Hypocalcaemia
Aetiology
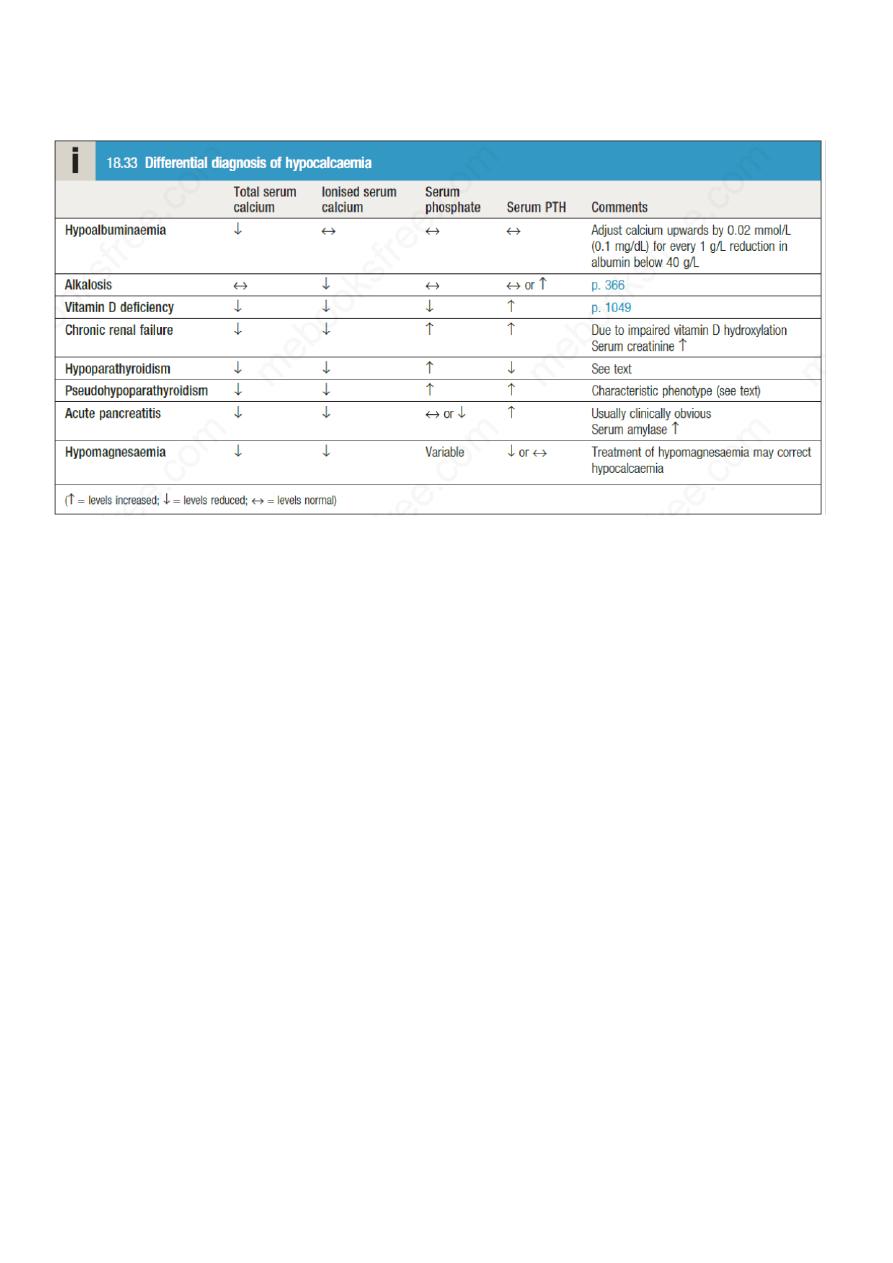
Hypocalcaemia is much less common than hypercalcaemia. The most common cause of
hypocalcaemia is a low serum albumin with normal ionised calcium concentration.
Conversely, ionised calcium may be low in the face of normal total serum calcium in patients
with alkalosis: for example, as a result of hyperventilation.
