
1
Babylon University Stage Fourth
College of Medicine Lecture 1
Dr. Athraa Falah
THE HEPATIC PATHOLOGY
General Features of Hepatic Disease
The major primary diseases of the liver are viral hepatitis, alcoholic liver disease, nonalcoholic
fatty liver disease (NAFLD), and hepatocellular carcinoma (HCC).
In the table below there are the most important laboratory tests that evaluate the liver function.
Table -- Laboratory Evaluation of Liver Disease
Hepatocyte integrity
Serum aspartate aminotransferase (AST)
Serum alanine aminotransferase (ALT)
Biliary function
Serum bilirubin (total and direct)
Serum alkaline phosphatase
Hepatocyte function
Serum albumin
Prothrombin time
Patterns of hepatic injury
• Hepatocyte degeneration and intracellular accumulations
• Hepatocyte necrosis and apoptosis
• Inflammation
• Regeneration
• Fibrosis
Clinical Syndromes related to liver disease
1. Jaundice and Cholestasis.
2. Hepatic Failure
3. Cirrhosis
4. Portal Hypertension
2
Jaundice and cholestasis
Alterations of bile formation become clinically evident as yellow discoloration of the skin and
sclera (jaundice and icterus, respectively) due to retention of bilirubin, and as cholestasis,
characterized by systemic retention of not only bilirubin but also other solutes eliminated in bile.
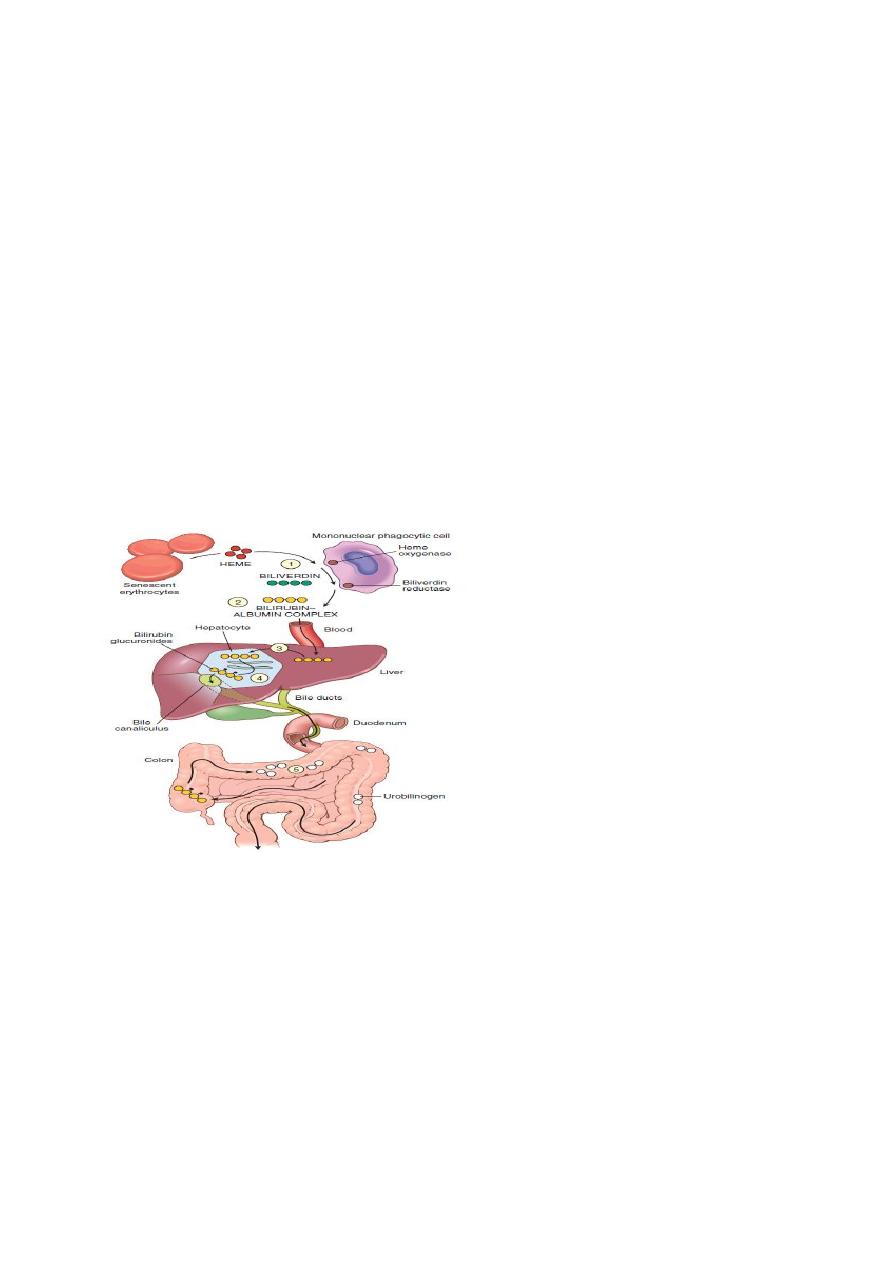
Bilirubin metabolism and elimination.
