaneous movements, irregularly timed, randomly
distributed and abrupt. “Queen square”
the word chorea is Greek in origin which means dance.
The word choreoathetosis is sometimes used when typical choreic
movements coexist with athetosis.
When chorea affect one side of body it indicate a lesion affecting the
contralateral basal nuclei (caudate) or their connections.
CLINICAL RECOGNITION OF CHOREA
Patient with chorea is usually unaware of this abnormal movement
especially HD and if they are aware of it they try to mask it by doing
voluntary movement (parakinesia).
Chorea is usually present at rest, may increase with maneuvers that
distract patient attention such as counting and usually disappears in sleep.
Chorea may interfere with respiration and vocalization, and affected
patients may have slurred or interrupted speech or involuntary
vocalizations.
Patients with chorea often demonstrate motor impersistence, the
inability to maintain an ongoing motor activity resulting in dropping
objects from hand, dipping during walking and difficulty holding the
tongue out or maintaining grip (milkmaid grip).
Ballism is a severe form of chorea, affecting proximal musculature such as the
hip or shoulder girdle. The resulting movements are high amplitude, kicking in
nature, indicate a lesion in the contralateral subthalamic nucleus.

2
CAUSES of Chorea
The differential diagnosis of chorea is broad but the most common causes
of chorea in order of
descending frequency are:
1. vascular causes were most common (40%).
2. drugs (14%).
3. HD (10%).
4. acquired immune deficiency syndrome (10%).
5. Unknown 6%.
6. Other causes, each accounting for fewer than 5% of cases, were
vasculitis, hypoxia, hyperglycemia, hyponatremia, Sydenham’s chorea,
Borrelia, and acanthocytosis
Neurometabolic Disorders
1. Lesch- Nyhan syndrome.
2. Lysosomal storage disorders.
Benign Choreas
• Hereditary Benign
familial chorea
• Sporadic Senile
chorea
Neurodegenerative Disorders
• Hereditary
AD
1. Huntington’s disease.
2. Fahr’s disease.
AR
1. Neuroacanthocytosis.
2. Wilson’s disease.
3. Neuronal degeneration with brain iron
accumulation type I (formerly atypical
Hallervorden- Spatz disease)
X-linked recessive
1. McLeod syndrome.
• Sporadic or unknown inheritance
1. Olivopontocerebellar atrophy.

3
3. Leigh’s disease.
4. Porphyria.
Acquired Structural Lesions
1. Vascular (infarction, hemorrhage).
2. Inflammatory (sarcoidosis).
3. Infectious ( AIDS, Creutzfeldt-Jakob disease).
4. Immune-mediated (Sydenham’s chorea, Postinfectious chorea,
paraneoplastic chorea, SLE, antiphospholipid syndrome, Behcet’s
disease, Polyarteritis nodosa, MS)
Systemic Metabolic Disorders
1. Hyperthyroidism.
2. Hypoparathyroidism.
3. Pregnancy.
4. Hypoxia.
5. Hyperglycemia.
6. Hypoglycemia.
7. Electrolyte disturbance (Hypernatremia, Hyponatremia
Hypomagnesemia, Hypocalcemia).
8. Nutritional disorders (Beriberi, Pellagra, Vitamin B6 deficiency in
infants.
Toxin-induced
1. Carbon monoxide
2. Mercury
3. Manganese
4. Thallium
5. Toluene

4
Drug-induced
1. Anticonvulsants.
2. Antiparkinsonian drugs.
3. Neurostimulant (Cocaine, Amphetamines, Alcohol intoxication and
withdrawal).
4. Neuroleptics (withdrawal, Tardive dyskinesia).
Pathophysiology of chorea
Chorea is thought to result from decreased inhibition of inhibitory
pallidothalamic pathways (either result from decrease activity of indirect
pathway or increase activity of direct pathway) and increase activity of
thalamocortical pathway.
Because dopamine increases activity of neurons” striatopallidal neurons”
in the direct pathway and decrease activity of neurons in the indirect
pathway, higher dopamine activity increases chorea and lower dopamine
activity decreases chorea. Thus, the most effective antichoreic agents
block dopamine receptors or deplete monoaminergic terminals of
dopamine.
Huntington’s disease
• Huntington’s disease (HD) is an autosomal dominant slowly progressive
neurodegenerative disorder and the most important inherited cause of
chorea.
• Onset is usually in adult life with a mean age of about 25-40 years.
although juvenile and elderly onset are well described. It is slowly
progressive causing death within 15-20 years from onset.
• Neuropathologically, in the early stages of disease, the brain can look
macroscopically normal, but later there is marked cortical atrophy with
ventricular dilatation, severe atrophy of the caudate more than the
putamen, with atrophy of the internal segment of the Globus pallidus
and substantia nigra.

