Congenital Anomalies of the Central Nervous System
DR.HAYDER QATRANNEUROSURGERY
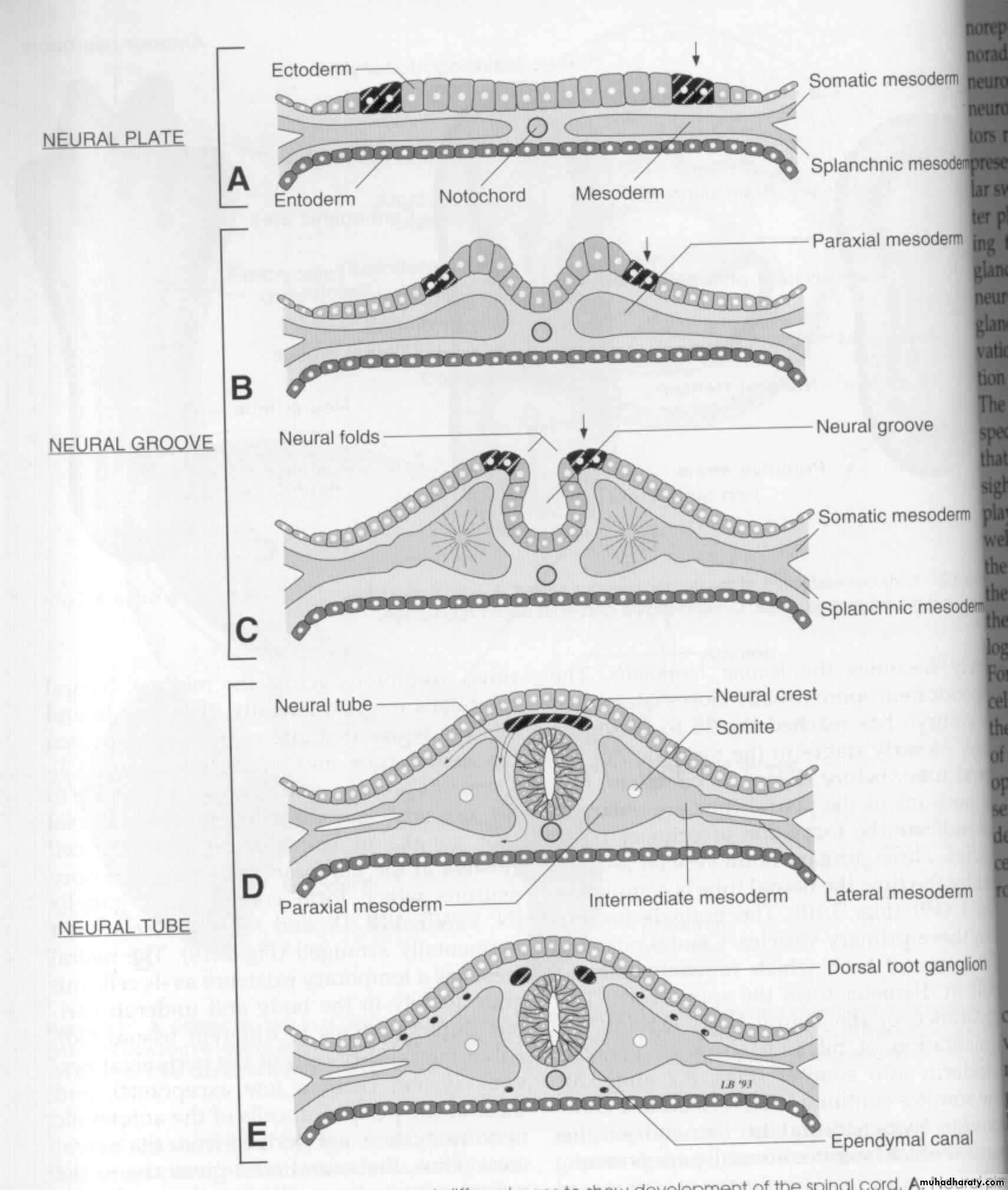
Transverse
sections ofembryos at
Different ages
to show the
Dev’t of
the spinal cord
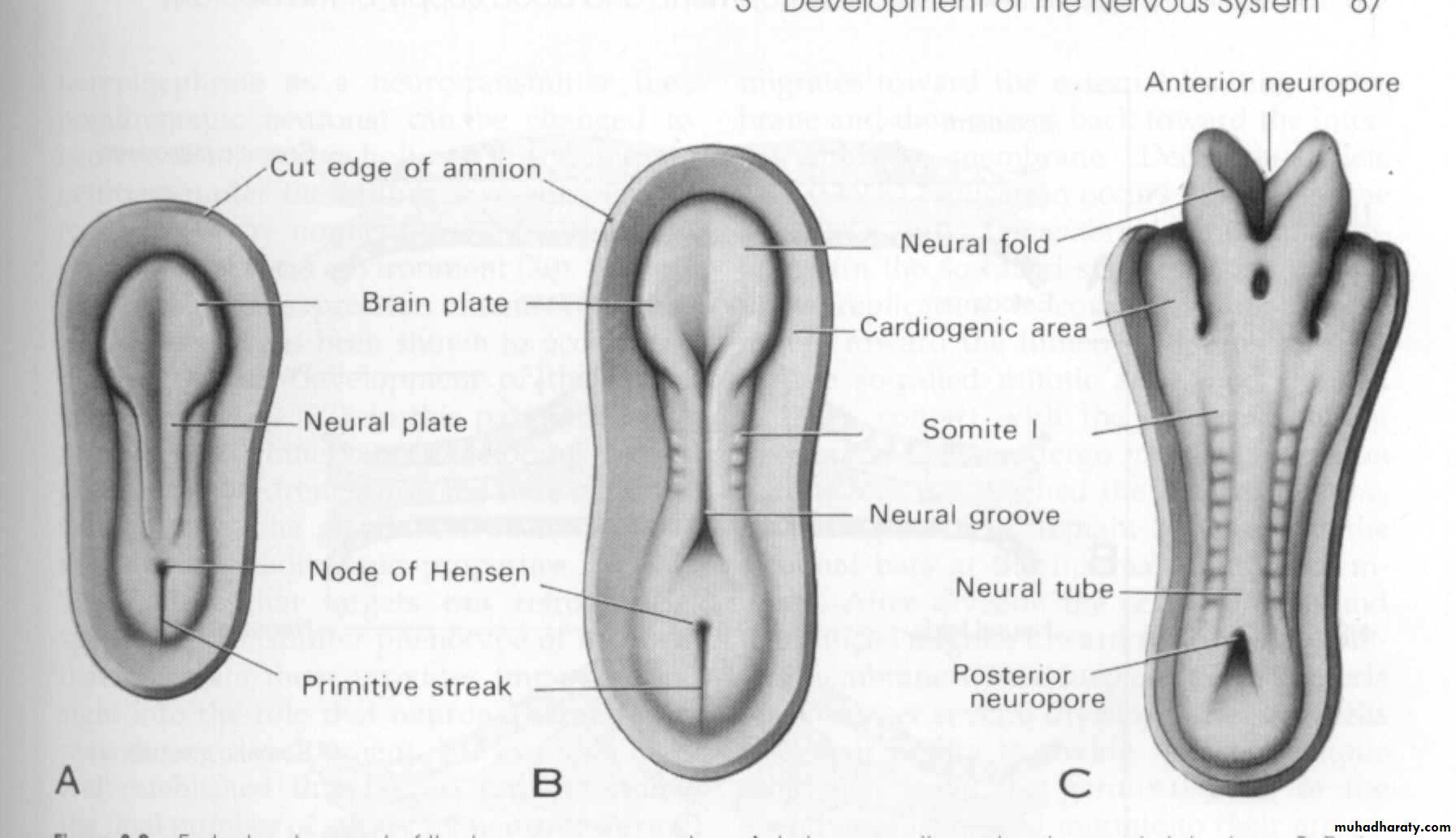
Early development of the human nervous system
late presomite and
early neural plate
stage
early somite and neural
groove stage
eight-somite and
early neural tube
stage
Neural Tube Defects (Posterior Midline Defects/Dysraphism)
Results from failure of the neural tube to close spontaneously between the 3rd-4th week of in utero developmentPossible etiologic factors:
Radiation
Drugs
Malnutrition
Chemicals
Genetic determinants (mutations in folate-responsive and folate-dependent pathways)
Neural Tube Defects
Spina bifida occultaMeningocoele/ Myelomeningocoele
Encephalocoele
Anencephaly
Dermal sinus
Tethered cord
Syringomyelia
Diastematomyelia
Neural Tube Defects
Diagnostic tool:
Failure of closure of the neural tube allows excretion of fetal substances (AFP, acetylcholinesterase) into the amniotic fluid
Prenatal screening of maternal serum for AFP during 16-18 week of gestation