(1) Normal bilirubin production from heme (0.2–0.3 gm/day) is derived primarily from the
breakdown of senescent circulating erythrocytes.
(2) Extrahepatic bilirubin is bound to serum albumin and delivered to the liver.
(3) Hepatocellular uptake and (4) glucuronidation in the endoplasmic reticulum generate bilirubin
monoglucuronides and diglucuronides, which are water soluble and readily excreted into bile.
(5) Gut bacteria deconjugate the bilirubin and degrade it to colorless urobilinogens. The
urobilinogens and the residue of intact pigments are excreted in the feces, with some reabsorption
and excretion into urine.
Pathophysiology of Jaundice
There are two important pathophysiologic differences between the two forms of bilirubin.
Unconjugated bilirubin is virtually insoluble in water at physiologic pH and exists in tight
complexes with serum albumin. This form cannot be excreted in the urine even when blood levels
are high.
Normally, a very small amount of unconjugated bilirubin is present as an albumin-free anion in
plasma. This fraction of unbound bilirubin may diffuse into tissues, particularly the brain in
infants, and produce toxic injury which may cause severe neurologic damage, referred to as
kernicterus.

3
In contrast, conjugated bilirubin is water-soluble, nontoxic, and only loosely bound to albumin,
and can be excreted in urine.
Serum bilirubin levels in the normal adult vary between 0.3 and 1.2 mg/dL, Jaundice becomes
evident when the serum bilirubin levels rise above 2.0 to 2.5 mg/dL;
Jaundice occurs when the equilibrium between bilirubin production and clearance is disturbed by
one or more of the following mechanisms ( Table below ):
(1) excessive extrahepatic production of bilirubin;
(2) reduced hepatocyte uptake;
(3) impaired conjugation;
(4) decreased hepatocellular excretion; and
(5) impaired bile flow.
The first three mechanisms produce unconjugated hyperbilirubinemia, and the latter two produce
predominantly conjugated hyperbilirubinemia.
TABLE -- Causes of Jaundice
PREDOMINANTLY UNCONJUGATED HYPERBILIRUBINEMIA
Excess production of bilirubin
Hemolytic anemias
Resorption of blood from internal hemorrhage (e.g., alimentary tract bleeding,
hematomas)
Ineffective erythropoiesis (e.g., pernicious anemia, thalassemia)
Reduced hepatic uptake
Drug interference with membrane carrier systems
Some cases of Gilbert syndrome
Impaired bilirubin conjugation
Physiologic jaundice of the newborn {decreased UGT1A1(uridine diphosphate–
glucuronyltransferase1A1) activity, decreased excretion}
Breast milk jaundice (β-glucuronidases in milk)
Genetic deficiency of UGT1A1 activity (Crigler-Najjar syndrome types I and II)
Gilbert syndrome
Diffuse hepatocellular disease (e.g., viral or drug-induced hepatitis, cirrhosis)
PREDOMINANTLY CONJUGATED HYPERBILIRUBINEMIA
Deficiency of canalicular membrane transporters (Dubin-Johnson syndrome, Rotor syndrome)
Impaired bile flow
4
Neonatal Jaundice.
Because the hepatic machinery for conjugating and excreting bilirubin does not fully mature until
about 2 weeks of age, almost every newborn develops transient and mild unconjugated
hyperbilirubinemia, termed neonatal jaundice or physiologic jaundice of the newborn. This may be
exacerbated by breastfeeding, as a result of the presence of bilirubin-deconjugating enzymes in
breast milk
Hereditary Hyperbilirubinemias.
Multiple genetic mutations can cause hereditary hyperbilirubinemia(Tablebelow).
In Crigler-Najjar syndrome type I hepatic UGT1A1 is completely absent. The liver is
morphologically normal by light and electron microscopy. However, serum unconjugated
bilirubin reaches very high levels, producing severe jaundice and icterus. Without liver
transplantation, this condition is invariably fatal.
