Endocrine system
Textbook nelson 20 ed.Essential nelson
Illustrated textbook(Tom lisawer)
Harriate lane
Pediatrics secret
BY PROF. RAZZAQ ALRUBAEE
THIQAR COLLEGE OF MEDICINE
2019
Endocrine system 2019… Hypothalamus &pituitary gland disorder & short stature
Thyroid ¶thyroid gland disordersAdrenal gland disorder
Type 1 diabetes
DKA
.complication
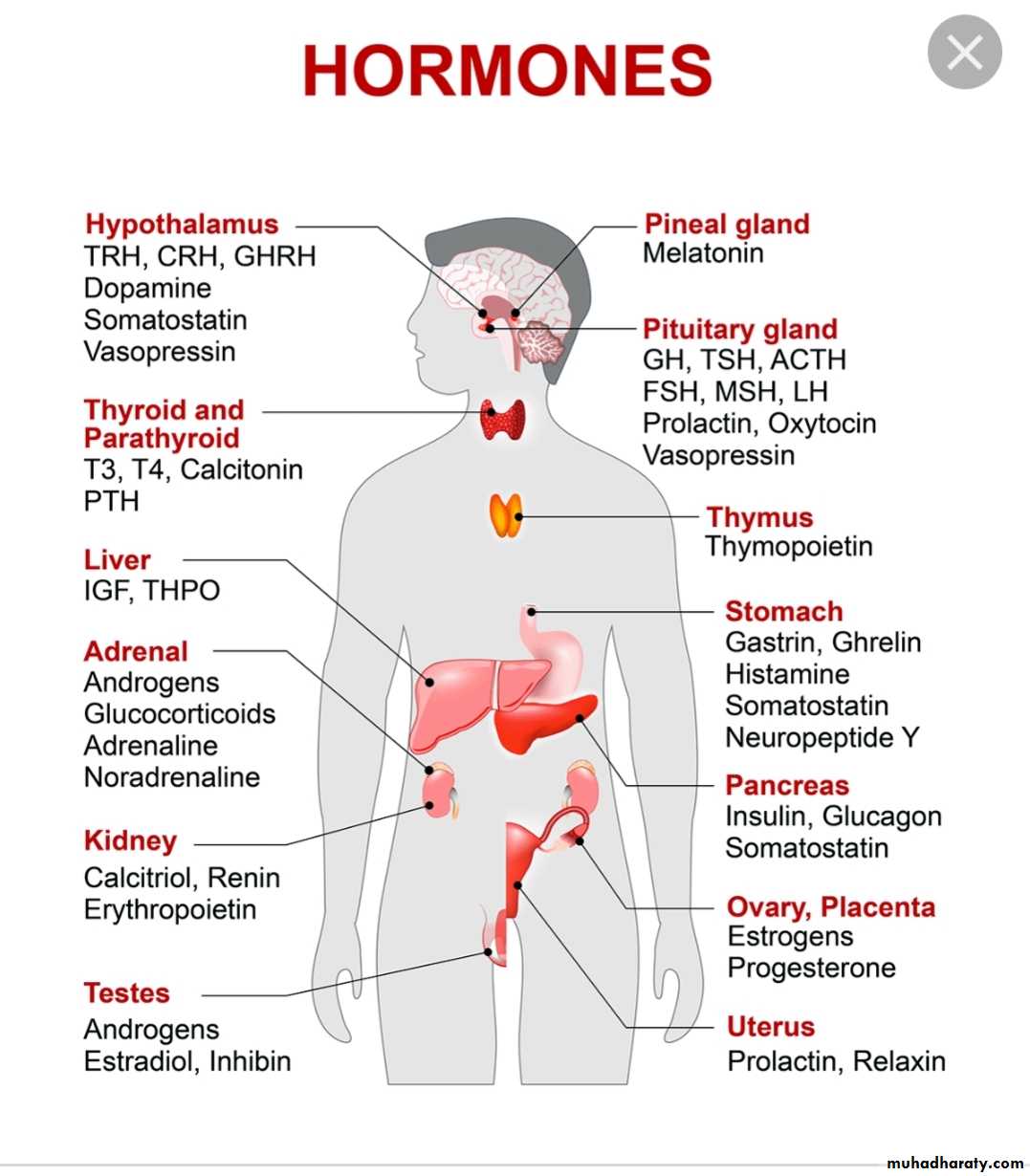
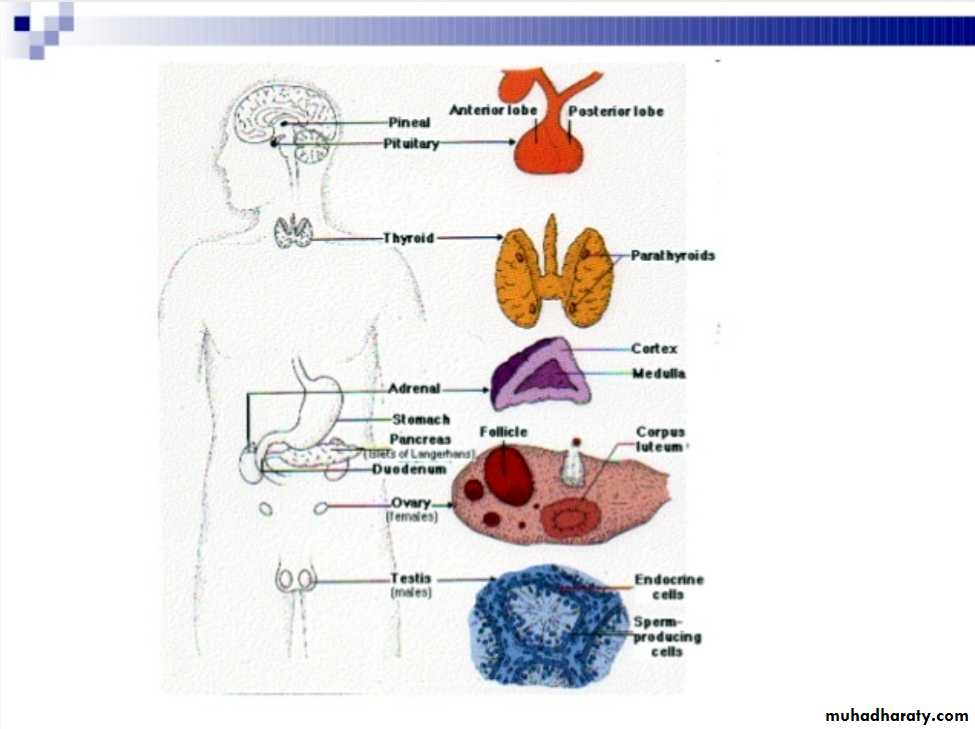
Hormones of hypothalamus and pituitary
The pituitary gland receive signals from hypothalamus & respond by sending pituitary hormones to target glands which produce hormones that provide negative feedback at level of hypothalamus and pituitaryHormones
Locations\I(stimulate .inhibit)
Function
ACTH
Ant. pituitary
S
Production of glucocorticoid MC.miniralocorticoid.