AFP obtained between 15-20 weeks’ gestation is most specific
Rostral end of the NT closes on the 23rd day and the caudal neuropore closes by the 27th day of development
Neural Tube Defects and FA
Maternal periconceptional use of folic acid supplementation reduces the incidence of NT defects by at least 50%US: recommends all women of childbearing age take 0.4 mg of folic acid daily, and women with previous pregnancy of NT defect should be treated with 4 mg of folic acid beginning one month before pregnancy is planned, until at least the 12th week of gestation when neurulation is complete
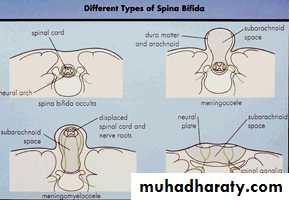
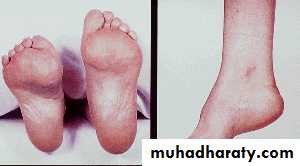
Spina Bifida Occulta
Midline defect of the vertebral bodies without protrusion of the SC or meninges
May be asymptomatic without neuro signs
In some, patches of hair, lipoma, discoloration of skin or dermal sinus may be present
Spina Bifida Occulta
Spine x-ray: defect in closure of the posterior vertebral arches and laminae, usually in L5 and S1May be associated with syringomyelia, diastematomyelia, and tethered cord
Recurrent meningitis of occult origin should prompt careful exam for dermal sinus tract