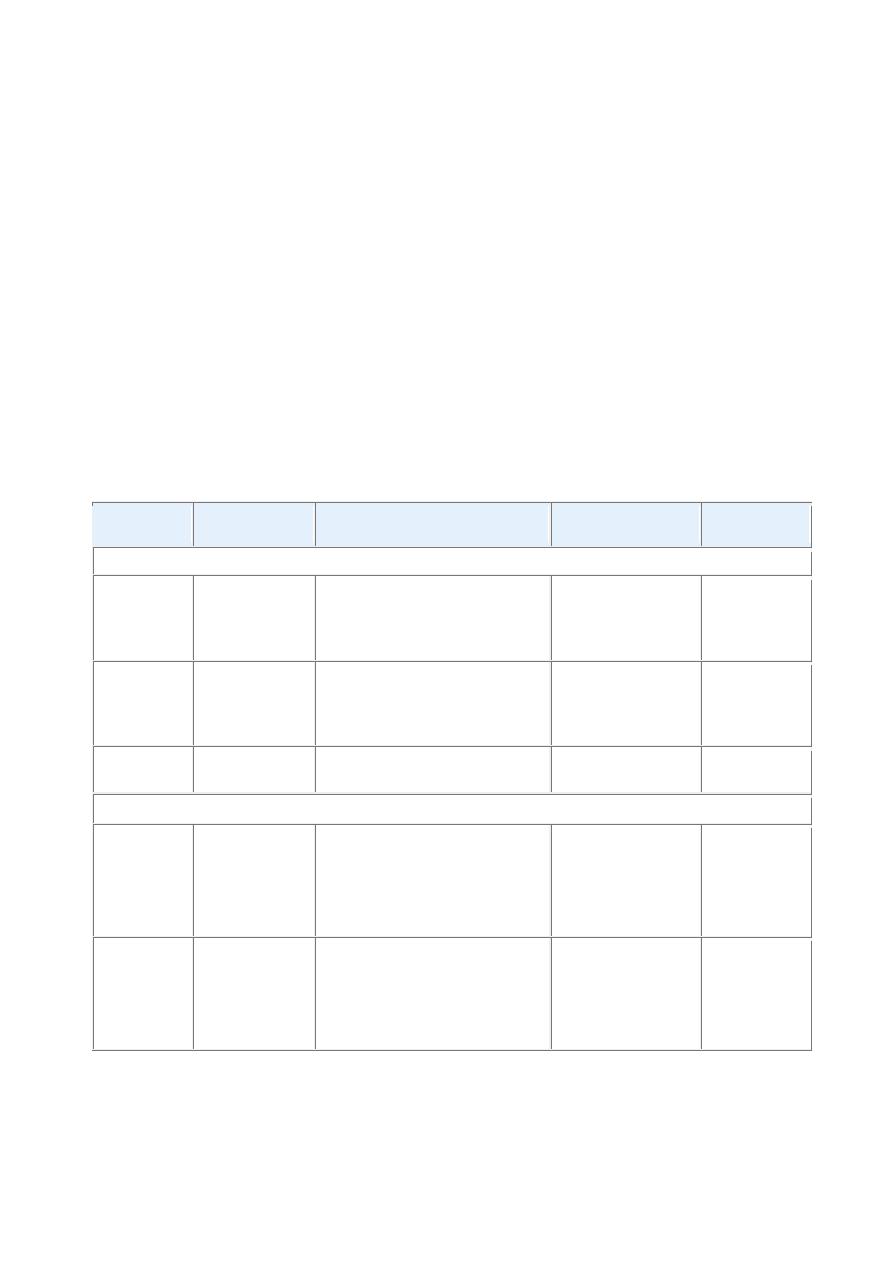
TABLE 18-3 -- Hereditary Hyperbilirubinemias
Disorder
Inheritance
Defects in Bilirubin Metabolism Liver Pathology
Clinical
Course
UNCONJUGATED HYPERBILIRUBINEMIA
Crigler-
Najjar
syndrome
type I
Autosomal
recessive
Absent UGT1A1 activity
None
Fatal
in
neonatal
period
Crigler-
Najjar
syndrome
type II
Autosomal
dominant
with
variable
penetrance
Decreased UGT1A1 activity
None
Generally
mild,
occasional
kernicterus
Gilbert
syndrome
Autosomal
recessive
Decreased UGT1A1 activity
None
Innocuous
CONJUGATED HYPERBILIRUBINEMIA
Dubin-
Johnson
syndrome
Autosomal
recessive
Impaired biliary excretion of
bilirubin glucuronides due to
mutation
in
canalicular
multidrug resistance protein 2
(MRP2)
Pigmented
cytopasmic
globules;
?epinephrine
metabolites
Innocuous
Rotor
syndrome
Autosomal
recessive
Decreased hepatic uptake
and storage?