androgen
ADH
Post. Pituitary
S
Water reabsorption from renal tubules
CRH. corticotropin releasing hor.
Hypothalamus
S
Secretion of ACTH
FSH (female)
Ant. Pituitary
I
Secretion of estrogen from ovary
FSH (male)
Ant. Pit
S
Production of sperm from testes
GnRH
Hypothalamus
S
Secretion of LH &FSH
LH(female )
Ant. Pit
S
Ovulation and development of corpus luteum
LH(male )
Ant. Pit
S
Production and secretion of testosterone
Oxytocin
Post. Pit
S
Contraction of uterus at birth &release of milk from breast
TSH
Ant. Pit
S
Secretion of T3.T4
PRL prolactin
Post. Pit
S
Promotion of milk synthesis
Somatostatin
Hypothalamus
I
Secretion of GH&TSH
TRH( thyrotropin releasing hormone)
Hypothalamus
S
Secretion of TSH & prolactin
Growth hormone deficiency &insensitivity (hypopituitarism )
Hypopituitarism : denotes underproduction of GH growth hormone alone or in combination with deficiency of other pituitary hormones .
• Congenital hypopituitarism growth hormone defciency )
Clinical features :The child usually normal size and weight at birth .may have neonatal emergencies like apnea .seizures .jaundice, cyanosis or Sever hypoglycemia with or without seizures prolonged neonatal jaundice is common .nystagmus may suggest septooptic dysplasia . micropenis in boys can be clue for GH def.
Physical exam:
head is round & face is short & broad ,prominent frontal bones eyes somewhat are bulging .mandible and chin are underdeveloped ,delayed teeth eruption and often crowded , short neck and small larynx , high pitch voice which remain high after puberty
Evaluation of suspected growth hor. Def.
Growth related history &physical exam• Growth failure
• Short stature
• GHD affect 1in 3500 child
Image & other evaluation
• Clinical diagnosis
• Bone age ( delayed )
• MRI,CT evaluate hypothamic pituitary region
Lab . finding
• Measures GH with stimulation , IGF-1 test
Rationale for treatment
• Replacement with GHTshould be started as soon as GHD is diagnosis
Diagnosis :
determined by low or absent level of GH in response to stimulation with insulin , arginine clonidine ,or glucagon to establish low level of GH <10 ng\ml and also necessary to evaluate others pituitary hormones deficiency like ACTH, TSH cortisol ,gonadotropin .
Treatment
recombinant hGH available since 1982 usually given in a dose 0.18-0.3 mg/kg/wk during childhood and higher dose needed during puberty. Therapy should continued until near final height is achieved and treatment discontinued if he or she tall enough or growth rate <I inch /year and bone age >14 yr in girls &>16 years in boys• Indication of GH therapy :
• 1-GHD 6-Prader willi syndrome• 2-Turner syndrome 7-SHOX gene abnormality
• ( short stature homebox)
• 8-Noonan syndrome
• 3-Chronic renal failure 5-Small for gestational age
• 4-Idiopathic short stature
Adverse effect of GH therapy .include
pseudotumor cerbri .gynecomastia ,slipped capital femoral epiphysis & worsening scoliosis
Growth hormone insensitivity (LARON syndrome)
autosomal recessive disease.Is a condition that occur when the body unable to utilize GH , and characterized by short stature ,hypoglycemia ,near normal at birth ,delayed puberty & short limbs (arms &legs) with obesity .other signs include small genitals ,thin fragile hair those people have low risk of cancer and type II diabetes .
• Diagnosis :
S&S , GH usually high and reduced level of IGF1 & genetic study to show abnormality in GH geneTreatment : no current cure for Laron syndrome & only available treatment is subcutaneously injection of IGF1
Prognosis :generally good d not affect life span
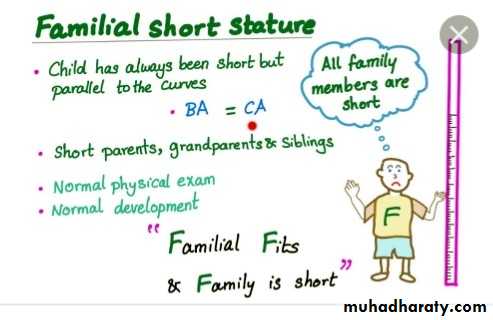
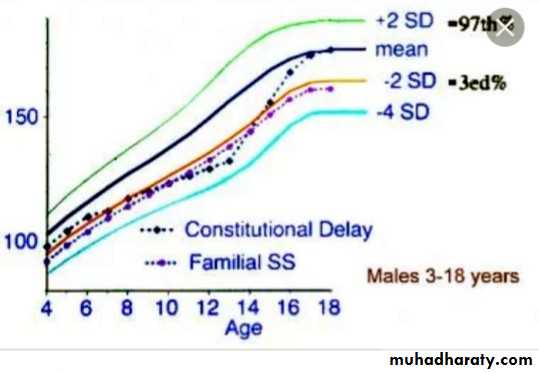
Constitutional Growth delay
: one of the variant of normal growth . length & weight normal at birth , growth is normal for the first 4-12 months , height sustained at low percentile during childhood .
