
Lec:3
Dr. Yasameen Alsaffar
Hyperprolactinaemia/galactorrhoea
Hyperprolactinaemia is a common abnormality which usually presents with
hypogonadism and/or galactorrhoea (lactation in the absence of breastfeeding).
Causes of hyperprolactinaemia include:
1- Physiological:
a- Stress. b- Pregnancy. c- Lactation.
2- Drug-induced
A- (Dopamine antagonists)
a- Antipsychotics (phenothiazines).
b- Antidepressants.
b- Antiemetic (e.g. metoclopramide, domperidone).
B-Dopamine-depleting drugs (Methyldopa).
B- Estrogens (Oral contraceptive pills).
3- Pathological: including
1- Disconnection hyperprolactinaemia.
2- Prolactinoma (usually microadenoma).
3- Primary hypothyroidism.
4- Polycystic ovarian syndrome.
5- Macroprolactinaemia (see below).
6- Renal failure.
Macroprolactinaemia
Prolactin usually circulates as a free (monomeric) hormone in plasma, but in
some individuals prolactin becomes bound to an IgG antibody. This complex is
known as macroprolactin and such patients have macroprolactinaemia. Since
macroprolactin cannot cross blood-vessel walls to reach prolactin receptors in
target tissues, it is of no pathological significance. Some commercial prolactin
assays do not distinguish prolactin from macroprolactin and so
macroprolactinaemia is a cause of spurious hyperprolactinaemia. Identification
of macroprolactin requires gel filtration chromatography or polyethylene glycol
precipitation techniques.
۱

Clinical assessment ((for hyperprolactinaemia)
In women, in addition to galactorrhoea, hypogonadism associated with
hyperprolactinaemia causes secondary amenorrhoea and anovulation with
infertility, in men there is decreased libido, reduced shaving frequency and
lethargy.
Investigation:
Pregnancy should first be excluded before further investigations are performed
in women of child-bearing potential. The upper limit of normal for many assays
of serum prolactin is approximately 500 mU/L (14 ng/mL). In non-pregnant and
non-lactating patients, monomeric prolactin concentrations of 500–1000 mU/L
are likely to be induced by stress or drugs, and a repeat measurement is
indicated. Levels between 1000 and 5000 mU/L are likely to be due to either
drugs, or a microprolactinoma or ‘disconnection’ hyperprolactinaemia.
Levels above 5000 mU/L are highly suggestive of a macroprolactinoma.
Patients with prolactin excess should have tests of gonadal function, and T4 and
TSH should be measured to exclude primary hypothyroidism causing TRH-
induced prolactin excess. MRI or CT scan of the hypothalamus and pituitary
should done for any patient with persistent hyperprolactinaemia. Patients with a
macroadenoma also need tests for hypopituitarism.
Prolactinoma
Most prolactinomas in pre-menopausal women are microadenomas because the
symptoms of prolactin excess usually result in early presentation.
In prolactinomas there is a relationship between prolactin concentration and
tumour size: the higher the level, the bigger the tumour.
Management
Medical
Dopamine agonist drugs are first-line therapy for the majority of patients, they
usually reduce serum prolactin concentrations and cause significant tumour
shrinkage after several months of therapy but visual field defects, if present,
may improve within days of first administration.
In microadenoma we can stop treatment after few years without recurrence. In
patients with macroadenomas, drugs can only be withdrawn after curative
surgery or radiotherapy and under close supervision.
It includes:
۲

1- Bromocriptine (2.5–15 mg/day):
Side effect:
1- Ergotamine-like side-effects (nausea, headache, postural hypotension,
constipation).
2- Rare reports of fibrotic reactions in various tissues.
3-
2- Cabergoline (250–1000
μg/week):
Side effect:
1- Limited data on safety in pregnancy.
2- Associated with cardiac valvular fibrosis
3- Ergot side effect (but less than bromocriptin).
3- Quinagolide (50–150
μg/day): non ergot medication with fewer side effects
but it untested in pregnancy.
Surgery and radiotherapy
Surgical decompression is usually only necessary when a macroprolactinoma
has failed to shrink sufficiently with dopamine agonist therapy, and this may be
because the tumour has a significant cystic component. Surgery may also be
performed in patients who are intolerant of dopamine agonists.
External irradiation may be required for some macroadenomas to prevent
regrowth if dopamine agonists are stopped.
Pregnancy:
For microprolactinoma: should withdraw dopamine agonist therapy as soon as
pregnancy is confirmed.