Decreased
biliary
excretion?
None
Innocuous

5
Crigler-Najjar syndrome type II is a less severe, nonfatal disorder in which UGT1A1 enzyme
activity is greatly reduced. The only major consequence is extraordinarily yellow skin.
Phenobarbital treatment can improve bilirubin glucuronidation .
Gilbert syndrome is a relatively common, benign, inherited condition presenting with mild,
fluctuating hyperbilirubinemia, in the absence of hemolysis or liver disease. It affects 3% to 10%
of the U.S. population. The mild hyperbilirubinemia may go undiscovered for years. When
detected in adolescence or adult life it is typically in association with stress, such as an intercurrent
illness, strenuous exercise, or fasting. Individuals who have Gilbert syndrome may be more
susceptible to adverse effects of drugs that are metabolized by UGT1A1.
Dubin-Johnson syndrome is an autosomal recessive disorder characterized by chronic
conjugated hyperbilirubinemia. It is caused by a defect in hepatocellular excretion of bilirubin
glucuronides across the canalicular membrane. The liver is darkly pigmented because of coarse
pigmented granules within the cytoplasm of hepatocytes . The liver is otherwise normal. Apart
from chronic or recurrent jaundice of fluctuating intensity, most patients are asymptomatic and
have a normal life expectancy.
Cholestasis
It is a pathologic condition of impaired bile formation and bile flow, leading to accumulation of
bile pigment in the hepatic parenchyma. It can be caused by:
-extrahepatic or intrahepatic obstruction of bile channels, or by
-defects in hepatocyte bile secretion.
Patients may have jaundice, pruritus, skin xanthomas (focal accumulation of cholesterol), or
symptoms related to intestinal malabsorption, including nutritional deficiencies of the fat-soluble
vitamins A, D, or K.
-A characteristic laboratory finding is elevated serum alkaline phosphatase and γ-glutamyl
transpeptidase (GGT) enzymes.
Morphology.
- accumulation of bile pigment within the hepatic parenchyma.
-Elongated green-brown plugs of bile are visible in dilated bile canaliculi.
-Rupture of canaliculi leads to extravasation of bile, which is quickly phagocytosed by Kupffer
cells.
-Droplets of bile pigment also accumulate within hepatocytes, which can take on a fine, foamy