., the pubertal growth spurt delayed and eventual normal stature , normal bone age . GH response to provocative test tend to be lower than in children with a more typical timing of puberty . the prognosis of those children to achieve normal adult height is guarded
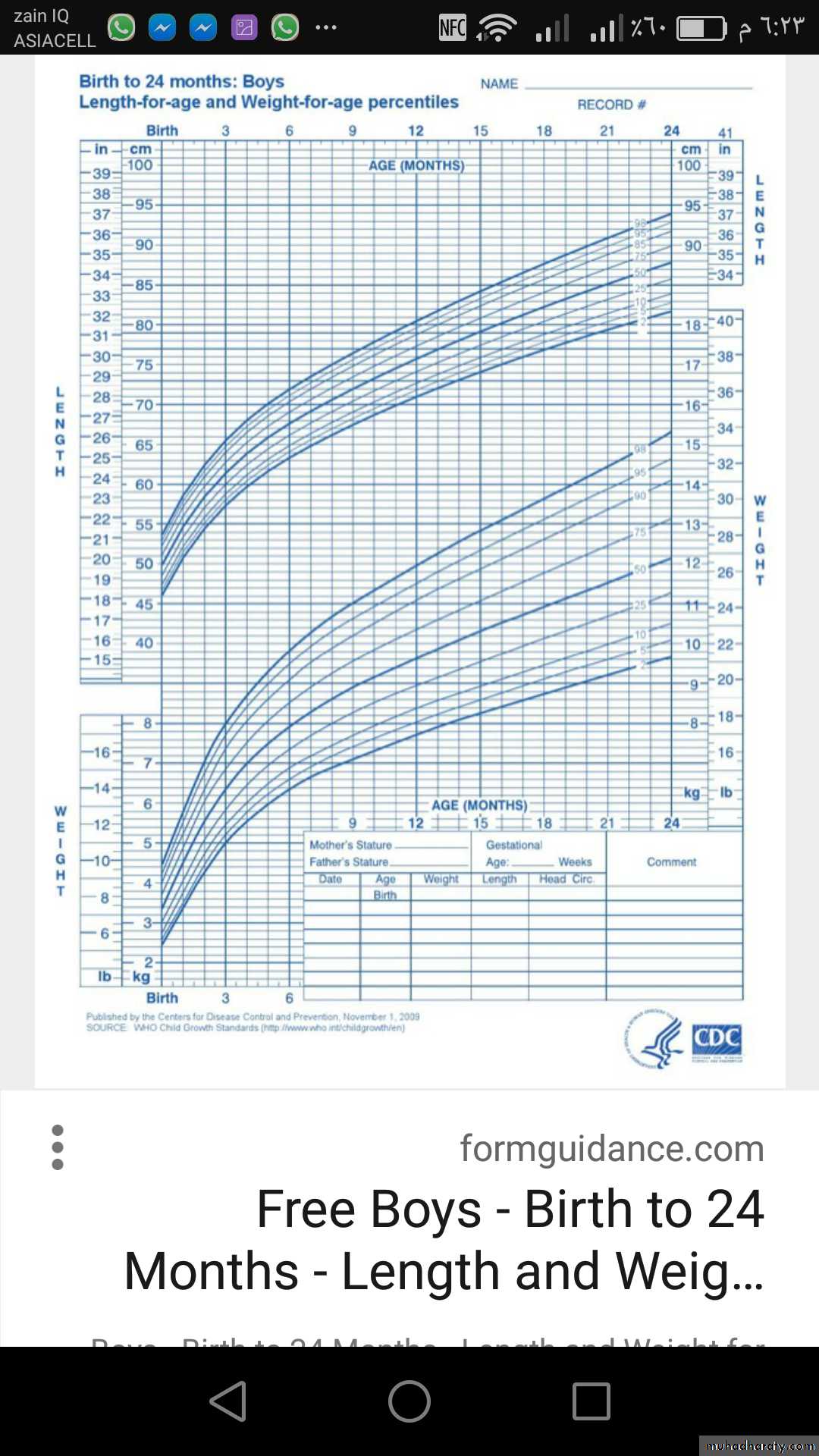
• HYPERPITUITARISM ( TALL STATURE )
Either primary or secondaryTable showed differential diagnosis of tall stature & overgrowth syndromes
FETAL OVERGROWTH :
Maternal DM
cerebral gigantisim (Sotos syndrome)
Beckwith Wiedemann
Postnatal overgrowth leading to childhood tall stature
• Nonendocrine causes
Familial (constitutional tall stature )
Exogenous obesity
Cerebral gigantism sotos syndrome
Marfan synd.
Bekwith wiedmann synd.
Klinfelter syndrome
Homocystinuria
• Endocrine cause
• Excess GH secretion• Precocious puberty
• Hyperthyroidism
• Mc Cune –Albright syndrome
•
Postnatal overgrowth leading to adult tall stature
• Familial
• Marfan
• Klinfelter
• Excess GH
• XXY
• ACTH or cortisol deficiency
Regarding Growth hormone
A - hypopitiutarism mean deficiency of all pituitary mormonesB- growth hormone deficient child usually small at birth
C- high pitch voice of GH deficient child usually corrected after puberty
D—treatment with GH usually stopped after 15 yr of age
E—laron syndrome have low risk of cancer

• THYROID GLAND DISORDER : L2
Etiological classification of congenital hypothyroidism:Primary hypothyroidism :
• Defect of fetal thyroid development (dysgenesis )
• Defect in thyroid hormones synthesis
• Defect in thyroid hormones transport
• Resistance tom thyroid hormones
• Maternal antibodies
• Iodine deficiency
• Maternal medication (iodides .amiodarone . methimazole ,radioiodide )
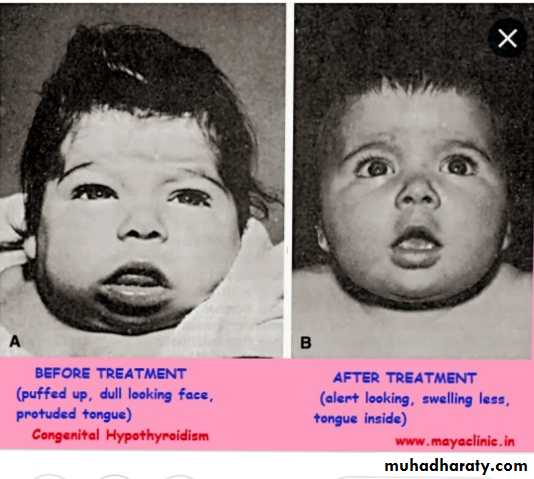
• Central hypo pituitary hypothyroidism
TSH deficiency• Isolated TRH deficiency
• Multiple congenital pituitary hormones deficiencies
Clinical features of congenital hypothyroidism
Most cases of congenital hypothyroidism are asymptomatic because partial trans placental passage of maternal T4.S of cong.hypothyroidsm &s
Normal weight & length at birthSlight enlargement of head because myxedema
Wide anterior &posterior fontanel
Prolonged physiological jaundice (early sign)
Feeding difficulties& choking &delayed teeth eruption
Cry little &sleep much poor appetite
Constipation not respond to treatment with umbilical hernia
Genital edema ,slow pulse ,cardiac murmur with macrocytic anemia
Broad hands &short fingers
Dry scaly skin &little perspiration
Developmental delay ,hoarse voice &they don’t learn to talk
Physical &intellectual impairment increase with age with delayed sexual maturation
Laboratory finding:
Heal prick between 2-5 days of age ( filter paper )
Serum level of T4 & free T4 usually low …..T3 may be normal & usually not helpful in diagnosis. ECG show slow voltage ,echo may show pericardial effusion .