1
2
3
4
5
6
7
8
9
Gestational Age in Months
Postnatal
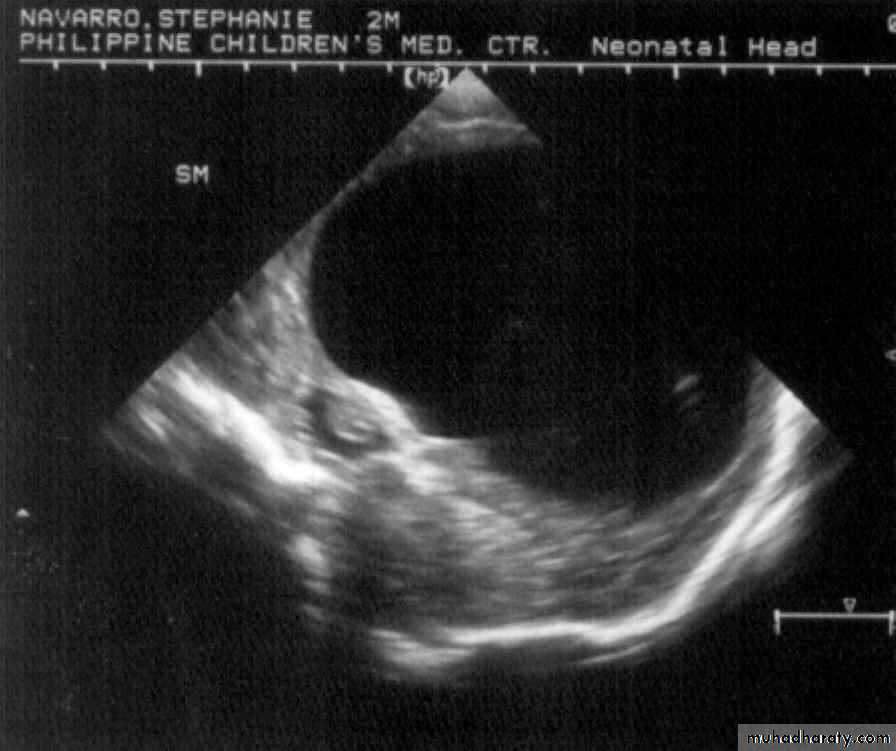
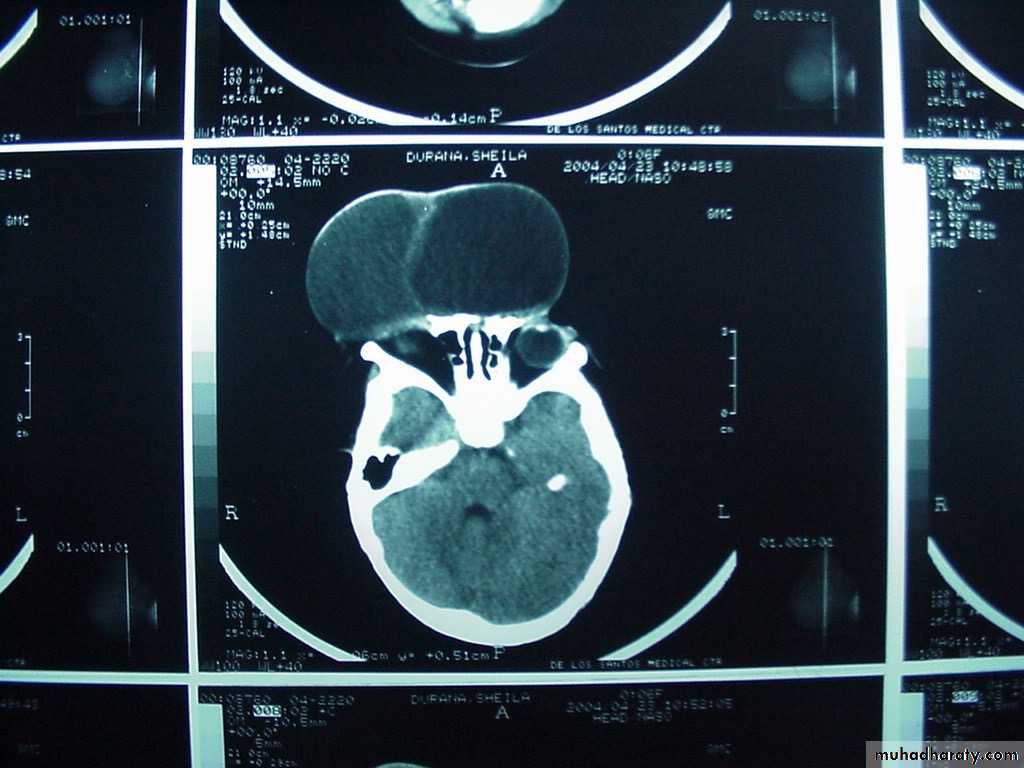
Neurulation PeriodS. N., 2 mos old, female
Marked obstructive Hydrocephalus
secondary to ARNOLD CHIARI II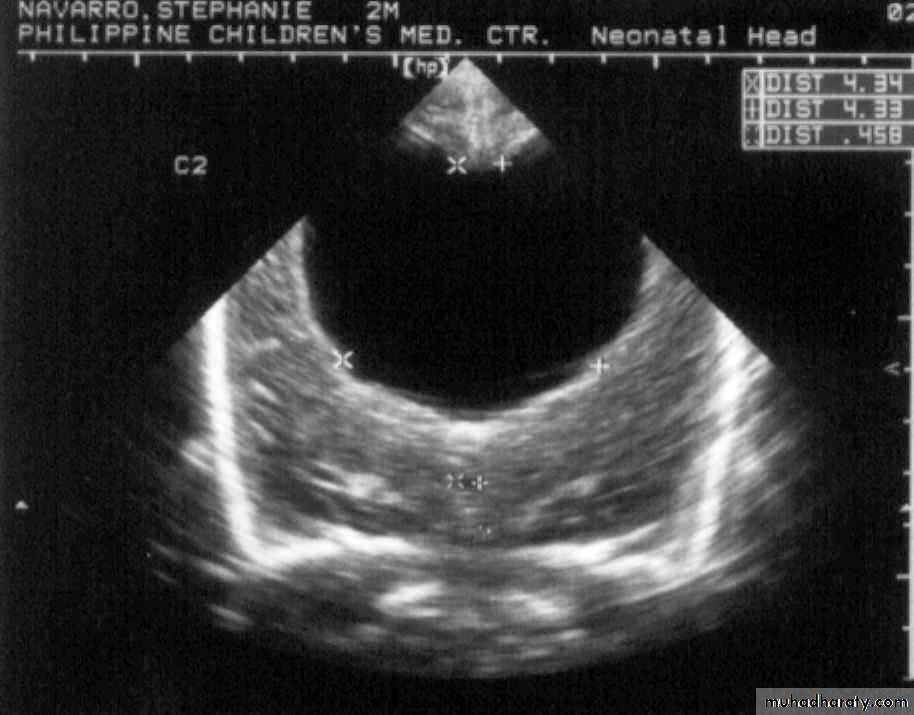
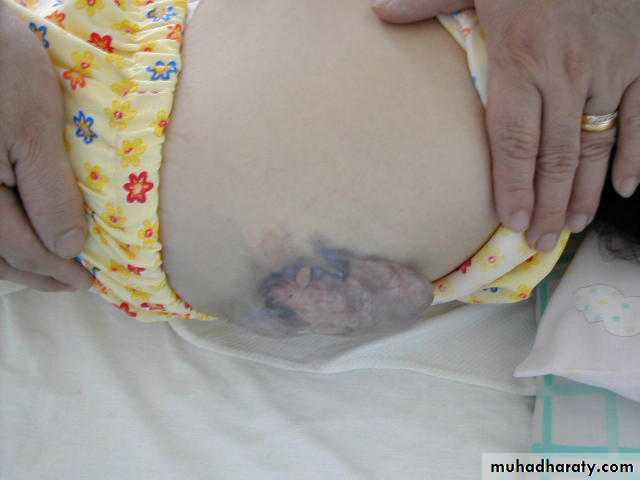
Meningocoele
Formed when the meninges herniate through a defect in the posterior vertebral archesSC may be normal, or may present with tethering, syringomyelia, or diastematomyelia
A fluctuant mass that may transilluminate along the vertebral column
Meningocoele
Dxtic: plain x-ray, u/s, MRI for the spine, CT of the head to R/O(role out) HCP (hydrocephalous)Txtic: Asymptomatic children with Normal neuro findings and full-thickness skin may have surgery delayed.
Patients with leaking CSF or a thin skin covering should undergo immediate repair to prevent meningitis.
Meningocoele
Anterior meningocoele may project into the pelvis through a defect in the sacrum causing symptoms of constipation and bladder dysfunction
Female patients may have associated anomalies of the genital tract (rectovaginal fistula, vaginal septa)