appearance (feathery degeneration).
Extrahepatic biliary obstruction is frequently amenable to surgical alleviation. In contrast,
intrahepatic cholestasis is not benefited by surgery (short of transplantation), and the patient's
condition may be worsened by an operative procedure.
6
Infectious Disorders of liver
VIRAL HEPATITIS
Unless otherwise specified, the term viral hepatitis is applied for hepatic infections caused by a
group of viruses known as hepatotropic virus (hepatitis viruses A, B, C, D, and E) that have a
particular affinity for the liver.
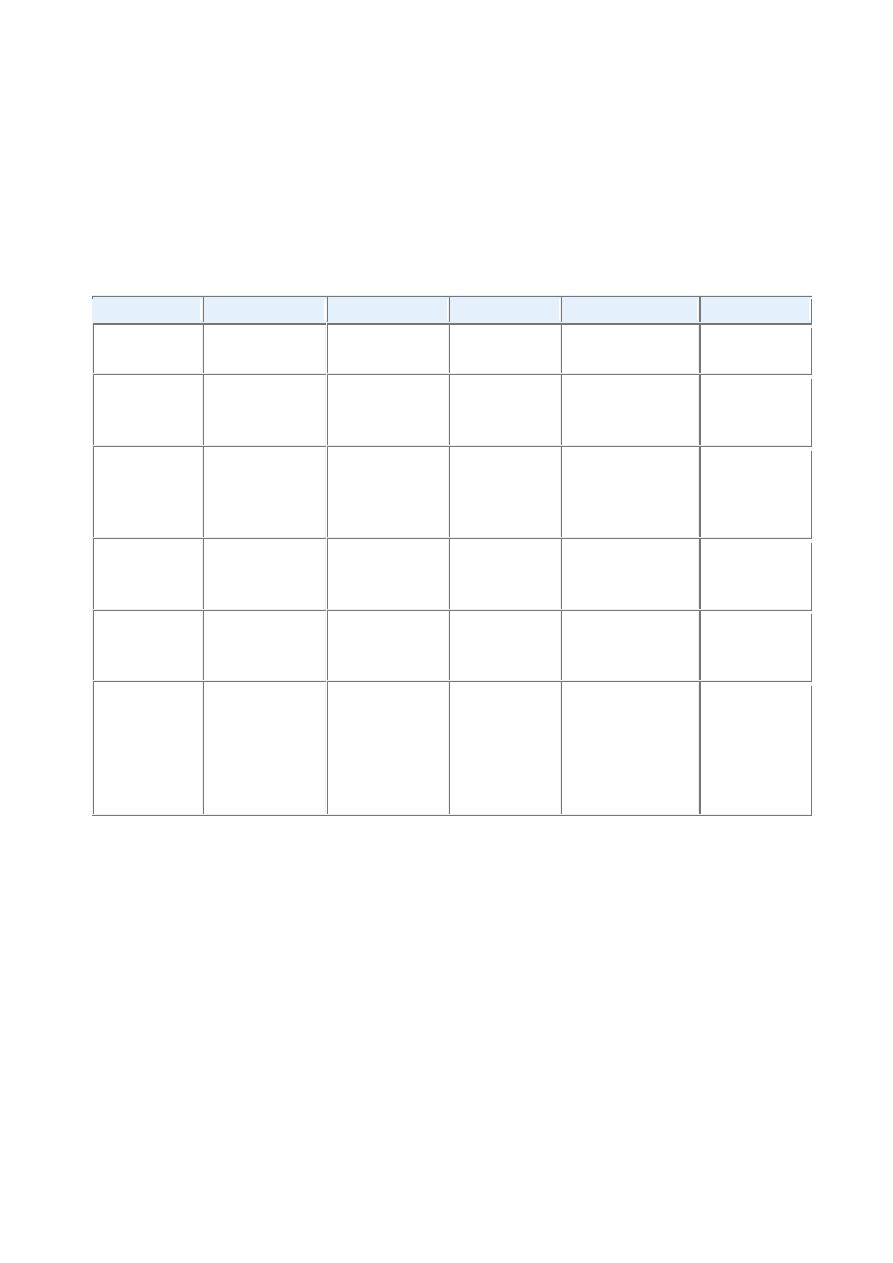
TABLE -- The Hepatitis Viruses
Virus
Hepatitis A
Hepatitis B
Hepatitis C
Hepatitis D
Hepatitis E
Type of virus SsRNA
partially
dsDNA
ssRNA
Circular
defective ssRNA
ssRNA
Viral family
Hepatovirus;
related
to
picornavirus
Hepadnavirus
Flaviridae
Subviral particle
in
Deltaviridae
family
Calicivirus
Route
of
transmission
Fecal-oral
(contaminated
food or water)
Parenteral,
sexual contract,
perinatal
Parenteral;
intranasal
cocaine use is
a risk factor
Parenteral
Fecal-oral
Mean
incubation
period
2–4 weeks
1–4 months
7–8 weeks
Same as HBV
4–5 weeks
Frequency of
chronic liver
disease
Never
10%
∼80%
5% (coinfection);
≤70%
for
superinfection
Never
Diagnosis
Detection
of
serum
IgM
antibodies
Detection
of
HBsAg
or
antibody
to
HBcAg
PCR for HCV
RNA;
3rd-
generation
ELISA
for
antibody
detection
Detection of IgM
and
IgG
antibodies; HDV
RNA
serum;
HDAg in liver
PCR for HEV
RNA;
detection
of
serum
IgM
and
IgG
antibodies
Hepatitis A Virus
Hepatitis A virus (HAV) is a benign, self-limited disease with an incubation period of 3 to 6
weeks. HAV does not cause chronic hepatitis or a carrier state and only rarely causes fulminant
hepatitis, so the fatality rate associated with HAV is about 0.1%.
HAV occurs throughout the world and is endemic in countries with substandard hygiene and
sanitation, where populations may have detectable antibodies to HAV by the age of 10 years.
Clinical disease tends to be mild or asymptomatic, and is rare after childhood.
Affected individuals have nonspecific symptoms such as fatigue and loss of appetite, and often
develop jaundice. Overall, HAV accounts for about 25% of clinically evident acute hepatitis
worldwide .