• Radiological finding :
retarded osseous maturation at birth in 60 % of cases .Distal femoral &proximal tibial epiphyses normally present at birth are often absent
Deformity( beaking )of the 12th thoracic or 1st or 2nd lumber vertebra is common , skull show wide sutures ,enlarge and round sella turcica
• Treatment :
Levothyroxine (L-T4)given orally at morningRecommended initial starting dose is 10-20 µg/kg/day [ serum T4 &freeT4 ,TSH ]should be checked every 1-2 mo.in 1st 6 mo. Of life .then every 2-4 mo. From 6mo. -3 yrs. Of age .
• Prognosis :
Early diagnosis and adequate treatment from 1st wk.of life result in normal linear growth &development . variable degree of brain damage may contribute to [delay diagnosis, inadequate treatment &poor compliance in first 2-3 years of life )• HYPERTHYROIDISM :
Excessive secretion of thyroid hormones it is caused by Graves diseaseClinical features of hyperthyroidism :
Symptoms :
• Hyperactivity .irritability ,altered mood, insomnia poor concentration
• Heat intolerance
• Fatigue ,weakness ,palpitation
• Dyspnea weight loss ,thirst &polyuria with loose stool

• Signs :
• Sinus tachycardia• Fine tremor , hyperreflexia moist &worm skin
• Palmer erythema ,
• Hair loss or thinning
• Chorea
• Osteoporosis ,hypokalemic periodic paralysis
• Psychosis
LAB. Finding :
High T3--T4 &free T4and low TSH and T3 usually elevated more than T4
• Treatment :
Medical treatment is recommended rather than radioiodine or surgeryPropylthiouracil(PTU) &methimazole[Tapazole ]( potent 10 time than PTU)
Initial dose of Methimazole :0.25mg-1mg /kg /24hr once or twice daily
Clinical response appear after 3-6 wks and adequate control evident in3-4 months
Beta blocker like propranolol in dose 0.5-2 mg/kg/day orally useful in toxic patients
Disorder of parathyroid gland:
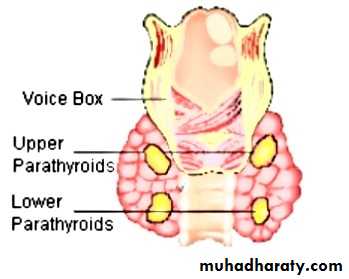
Parathyroid hormones
: PTH also called parathemone secreted by parathyroid gland which is important in bone remodeling .PTH secreted in response to low blood serum calcium level .calcitonin : also called thyrocacitonin it is hormone produced primarily by Para follicular cell in thyroid gland and act to reduce level of calcium and opposing the effect of PTH hormone .HYPORARATHYROIDISM :
• Etiology:
A: congenital• Transient neonatal
• Familial isolated hypopara .(AR,AD.Xlinked )
• Di George syndrome & sanjad –sakati syndrome (short stature, retardation .dysmorphism )
• Barakat syndrome (sensorineural hearing loss with renal dysplasia )
B :acquired (autoimmune ,infiltrative ,maternal hyperparathyroidism ,hypomagnesemia)
Clinical features:
Vary from asymptomatic to sever form of hypocalcemiaMuscular pain &cramps are early signs
Numbness stiffness &tingling of hands and feet
Chevostek or trousseau sign or carpopedal spasm may be only signs
Convulsion with or without loss of consciousness (mistaken as epilepsy )
Headache & vomiting because raised ICP
Delayed teeth eruption &dry scaly skin
Mucocutaneous candidiasis
Cataract .addison diseasepernicius anemia ,alopecia