Dxtic: plain x-ray, CT, MRI
Myelomeningocoele
• Most severe form of dysraphism involving the vertebral column with an incidence of ∼1/4000 LB• Risk of recurrence after one affected child increases to 3-4% and increases to ~10% with 2 previous abnormal pregnancies
• Certain drugs that antagonize folic acid ( AEDs: tegretol, phynitoin,primidone) increase the risk of myelomeningocoele
• Valproic acid(depakine) cause NT defects in ~1-2% of pregnancies
Myelomeningocoele
• May be located anywhere along the neuraxis but the LS (lumbo-sacral) region accounts for 75% of the cases• Extent and degree of the neuro deficit depend on the location (more high up more damage)
• flaccid paralysis, absent DTRs(deep tendon reflexes), sensory deficit below the affected level, postural abnormality of the LE (clubfeet, subluxation of the hips), constant urinary dribbling and a relaxed anal sphincter
Myelomeningocoele
• HCP (hydrocephalous) in association with a type II Chiari defect develops in at least 80% with myelomeningocoele• Infants with HCP and Chiari II develop symptoms of hindbrain dysfunction: difficulty feeding, choking, stridor, apnea, VC paralysis, pooling of secretions, spasticity of UEs
• Chiari crisis is due to downward herniation of the medulla and cerebellar tonsils
Myelomeningocoele
• Requires a multidisciplinary approach: surgeon, therapist, pediatrician
• Surgery: repair and shunting; orthopedic procedure, urologic evaluation
• GUT: regular catheterization to prevent UTI and reflux leading to PN (pyeloniphritis) and hydronephrosis, urine cult, serum electrolytes, creatinine, renal scan, IV pyelogram, U/s
• Rehab: functional ambulation (sacral or LS lesion)
Myelomeningocoele
Prognosis:MR- 10-15%
Most deaths occur before age 4 years
70% have normal intelligence, but learning problems and seizure disorders are common
History of meningitis or ventriculitis adversely affect the ultimate IQ
1
2
3
4
5
6
7
8
9
Gestational Age in Months
Postnatal
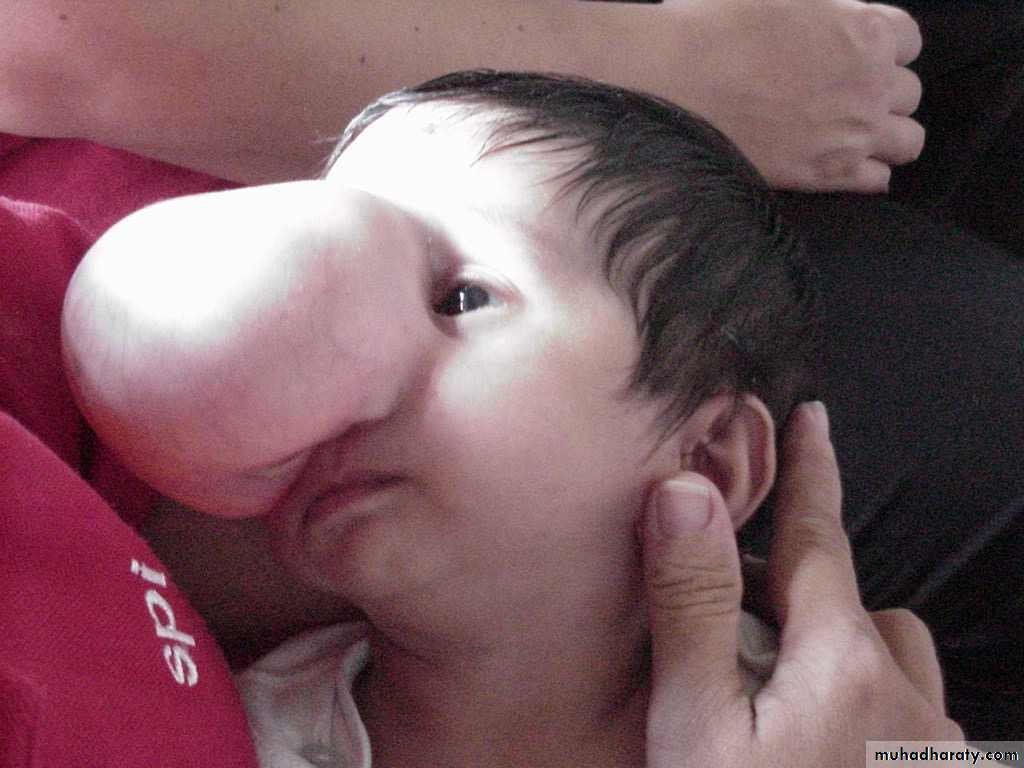
Neurulation Period11 mos old, male
Encephalocoele
• Cranium meningocoele: CSF-filled meningeal sac only• Cranial encephalocoele: contains the sac plus cerebral cortex, cerebellum or portions of the brainstem usually with abnormalities