7
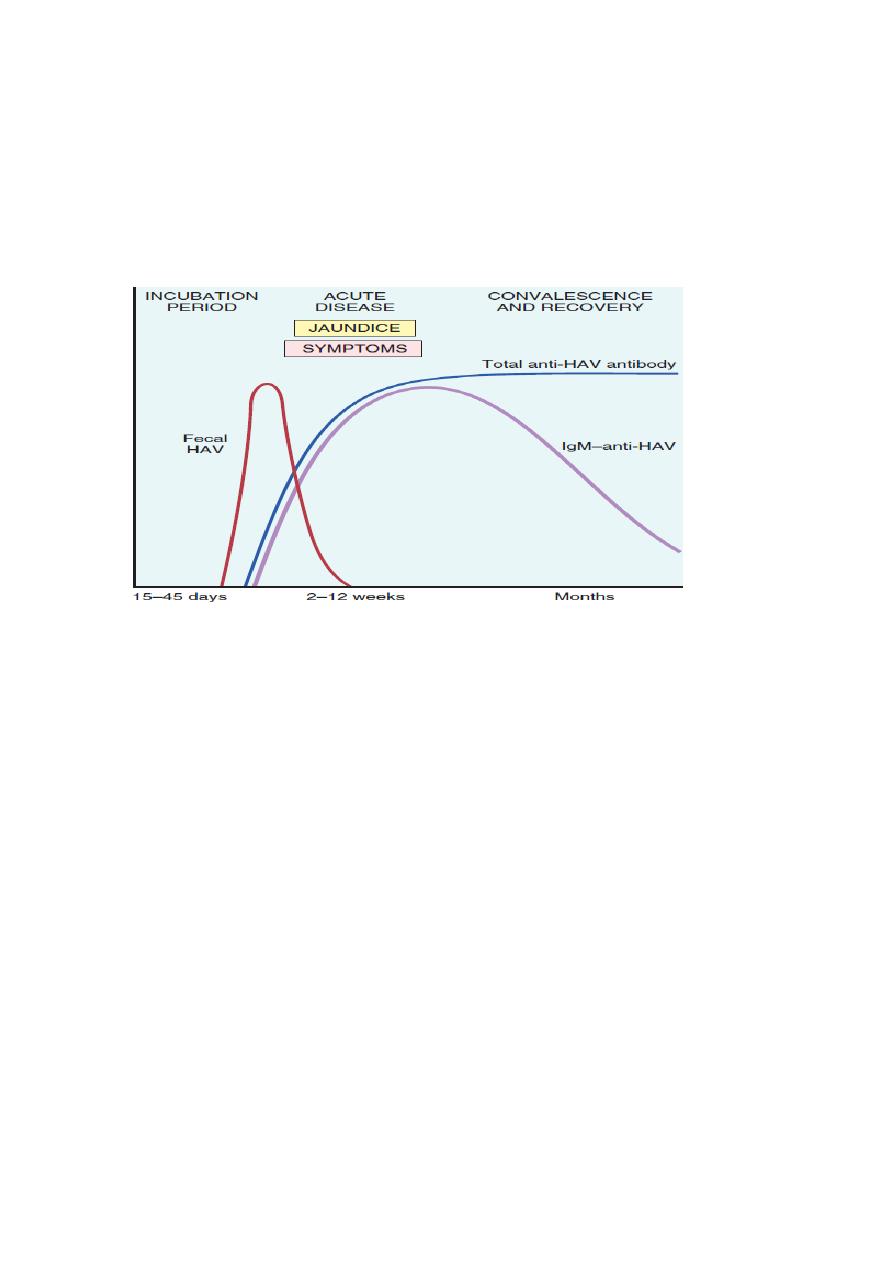
Specific IgM antibody against HAV appears in blood at the onset of symptoms, constituting a
reliable marker of acute infection ( Fig. below ). Fecal shedding of the virus ends as the IgM titer
rises. The IgM response usually begins to decline in a few months and is followed by the
appearance of IgG anti-HAV. The latter persists for years, perhaps conferring lifelong immunity
against reinfection by all strains of HAV. However, there are no routinely available tests for IgG
anti-HAV. The presence of this antibody is inferred from the difference between total and IgM
anti-HAV.
The sequence of serologic markers in acute hepatitis A infection
Hepatitis B Virus (HBV)
HBV can produce:
(1) acute hepatitis with recovery and clearance of the virus,
(2) nonprogressive chronic hepatitis,
(3) progressive chronic disease ending in cirrhosis,
(4) fulminant hepatitis with massive liver necrosis, and
(5) an asymptomatic carrier state.
HBV-induced chronic liver disease is an important precursor for the development of
hepatocellular carcinoma. The clinical outcomes of HBV infection are show in Figure below .