Murtada
With DM
On carbimazole
Lab. Finding :
serum calcium is low5-7mg/dl
phosphorus level is elevated
(7-12mg/dl)
Ionized Ca which reflect 45% of the total
Low
Alkaline phosphatase
normal or
low
1,25(OH)2D3
Usually low& high in sever hypocalcemia
Magnesium
Normal
PTH
Low
• Treatment :
Emergency treatment of neonatal tetany consist of injection of 5-10 ml or 1-3 mg/kg of 10%solution calcium gluconate (elemental 9.3 mg/ml )
D3 : also should be given (initial dose .25µg/24 hr .with maintenance dose 0.01-0.1 µg/kg/24hr
Supplemental calcium in form of calcium gluconate provide 800 mg elemental calcium daily
PSUODOHYPOPARATHYROIDSM( PHP)
:{Albright hereditary osteodystrophy )Parathyroid hormone here are normal and serum level of PTH are elevated even when patient is hypocalcemic(peripheral resistance to PTH rather than deficiency ) .we have type 1A (common ) &type 1B less common & type 2.(tissue specific resistant to PTH)
Hypocalcemia ,high PTH, hyperphosphotemia
Type 1A : account for majority of cases• Tetany is often presenting sign
• Short stocky build &round face
• Brachydactyly with dimpling dorsum of the hand
• 2nd metacarpal bone involve result in index finger longer than middle
• Short and wide phalanges .bowing ,exostoses &thickening of calvaria
• Moderate degree of cognitive impairment
• Calcification of basal ganglia with lenticular cataract

Hypoparathyroidism low ca.high phosphorous low PTH
Sudospodohypoparathyroidism PPHPInherited disorder ,it is similar to pseudohypoparathyroidism (PHP) in presentation
serum level of calcium and phosphorus usually normal with PTH hormone slightly elevated & no resistant to PTH
Summary
ConditionAppearance
PTH
Calcium
Phosphates
Hypoparathyroidism
Normal
Low
Low
High
Pseudohypoparathyroidism type 1A
Skeletal defect
High
Low
High
Pseudohypoparathyroidism type 1B
Normal
High
Low
High
Pseudohypoparathyroidism type 2
Normal
High
Low
High
Pseudopseudohypoparathyroidism
Skeletal defect
Normal
Normal
Normal

DISORDER OF ADRENAL LAND L3
Adrenal gland consist of cortex & medulla1-Cortex
outer layer zona glomerularis(aldosterone secretion )
middle layed :zona fasiculata (glucocorticoid &cortizole )
inner layer :zona reticularis (androgen secretion )
2- medulla (epinephrine &norepinephrine )
CONGENITAL ADRENAL HYPERPLASIA (adrenal insufficiency ):
Adrenal steroidoenesis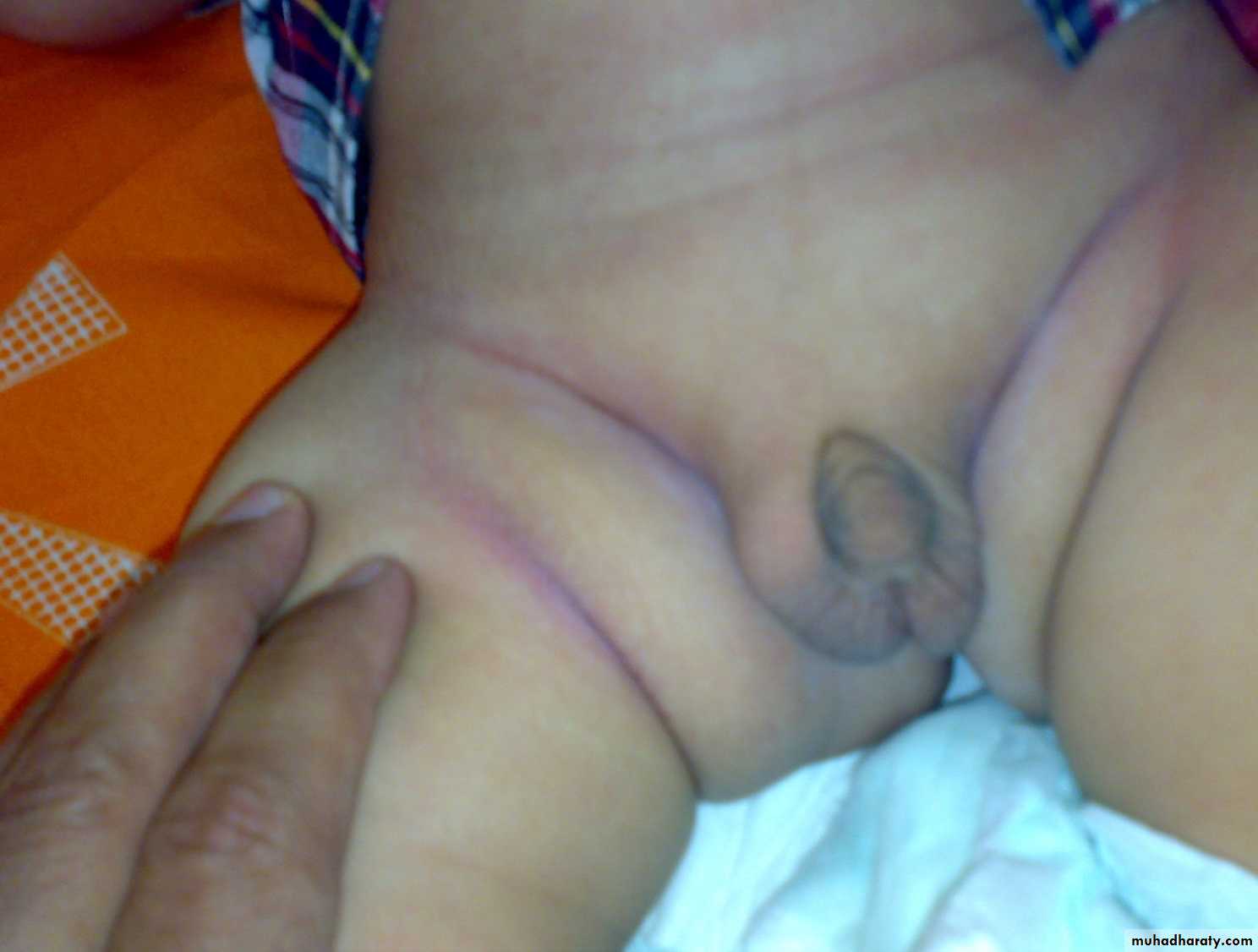
Ambiguous genitalia
Adrenal steroidogenesis
Cholesterol
Cholesterol desmolase
Pregninolone 17 OH pregninolone dehydroepiandrosterone
3 beta hydroxylsteroid dehydrogenasae DHEA
Progesterone
deoxycortisole Androsterone21 hydroxylase
11 beta hydroxylase 17 OH steroid deh ydrogenase
DeoxycorticosteroneCortisole testesterone
•
•
Corticosterone
18 oxydase
aldosterone
A several of autosomal recessive disorder result from mutation of gene for enzymes mediating biochemical steps of production of mineralocorticoids ,glucocorticoids & sex hormones from cholesterol by adrenal gland
Most common types :
21 hydroxylase deficiency ,
11β hydroxylase def..