• Usually occurs in the occipital region or below the inion, although in some countries, frontal or nasofrontal encephalocoeles are more prominent
Encephalocoele
Dxtic:Plain x-ray of the skull and cervical spine
Cranial u/s
In utero: AFP, biparietal diameter
Prognosis:
Encephalocoele- at risk for visual problems, microcephaly, Mental retardation, seizures
Meckel-Gruber syndrome: occipital encephalocoele, cleft lip or palate, microcephaly, microphthalmia, abnormal genitalia, polycystic kidneys, and polydactyly
Anencephaly
• Large defect of the calvarium, meninges, and scalp associated with a rudimentary brain which results from failure of closure of the rostral neuropore
• Primitive brain consists of portions of connective tissue, vessels and neuroglia
• The cerebral and cerebellar hemispheres are usually absent, and only a residue of the brainstem can be identified; the pituitary gland is hypoplastic, and the SC pyramidal tracts are absent
Anencephaly
• Associated anomalies: folding of the ears, cleft palate, congenital heart defects (10-20%)• Die within several days of birth
• Frequency: 1/1000 LB
• Recurrence risk: 4% and increases to 10% with 2 previously affected pregnancies
• Monitoring of succeeding pregnancies: amniocentesis, AFP levels, U/s between 14-16th week of gestation
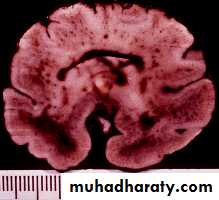
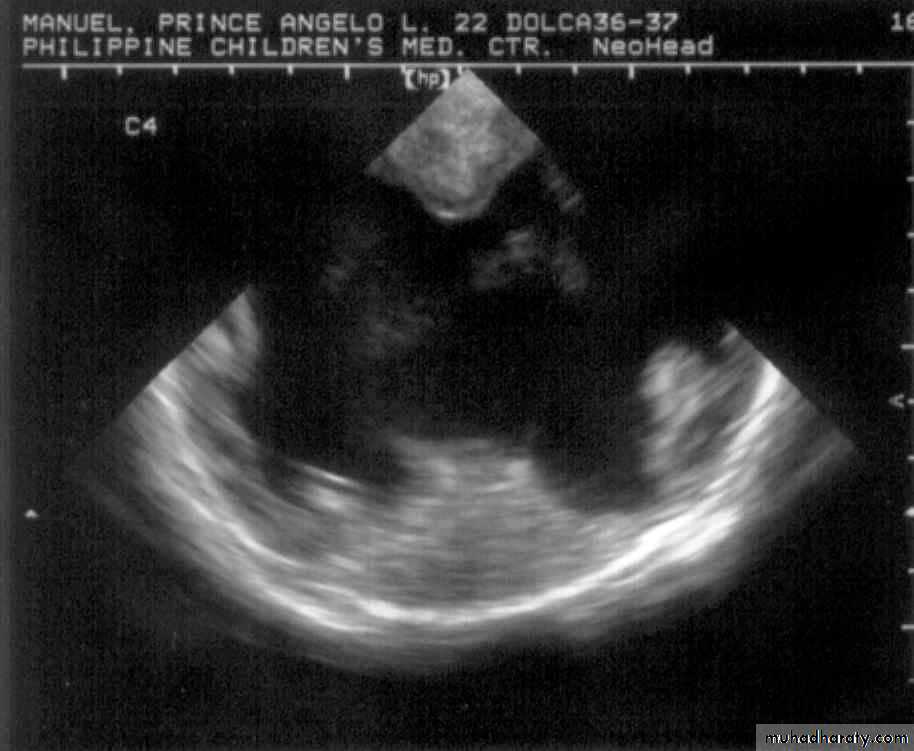
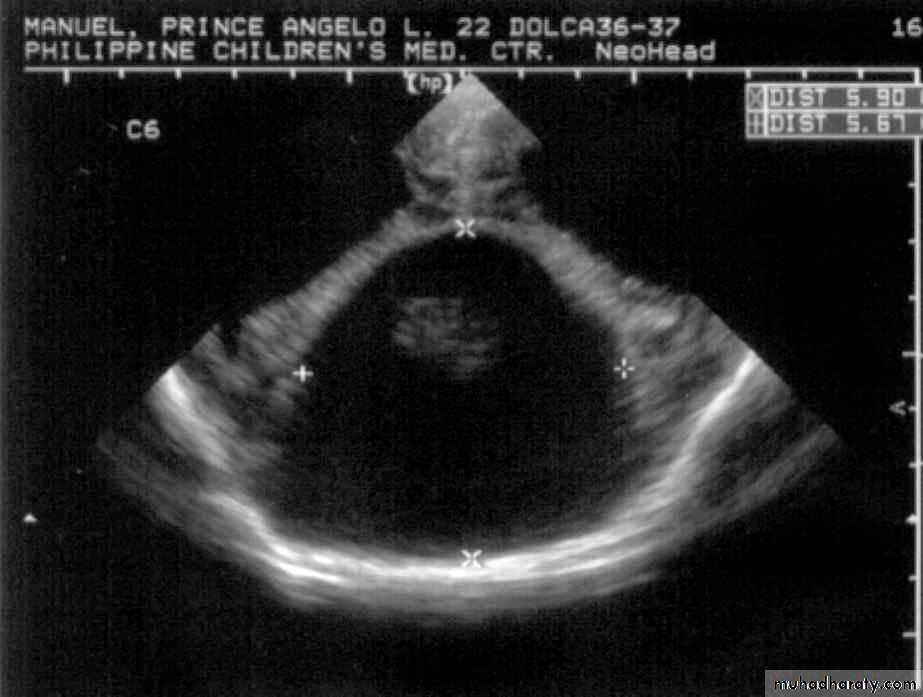
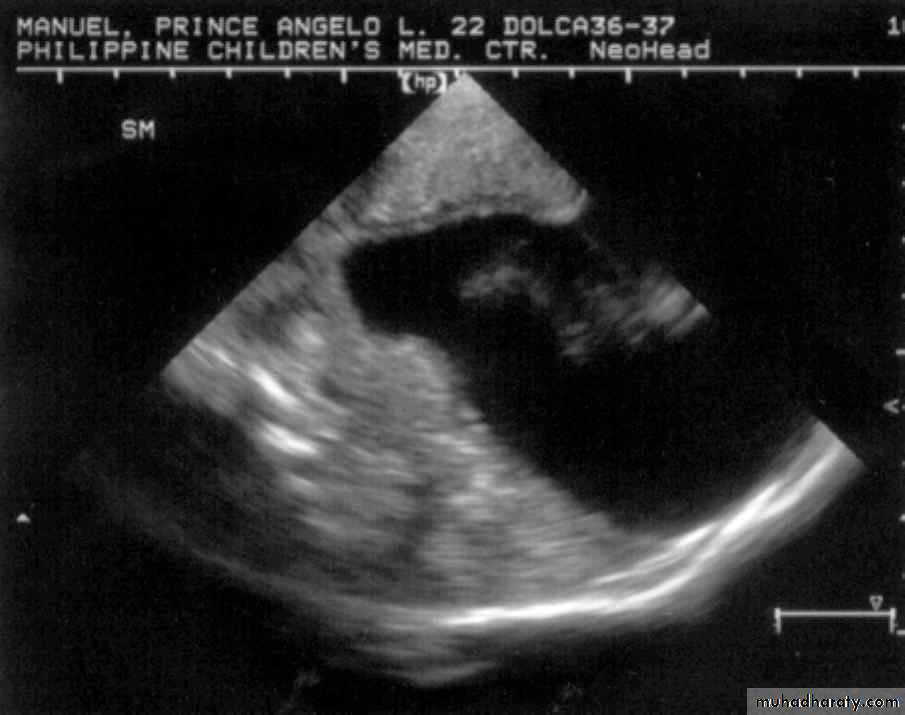
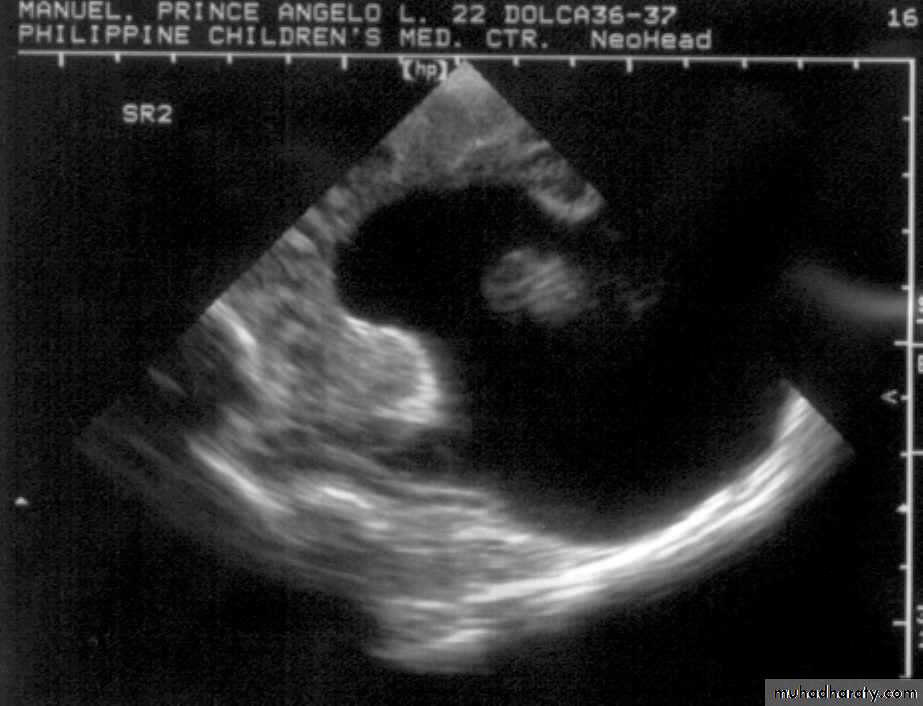
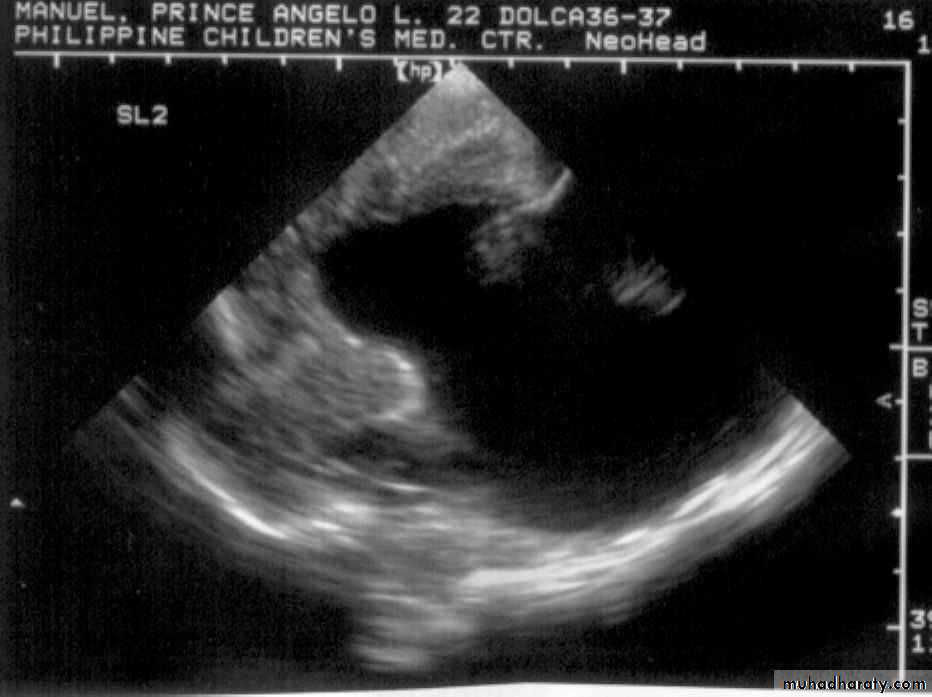
P.M, 22 days old CA 36-37 wks, with cleft lip and palate
Holoprosencephaly, semilobarMacrogyria
Absent Septum Pellucidum
Dysgenesis of the Corpus Callosum
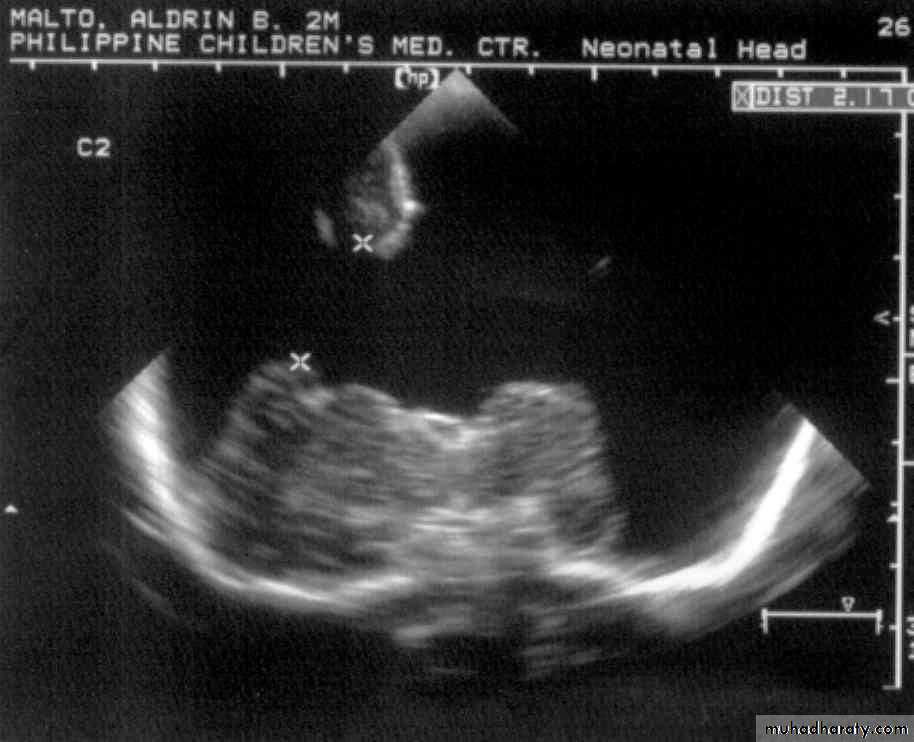
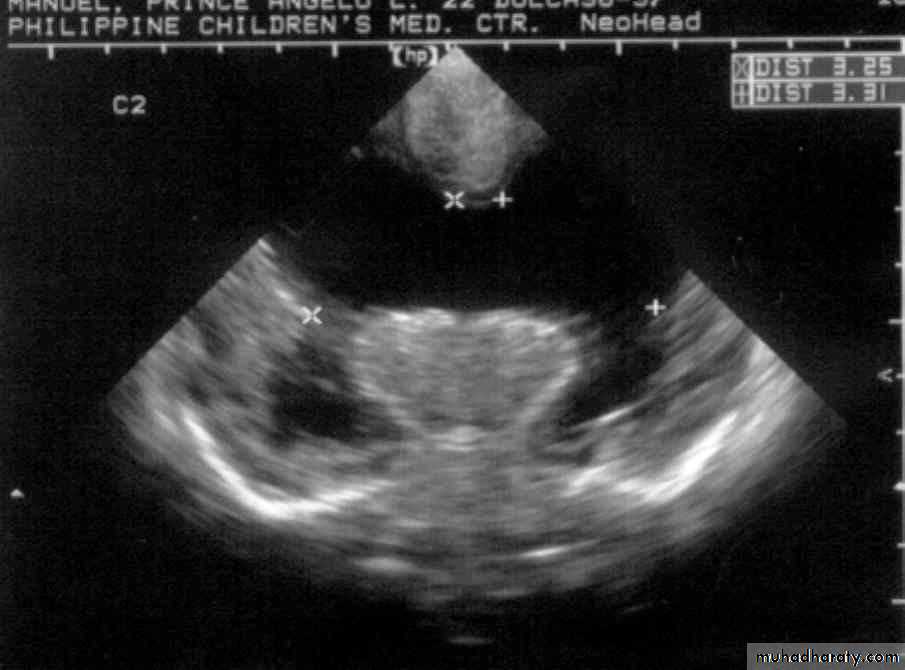
A.M., 2 months old
was noted to be microcephalic w/ szPorencephalic Cyst
SchizencephalyAbsent Septum Pellucidum
Dysgenetic Corpus Callosum
Aqueductal Stenosis
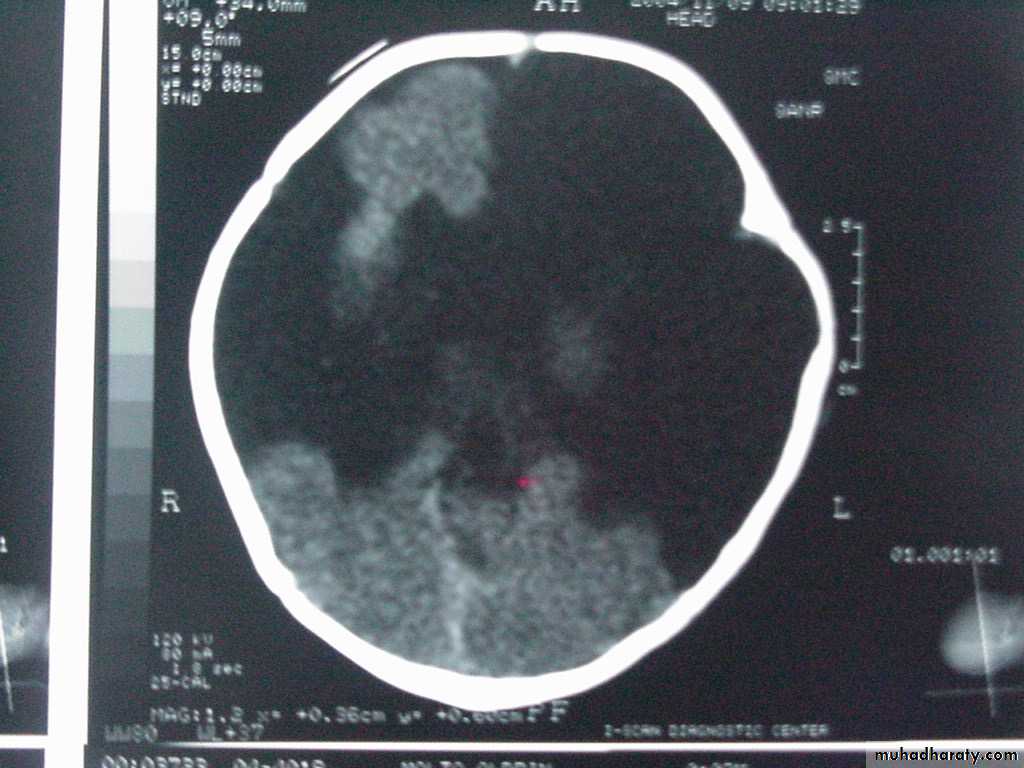
1
2
3
4
5
6
7
8
9
Gestational Age in Months
PostnatalNeuronal Migration
Porencephaly
Cysts or cavities within the brain that may or may not communicate with the ventricular system, resulting from vascular or infectious results during late fetal or early infantile lifeUsually present with hemiparesis and focal seizures during the 1st year of life
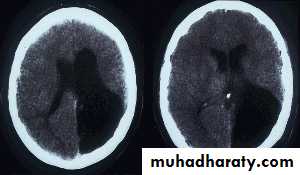
Porencephaly
Agenesis of the Corpus CallosumResults from an insult to the commissural plate during embryogenesis
When it appears as an isolated phenomenon, the patient may be normal; but those with associated migration defects may present with Mental Retardation, microcephaly, hemiparesis, diplegia and seizures
CT/MRI: widely separated frontal horns with an abnormally high position of the 3rd ventricle
Agenesis of the Corpus Callosum
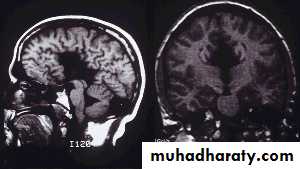
Agenesis of the Corpus Callosum
Aicardi SyndromePatients are almost all females (may be lethal in males)
Characterized by severe MR, intractable seizures with onset between birth and 4 mons of age, and chorioretinal lacunae. Hemivertebrae and costovertebral anomalies are common.
EEG: independent activity from both hemispheres as a result of the absence of the CC
Microcephaly
HC >3 SDs below the mean for age/sexPrimary (genetic) Microcephaly
Secondary (non-genetic) Microcephaly
Dxtic: mother’s serum phenylalanine, karyotype (abnormal facies, short stature, associated congenital abn), CT/MRI (TORCH), fasting plasma and urine amino acid analysis, serum NH4.