For macroprolactinomas: it may enlarge rapidly under oestrogen stimulation
and these patients should continue dopamine agonist therapy and need
measurement of prolactin levels and visual fields during pregnancy.
Acromegaly
Acromegaly is caused by growth hormone (GH) secretion from a pituitary
tumour, usually a macroadenoma.
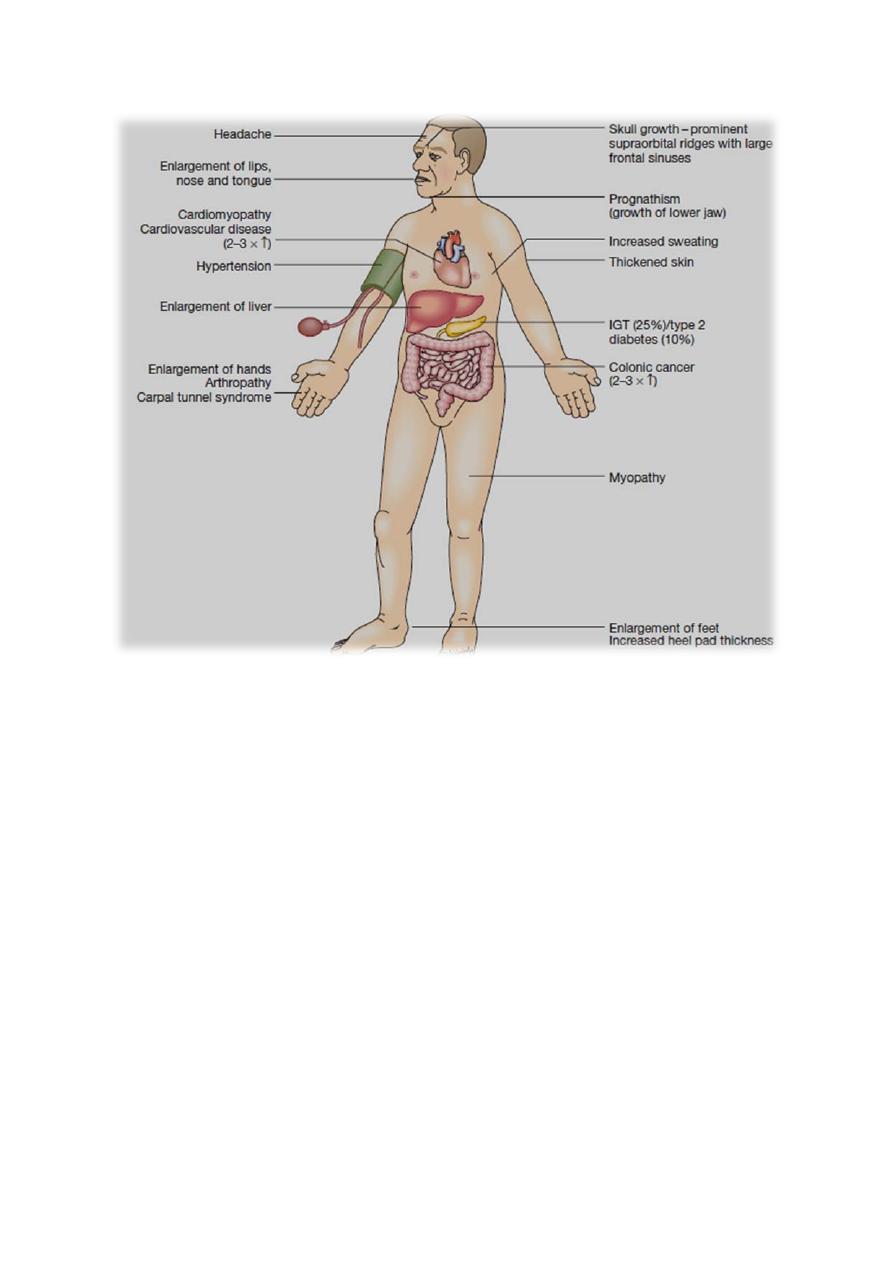
Clinical features
If GH hypersecretion occurs before puberty, then the presentation is with
gigantism. More commonly, GH excess occurs in adult life and presents with
acromegaly. If hypersecretion starts in adolescence and persists into adult life,
then the two conditions may be combined.