3βhydroxysteroid dehydrogenase def.
Symptoms (depend on the form of CAH &sex of patient
Mineralocorticoid deficiency :
• Vomiting due to salt loss(dehydration &death )
Excess androgen :
• Average size penis• Ambiguous genitalia in females
• Early pubic hair & rapid growth in childhood
• Precocious puberty
• Excessive facial hair virilization
• Infertility due to anovulation
• Clitoromegaly
Undervirilization in XY male which can result in apparently female external genitals
Investiations
• Hypoglycemia ,hyperkalemia. hyponatremia (due to hypoaldosteronism )• Low cortisole level
• High ACTH
• High 17 α hydroxyprogesterone in blood
• High 17 ketosteroid in urine
• Most definite (measure serum cortisol pre & after ACTH administration
Treatment
• Immediate & vigorous fluid &electrolytes replacement
• supply enough glucocorticoid to reduce hyperplasia & overproduction of androgen or mineralocorticoid
• Replace enough mineralocorticoid
Treatment
• Provide testesterone or estrogen at puberty if deficient• Optimize growth &bone maturation
• Hydrocortisone Na succinate 10-15 m \m²\24hr TID
fludrocortisone (synthetic mineralocorticoid ) orally in dose 0.05-0.2 mg/day
• Addison dsiaese : (acquired adrenal insufficiency ):
Occur as a part of autoimmune polyendocrinopathy syndrome type-2(APS-2): which consist of Addison dis.autoimmune thyroiditis , or type 1 DM .Type 1 polyendocrinopathy : consist of : mucocutaneous candidiasis & various autoimmune endocrinopathies like hypoparathyroidism ,Adrenal insufficiency
Clinical features
Symptoms :• fatigue ,anorexia ,weight loss,myalgia &joint pain
• nausea ,vomiting
• Glucocorticoid deficiency
•
•
• Both glucocorticoid &mineralocorticoid
Signs
Signs :
• Low blood pressure
• Skin or mucosal hyperpigmentation
•
• Both mineralo&glucocorticoid lack
• Excess of proopiomelanocortin –derived peptide
Lab.findings
HyponatremiaHypoglycemia &ketosis
Hyperkalemia
Low cortisol level
Eosinophilia &lymphocytosis
High ACTH level
High plasma renin activty
• Treatment :
Correction of electrolyte abnormality with hypoglycemiaSteroid replacement. Hydrocortisone orally in daily dividing dose of 10-15 mg/m²/day
• Cushing syndrome:
• excess of cortisol or other glucocorticoid due to either adrenal tumors or central pituitary involvement and or exogenous steroid usage (details in internal medicine lectures ).
• Physiology of puberty L4
Between early childhood and 8-9 yr of age (prepubertal stage )the hypothalamic –pituitary –gonadal axis is dormant as reflected by undetected level of LH & sex hormones testosterone in boys and Estradiol in girls .1-3 yr before onset of puberty low serum level of LH demonstrate during sleep in pulsatile fashion. the pulsatile secretion of gonadotropins is responsible for enlargement and maturation of the gonads and the secretion of the sex hormones
in mid puberty the level of LH become evident even during day time and occur at about 90-120 minutes intervals
It is clear that GnRH is the primary if not the only , hormone responsible for the onset and progression of puberty
The effect of gonadal steroid (testosterone and estradiol )on bone growth and osseous maturation are critical , both aromatase deficiency and estrogen receptor defect result in delayed epiphyseal closure and tall stature in affected boys,
that is mean estrogen rather than androgen are responsible for process of bone maturation and epiphyseal fusion and cessation of growth
Estrogen also mediate the increased production of growth hormone which also responsible with sex hormone for pubertal growth spurt.
The onset of puberty vary and more correlated with osseous than with chronological maturation age
In female, breast bud(thelarche ) is first sign of puberty (10-11yrs) followed by pubic hair (6-12 months later ), the interval to menarche usually 2-2.5 yrs. peak height velocity usually start early 12 yr of age in girls &always precedes menarche (12.75yr)
In male growth of testes(>2.5 cm) and thinning of scrotum is the first sign of puberty followed by pigmentation of scrotum and enlargement of penis .pubic hair then appeared . axillary hair appear in mid puberty in male unlike female growth acceleration appears after puberty . in male growth spurt usually 2 yrs later than in females .and growth may continue beyond 18 yrs .of age
Precocious puberty :
Definition : onset of breast development before age of 8 yrs. In girls & onset of testicular development before age of 9 years in boys .Etiology
Central (gonadotropin dependent true precocity )• Idiopathic
• Brain lesions
• Hypothalamic hamertoma
• Brain tumors
• Prolonged untreated hypothyroidism
Combined peripheral ¢ral
• Treated CAH congenital adrenal hyperplasia
• Familial male precocity
Peripheral (gonadotropin independent precocious puberty)
• Girls
• Isosexual (feminizing )• Ovarian tumors
• Teratoma
• Exogenous androgens
• Mc Cune –Albright syndrome
• Heterosexual (masculinizing)
• CAH
• Exogenous androgen
• Adrenal &ovarian tumors
• Boys
• Isosexual ( Masculinizing)
• CAH
• Leydig cell tumor
• hCG secreting tumors
• teratoma
• exogenous androgen
• heterosexual (feminizing )
• exogenous androgen

4.5 yrs
Bone ae 9 yrsClinical features
• hair underarm &genitalia &for boys on face• acne
• adult body odor
• sexual development ( breast & testes )
• emotional changes
• mood swing
diagnosis
• Hormonal levels(sex hormones )
• Low estradiol level or undetected in girls
• High testosterone (detectable in boys
• Detectable level of LH in girls & boys
• bone age (advance) osseous maturation
• ultrasound for adrenal &uterus size
• MRI or CT scan role out tumors
• Treatment :
GnRH .analogs are effective in arresting pubertal progression in patient with central precocity like decapeptyle (zoladex ) monthly IM injection .Meroxyproesterone acetate some time used to :suppress puberty and arrest menses
• Peripheral precocity :
• Testolactone which inhibit conversion of testosterone to estrogen• Ketoconazole inhibit steroid biosynthesis 200mg tds
• Cyproterone acetate Potent progestin & antiandrogen, inhibit androgens at the receptor level / supress gonadal & adrenal steroidogenesis : antigonadotrophic
• 100 mg/m2,, 2 divided doses
Diabetes mellitus in children (type 1 DM ): L5
Definition :Common chronic metabolic disease characterized by hyperglycemia as a cardinal biochemical features .Etiological classification of DM
1-Type 1 DM (beta cell destruction )• Immune mediate
• Idiopathic
2-type 2DM (insulin resistance &deficiency )
• Typical
• Atypical )
3-enetic defect of beta cell
• Mody (maturity onset diabetes of young )
• Wolfram syndrome (DDMOD diabetes mellitus ,diabetes insipidus optic atrophy &deafness)
• Mitochondrial DNA mutation
• Thiamine responsiveness megaloplastic anemia with diabetes
4-drug or chemical induced• L-asparginase
• Anti rejection .cyclosporine …..