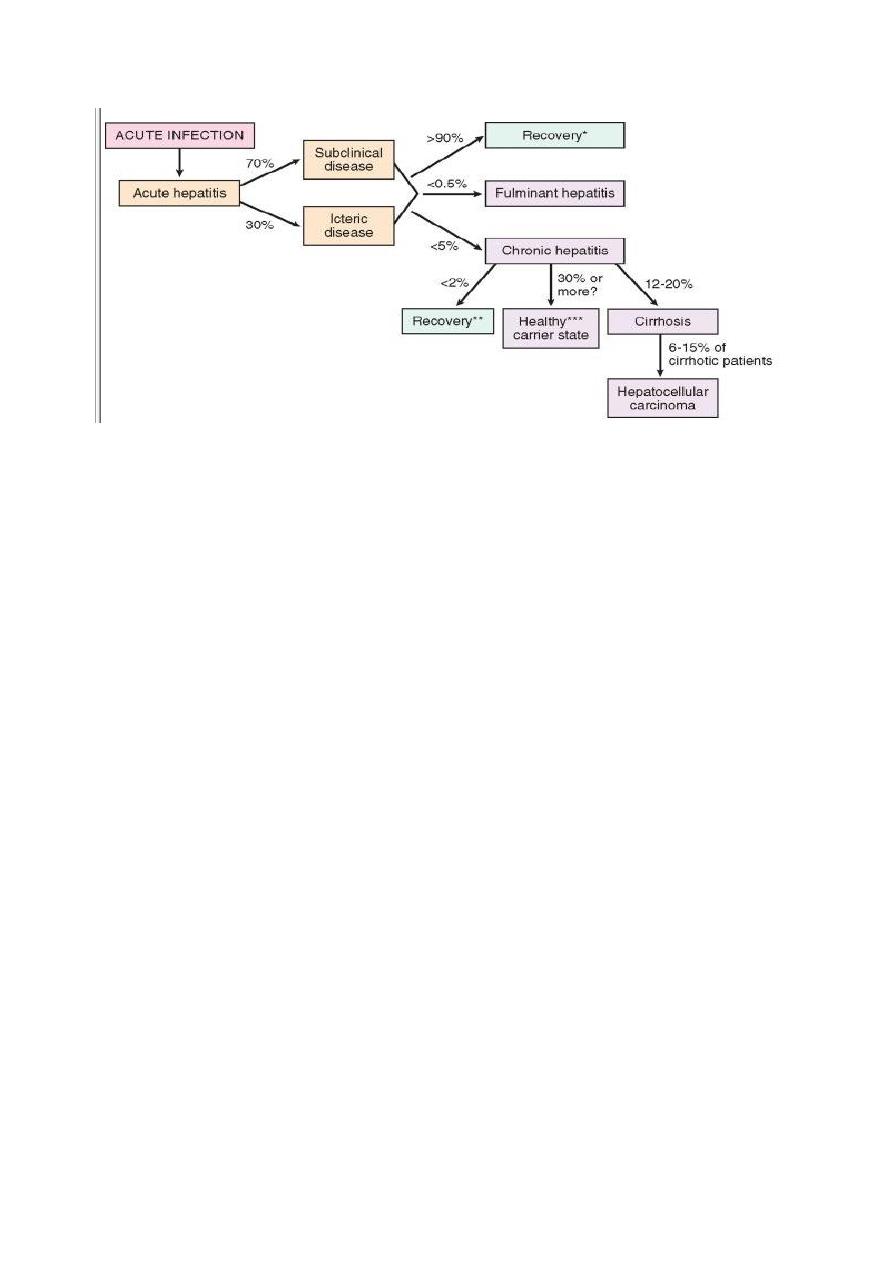
8
The potential outcomes with hepatitis B infection in adults
Liver disease due to HBV is an enormous global health problem. One third of the world
population (2 billion people) have been infected with HBV, and 400 million people have chronic
infection. The carrier rate is largely dictated by the age at infection, being the highest when
infection occurs in children perinatally and the lowest when adults are infected.
The mode of transmission of HBV occurs in different ways:
-perinatal transmission during childbirth .
-horizontal transmission (through minor cuts and breaks in the skin or mucous membranes among
children with close bodily contact),
-unprotected heterosexual or homosexual intercourse and intravenous drug abuse (sharing of
needles and syringes) .
The incidence of transfusion-related spread has decreased greatly in recent years due to screening
of donated blood.
Approximately 70% have mild or no symptoms and do not develop jaundice. The remaining 30%
have nonspecific constitutional symptoms such as anorexia, fever, jaundice, and upper right
quadrant pain. In almost all cases the infection is self-limited and resolves without treatment.
Chronic disease rarely occurs in adults in non-endemic areas. Fulminant hepatitis is also rare,
occurring in approximately 0.1 to 0.5% of cases.
The natural course of the disease can be followed by serum markers ( figure below).