5
Genetics HD is caused by an expanded and unstable CAG trinucleotide
repeat in the huntingtin gene on chromosome 4.
More than 40 patient develop dis
more than 60 juvenile HD
more than 80 HD 1 deca
Clinical features
Many patients report psychiatric problems or mild cognitive symptoms
before developing any motor problems. However, the definitive diagnosis
of HD is usually made when motor abnormalities are noted on
examination.
Psychiatric features
Psychiatric symptoms are common, particularly depression and anxiety,
irritability and sometimes the patient become aggressive. As the disease
progresses there will be obsessive compulsive behavior that disturb the
life of the patient and the relatives with psychoses and higher suicide rate
“7.5% live end with suicide”
Cognitive features
The key cognitive abnormalities seen are:
1. impaired executive function with poor planning and judgment.
2. visual and verbal memory problems.
3. poor concentration and attention.
Motor features Clumsiness is often the earliest subjective motor symptom
of HD. Over time, clumsiness evolve into frank chorea which appears early
as abnormal movement of hands, fingers and toes during stress or walking.
As the illness progresses over years, chorea may lessen and patients
typically become more bradykinetic with dystonia, dysarthria and
dysphagia.

6
Oculomotor abnormalities are a cardinal feature of the disease, early in
the coarse of the disease there is hesitation in generation of saccades/
saccadic slowing and occur in about 75% of patient during the coarse of
disease and in late-stage disease, voluntary eye movements are limited
(vertical more than horizontal). “Gaze impersistence”
Gait disturbance is common with progressive widening of base and
increasing postural instability with falls and injury are common.
Dysarthria and dysphagia are common and should be asked about.
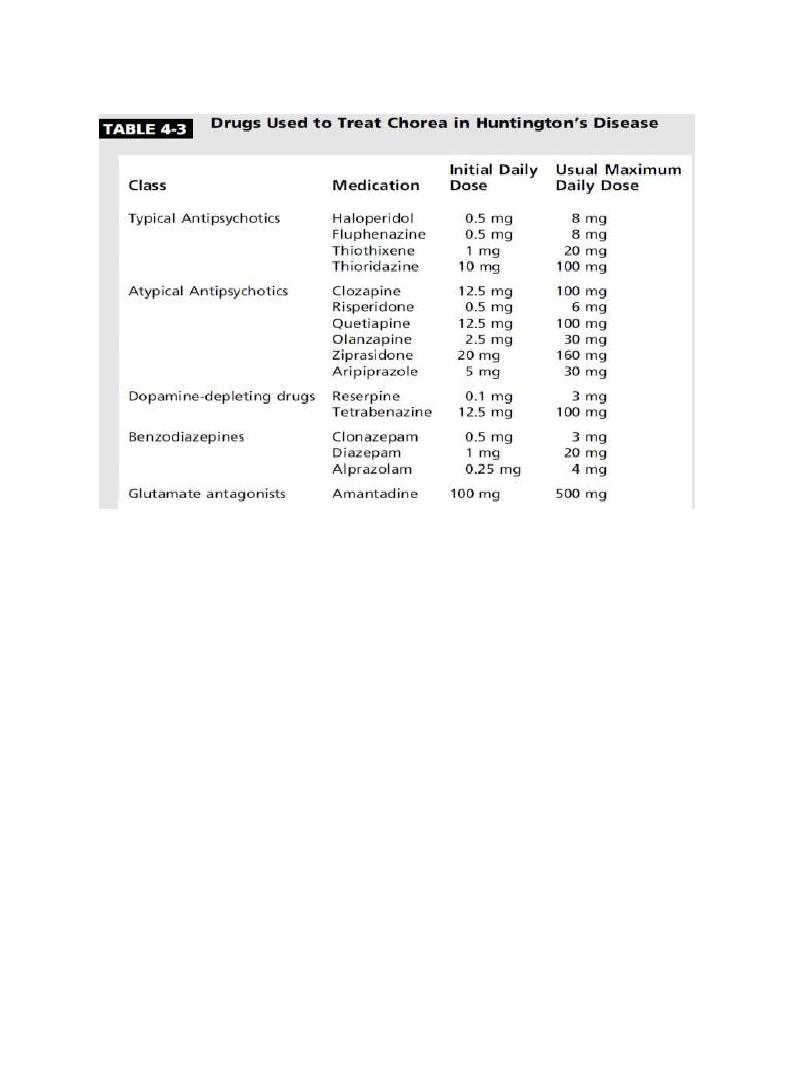
Treatment
• no therapy has yet been shown to slow the progression of HD, although
coenzyme Q10 suggested to slow disease progression in some trials.
• Otherwise the treatment is symptomatic to ameliorate psychiatric,
cognitive and motor features.
• Psychiatric features can be treated with selective serotonin reuptake
inhibitors, serotonin norepinephrine reuptake inhibitors, and other
antidepressants.
• Antidopaminergic therapies are the mainstay for reducing the severity
of choreic movements.
• For many years, typical neuroleptics such as haloperidol at doses
ranging up to 100 mg/d. Common or serious adverse effects, including
depression, suicide, restlessness, akathisia.
• Benzodiazepines (Diazepam, clonazepam, alprazolam) have a mild
antichorea effect and may be helpful in the management of anxious
patients.
7
Juvenile Huntington’s disease
Juvenile HD cases are defined as onset before age 20 years.
It account for 5% of patients with HD.
They have more severe disease and shorter life expectancy.
The most common form is The akinetic-rigid form of the disease (Westphal
variant): patients typically have little chorea and are predominantly
parkinsonian and dystonic features.
Juvenile HD patients also have a higher incidence of seizures and
myoclonus than adult onset patients.