• Phenytoin
• Diazoxide
• Beta blockers
• α interferone
5- disease of exocrine pancreas
• Cystic fibrosis
• Trauma to pancreas
• Pancreatitis
6-infection (CMV ,Rubella .HUS)
7-genetic syndromes
• Prader willi syndrome
• Downs syndrome
• Turner syndrome
• Klinefelter syndrome
8-Gestational diabetes
9-Neonatal diabetes
• Transient
• Permanent
• Type 1 diabetes mellitus :
Insulin dependent or juvenile diabetes characterized by low or absent level of endogenously produced insulin & by dependence on exogenous insulin ; insulin act on movement of glucose into cells to subdue hepatic glucose production & halt movement of fatty acid from periphery to liver
the natural history include 4 stages :
• Preclinical Beta cell autoimmunity with progressive defect of insulin production• Clinical diabetes
• Transient remission ,honeymoon period
• Established diabetes
Its account about 10 % of cases of diabetes affecting >10 million people in the world ,over all incidence of type 1 DM varies from 0.7/100 000 /year [2hin Pakistan to 40/100 000/year In Finland. Girls and boys are equally effected ,no apparent correlation with socioeconomic status .
Age incidence ,2 peak group 5-7 yrs and time of puberty .there familial clustering inT1DM with prevalence in sibling approaching 6% but in general population is 0.4%in US .
HLA system mostly associated with DR3/4-DQ2/8
Natural history of diabetes involve some or all of the following stages :
• Initiation of autoimmunity• Preclinical of autoimmunity with progressive loss of beta cell function
• Onset of clinical disease
• Transient remission
• Established disease
• Development of complications
Influence of high insulin vs low insulin on some metabolic processes in liver, muscle &adipose tissues :
High plasma insulin(postprandial state )
Low plasma insulin (fast state )
Liver
Glucose uptake
Glycogen synthesis .lipogenesis
Absence of ketognesis
Glucose production
Glycogenolysis
Gluconeogenesis
Muscles
Glucose uptake
Glucose oxidation
Glycogen synthesis
Protein synthesisAbsence of glucose uptake
Fatty acid &ketone oxidation
Glycogenlysis
ProteolysisAdipose tissues
Glucose uptake
Lipid synthesis
Absence of glucose uptake
Lipolysis & fatty acid release
Diagnosis
Impaired glucose tolerance
Diabetes mellitus
Fasting glucose 100-125mg/dl
Or
2hrs plasma glucose during OGTT more or equal 140mg/dl but <200mg/dlSymptoms of diabetes +random plasma glucose more or equal to 200mg/dl
Or
Fasting (at least 8 hr )plasma glucose more or equal to 126mg/dl
or
2hrs plasma glucose during OGTT more or equal to 200mg/dl
Or HA1c >or equal to 6.5%
DM should suspected in any child with polyuria & dehydration ,poor weight gain ,hyperglycemia ,glucosuria & ketonuria
Random serum sugar >200mg/dl with typical symptoms with or without ketonuria is diagnostic
• Initial management of type1 DM
Most newly cases of DM are alert and able to eat and drink and can manage with subcutaneous insulin aloneiv fluid required if the child vomiting or dehydrated . intensive educational programme is needed for the parents and child to cover
• Basic understanding of pathophysiology ofDM
• Insulin injection technique &sites
• Diet , regular meal & snacks ,reduced refined CHO .healthy diet no >than 30% fat intake
• 4-match food intake with insulin &exercise
• 5-blood glucose monitoring• 6-recognition and treatment of hypoglycemia
• 7-the psychological impact of lifelong condition with serious short & long term complications
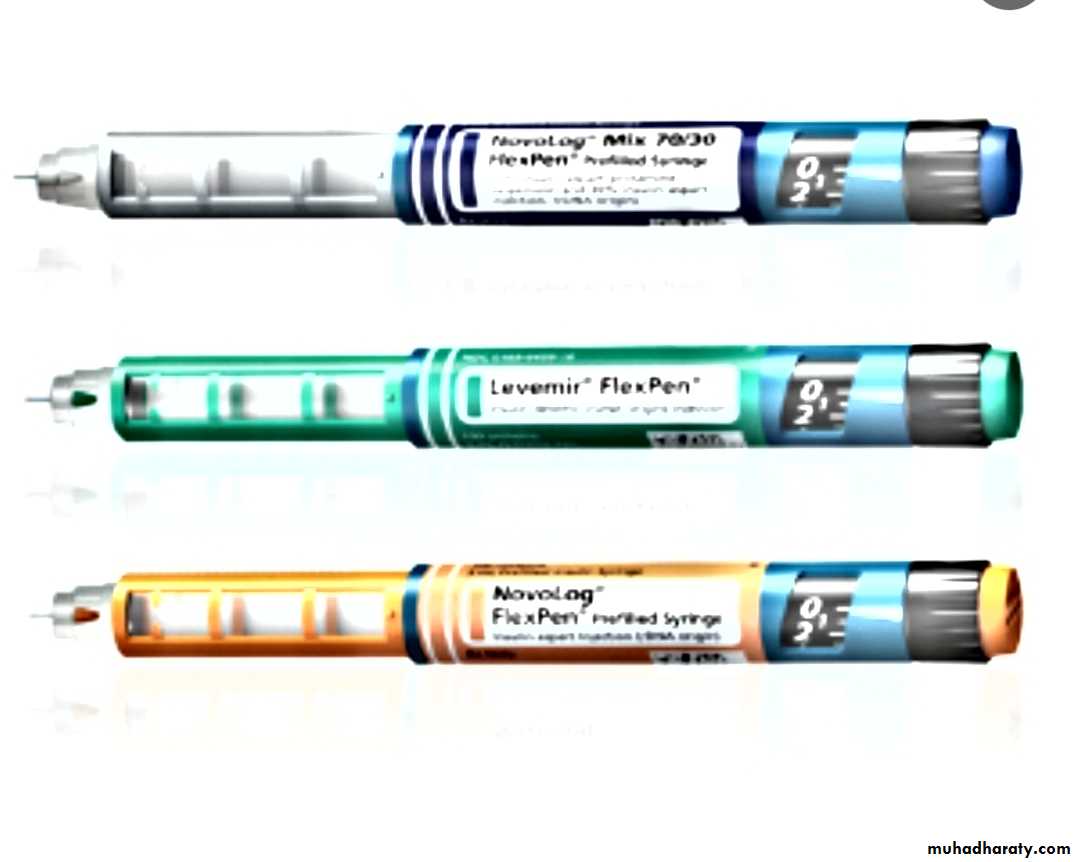
• INSULIN THERAPY
• Most insulin used in Iraq for children is humen with concentration 100U/ml with different types includes• human insulin analogues . rapid acting like lispro &aspart within few minutes
• short acting soluble insulin onset 30-60 min. peak 2-4 hrs. duration up to 8 hrs. given 15-30 min.before meal
• intermediate acting insulin onset 1-2 hrs . peak 4-12 hrs( insulin with protamine )