۳
Investigations
The clinical diagnosis must be confirmed by measuring GH levels during an
oral glucose tolerance test and measuring serum IGF-1. In normal subjects,
plasma GH suppresses to below 0.5
μg/L (approximately 2 mU/L). In
acromegaly, GH does not suppress and in about 30% of patients there is a
paradoxical rise; IGF-1 is also elevated. The rest of pituitary function should be
investigated. Prolactin concentrations are elevated in about 30% of patients due
to co-secretion of prolactin from the tumour.
Management
The main aims are to improve symptoms and to normalize serum GH and IGF-1
to reduce morbidity and mortality.
Surgical
Trans-sphenoidal surgery is usually the first line of treatment and may result in
cure of GH excess, especially in patients with microadenomas.
Radiotherapy
External radiotherapy is usually employed as secondline treatment if
acromegaly persists after surgery, to stop tumour growth and lower GH levels.
٤

Medical
If acromegaly persists after surgery, medical therapy is usually employed to
lower GH levels to below 1.5
μg/L (below approximately 5 mU/L) and to
normalize IGF-1 concentrations. Medical therapy may be discontinued after
several years in patients who have received radiotherapy. Somatostatin
analogues (such as octreotide or lanreotide) can be administered as slow-release
injections every few weeks. Somatostatin analogues can also be used as primary
therapy for acromegaly (without surgery).
Dopamine agonists are less effective at lowering GH. Pegvisomant is a peptide
GH receptor antagonist administered by daily self-injection and may be
indicated in some patients whose GH and IGF-1 concentrations fail to suppress
sufficiently following somatostatin analogue therapy.
Craniopharyngioma
Craniopharyngiomas are benign tumours, may be located within the sella
turcica, or commonly in the suprasellar space. They are often cystic, with a solid
component that may or may not be calcified, in young people; they are
diagnosed more commonly than pituitary adenomas.
They may present with pressure effects on adjacent structures, hypopituitarism
and/or cranial diabetes insipidus. Other clinical features directly related to
hypothalamic damage may also occur. These include hyperphagia and obesity,
loss of the sensation of thirst and disturbance of temperature regulation.
Treatment:
By surgery either transsphenoidal approach or craniotomy, by removal of tumor
or just insertion of catheter for drain in the cystic part of tumor.
Radiotherapy is to decrease the chance of recurrence.
Diabetes insipidus
This uncommon disorder is characterised by the persistent excretion of
excessive quantities of dilute urine and by thirst. It is classified into two types:
• Cranial diabetes insipidus, in which there is deficient production of ADH by
the hypothalamus
• Nephrogenic diabetes insipidus, in which the renal tubules are unresponsive to
ADH.
Causes
:
A- Cranial:
1- Structural hypothalamic or high stalk lesion.
2- Idiopathic.
3- Genetic defect.
B- Nephrogenic:
1- Genetic defect.
٥

2- Metabolic abnormality (Hypokalaemia or Hypercalcaemia).
3- Drug therapysuch as Lithium.
4- Poisoning such as heavy metals.
5- Chronic kidney disease.
Clinical feature:
The most marked symptoms are polyuria and polydipsia. The patient may pass
5–20 L or more of urine in 24 hours. If there is associated cortisol deficiency,
then diabetes insipidus may not be manifest until glucocorticoid replacement
therapy is given.
The most common differential diagnosis is primary polydipsia, caused by
drinking excessive amounts of fluid in the absence of a defect in ADH or thirst
control.
Investigations
Diabetes insipidus can be confirmed if serum ADH is undetectable (although
the assay for this is not widely available) or the urine is not maximally
concentrated (i.e. is below 600 mOsm/kg) in the presence of increased plasma
osmolality (i.e. greater than 300 mOsm/kg), the sodium level elevated or in the
upper limit.
Sometimes, the diagnosis can be confirmed or refuted by random simultaneous
samples of blood and urine, but more often a dynamic test is required;1- The
water deprivation test, or 2- infuse hypertonic (5%) saline.
Anterior pituitary function and suprasellar anatomy should be assessed in
patients with cranial diabetes insipidus.
In primary polydipsia, the urine may be excessively dilute but plasma
osmolality and sodium are low rather than high.
Management
Treatment of cranial diabetes insipidus is with desamino- des-aspartate-arginine
vasopressin (desmopressin, DDAVP), an analogue of ADH which has a longer
half-life. DDAVP is usually administered intranasally.(oral and intramuscular
formula are also available).
The polyuria in nephrogenic diabetes insipidus is improved by thiazide,
amiloride and NSAIDs.
With best regard
٦