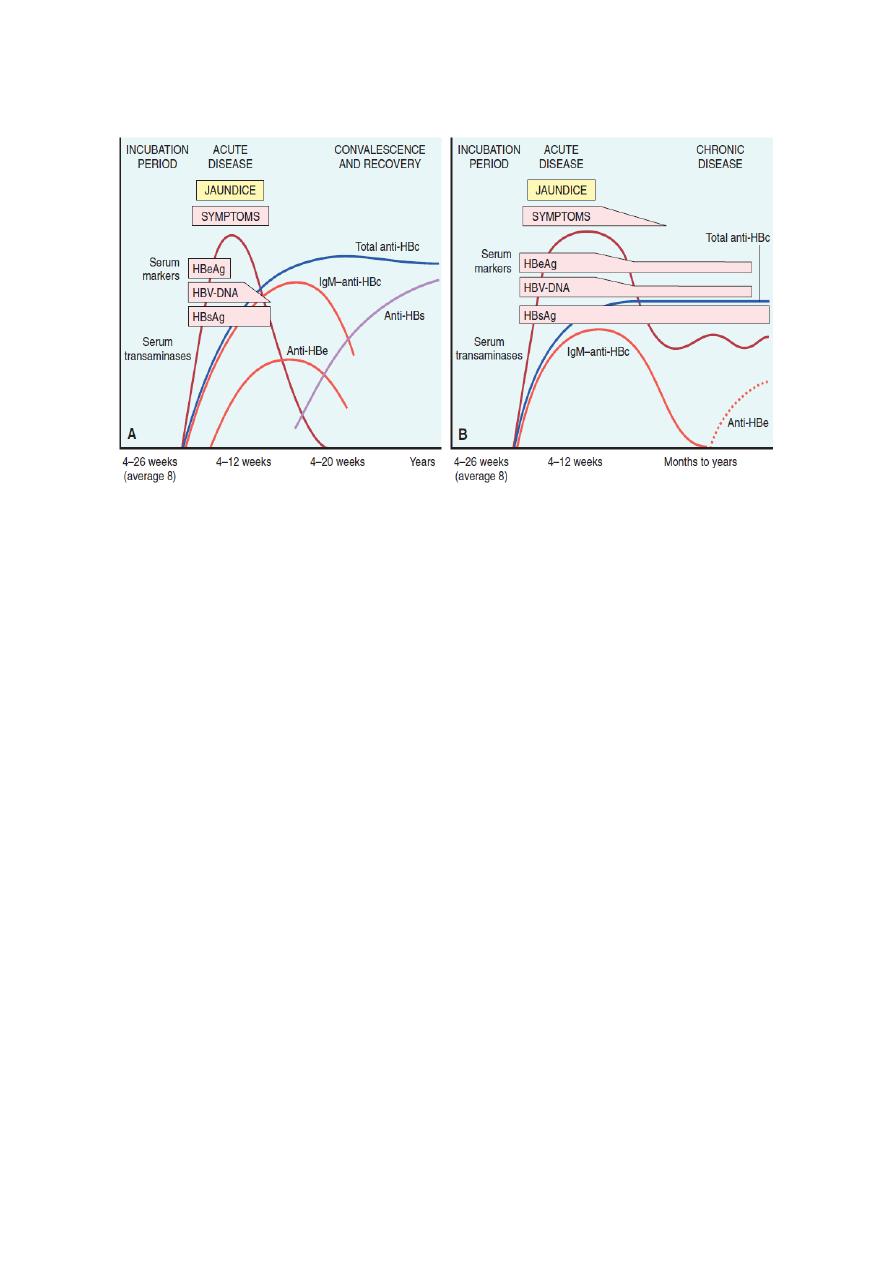
9
Sequence of serologic markers for hepatitis B viral hepatitis demonstrating (A) acute infection with resolution and
(B) progression to chronic infection
.
• HBsAg appears before the onset of symptoms, peaks during overt disease, and
then declines to undetectable levels in 3 to 6 months.
• Anti-HBs antibody does not rise until the acute disease is over and is usually not
detectable for a few weeks to several months after the disappearance of HBsAg.
• HBeAg, HBV-DNA, and DNA polymerase appear in serum soon after HBsAg,
and all signify active viral replication. Persistence of HBeAg is an important
indicator of continued viral replication, infectivity, and probable progression to
chronic hepatitis. The appearance of anti-HBe antibodies implies that an acute
infection has peaked and is on the wane.
• IgM anti-HBc becomes detectable in serum shortly before the onset of symptoms,
concurrent with the onset of elevated serum aminotransferase levels (indicative of
hepatocyte destruction). Over a period of months the IgM anti-HBc antibody is
replaced by IgG anti-HBc.
Hepatitis B can prevented by vaccination and by the screening of donor blood, organs, and tissues.