• mixed short & intermediate 30/70 mixtard• very long acting insulin analogues e.g glargine (lantus)
teenager preferable to use bolus & basal (basal .lantus at night and short acting before each meal )
Factors affecting blood glucose
Increase blood glucoseDecrease blood glucose
• omission of insulin
• refined food
• illness
• menstruation
• growth hormone
• corticosteroids
• sex hormones at puberty
• stress of an operation
• insulin
• exercise
• anxiety (marked )
• some drugs
•
DIET : healthy diet recommended with high complex CHO &relatively low fat content ,diet should be high in fiber
• Diabetic ketoacidosis DKA :
End result of metabolic abnormalities result from sever deficiency of insulin or insulin ineffectiveness. It is occur in 20-40 % of children with newly diagnosed diabetes & DKA consider when serum sugar >300 mg .acidosis .+S/S of DM with ketosis .
Classification of DKA
Normal
Mild
Moderate
Sever
Co2)meq/l venus
20-30
16-20
10-15
<10
pH venous
7.35-7.45
7.25-7.35
7.15-7.25
<7.15
Clinical
No changes
Only fatigue
Kussmaul ,oriented but sleepy
Kussmaul or depress respiration ,sleepy to coma
Treatment
Time
Therapy
Comment
1st hr.
10-20ml/kg IV bolus 0.9%NaClor LR
Insulin drip at 0.05-0.1unit/lg/hr
Volume expansion ,NPO monitor I/O,use flow sheet prepare manitol 1g/kg at bed side if cerebral edema developed
2nd hr. until DKA resolution
0.45% Nacl plus continue insulin drip
20meq/l KPhos &20 meq/l K Ac….5%glucose if blood >250mg/dl
85ml/kg +maintenance -bolus
IV rate =
23 hr
If K <3meq give 0.5-1 meq as oral solution or increase iv K to 80meq/l
Maintenance =100ml/kg for 1st 10 kg+50ml/kg for 2nd 10 kg +25 ml/kg for remaining kg
Initial bolus fluid consider part of total fluid allowed & subtracted before calculating iv rate
Sample calculating for 30 kg child
1st hr 300 ml iv bolus 0.9% NaCl or LR2nd & subsequent hrs= (85ml × 30)+1750ml -300ml =
23hr
= 175 ml
hr
I/O input output, NPO nothing by mouth ,KAc potasium acetate ,kphos=potasium phosphate ….LR lactated Ringer..NaCl sodium chlodide
• Long term management of DM
Aim of long term management :• normal growth & development
• normal home & school life as possible
• good diabetic control through knowledge & technique
• encourage children to be self- reliant
• avoidance of hypoglycemia
Assesment of a child with DM summary
Assessment of diabetic control :• any episode of hypoglycemia
• school absence
• interference with normal life
• HbA1C result
• Insulin regimen ---appropriate
• Diet –healthy diet
General overview :
• Normal growth and pubertal development , ovoid obesity
• Blood pressure checking
• Renal for microalbuminuria
• Eye ---cataract
• Feet –care
• Screening for celiac and thyroid disease
Knowledge &psychological aspects
• Good understanding of diabetes• Becoming self-reliant but appropriate supervision at home
• Taking exercise ,sport?
• Smoking ???
• Is hypo treatment readily available ?
•

Ddiabetic lipohypertrophy

What shall I eat?
Breakfast• a bowl of cereal with semi-skimmed milk
• wholegrain toast with spread and/or jam
• yogurt and fruit
• a cereal bar and a glass of milk.
Lunch
• a chicken or ham salad sandwich...• a small pasta salad...
• soup and a roll...
...with a piece of fruit and a yogurt.
Dinner
• salad• roast chicken with potatoes and
• vegetables
• beef stir fry, vegetables and rice
• chicken tortillas and salad
• salmon and noodles
• curry and rice
• What sort of snacks do I need to eat?
.
The healthiest snack choice is definitely a piece of fruit, but rice cakes, crackers, a couple of biscuits, a small bag of crisps, a cereal bar, or a yogurt are good snack choices too.
• Foods to avoid for a type 1 diabetes diet include
sodas (both diet and regular),simple carbohydrates - processed/refined sugars (white bread, pastries, chips, cookies, pastas),
trans fats (anything with the word hydrogenated on the label), and high-fat animal products.