Hypocalcaemia may also develop as a result of magnesium depletion and should be
considered in patients with malabsorption, those on diuretic or proton pump inhibitor
therapy, and/or those with a history of alcohol excess. Magnesium deficiency causes
hypocalcaemia by impairing the ability of the parathyroid glands to secrete PTH (resulting in
PTH concentrations that are low or inappropriately in the reference range) and may also
impair the actions of PTH on bone and kidney.
Clinical assessment
Mild hypocalcaemia is often asymptomatic but, with more profound reductions in serum
calcium, tetany can occur. This is characterised by muscle spasms due to increased
excitability of peripheral nerves.
Children are more liable to develop tetany than adults and present with a characteristic triad
of carpopedal spasm, stridor and convulsions, although one or more of these may be found
independently of the others. In carpopedal spasm, the hands adopt a characteristic position
with flexion of the metacarpophalangeal joints of the fingers and adduction of the thumb
(‘main d’accoucheur’). Pedal spasm can also occur but is less frequent. Stridor is caused by
spasm of the glottis.

Adults can also develop carpopedal spasm in association with tingling of the hands and feet
and around the mouth, but stridor and fits are rare.
Latent tetany may be detected by eliciting Trousseau’s sign: inflation of a
sphygmomanometer cuff on the upper arm to more than the systolic blood pressure is
followed by carpal spasm within 3 minutes. Less specific is Chvostek’s sign, in which
tapping over the branches of the facial nerve as they emerge from the parotid gland produces
twitching of the facial muscles.
Hypocalcaemia can cause papilloedema and prolongation of the ECG QT interval, which
may predispose to ventricular arrhythmias.
Prolonged hypocalcaemia and hyperphosphataemia (as in hypoparathyroidism) may cause
calcification of the basal ganglia, grand mal epilepsy, psychosis and cataracts.
Hypocalcaemia associated with hypophosphataemia, as in vitamin D deficiency, causes
rickets in children and osteomalacia in adults .
Management
Emergency management of hypocalcaemia associated with tetany
is
Hypoparathyroidism
The most common cause of hypoparathyroidism is damage to the parathyroid glands (or their
blood supply) during thyroid surgery. Rarely, hypoparathyroidism can occur as a result of
infiltration of the glands with iron in haemochromatosis or copper in Wilson’s disease .
There are a number of rare congenital or inherited forms of hypoparathyroidism. One form is
associated with autoimmune polyendocrine syndrome type 1 and another with DiGeorge
syndrome . Autosomal dominant hypoparathyroidism is the mirror image of FHH , in that an
activating mutation in the calcium-sensing receptor reduces PTH levels, resulting in
hypocalcaemia and hypercalciuria.

Pseudohypoparathyroidism
In this disorder, the individual is functionally hypoparathyroid but, instead of PTH
deficiency, there is tissue resistance to the effects of PTH, such that PTH concentrations are
markedly elevated. The PTH receptor itself is normal but the downstream signalling
pathways are defective due to mutations that affect GNAS1. There are several subtypes but
the most common (pseudohypoparathyroidism type 1a) is characterised by hypocalcaemia
and hyperphosphataemia, in association with short stature, short fourth metacarpals and
metatarsals, rounded face, obesity and subcutaneous calcification; these features are
collectively referred to as Albright’s hereditary osteodystrophy (AHO). Type 1a
pseudohypoparathyroidism occurs only when the GNAS1 mutation is inherited on the
maternal chromosome (maternal imprinting).
The term pseudopseudohypoparathyroidism is used to describe patients who have clinical
features of AHO but normal serum calcium and PTH concentrations; it occurs when the
GNAS1 mutation is inherited on the paternal chromosome. The inheritance of these
disorders is an example of genetic imprinting.
The difference in clinical features occurs as a result of the fact that renal cells exclusively
express the maternal GNAS1 allele, whereas both maternal and paternal alleles are expressed
in other cell types; this explains why maternal inheritance is associated with hypocalcaemia
and resistance to PTH (which regulates serum calcium and phosphate levels largely by an
effect on the renal tubule), and why paternal inheritance is associated with skeletal and other
abnormalities in the absence of hypocalcaemia and raised PTH values.
Management of hypoparathyroidism
Persistent hypoparathyroidism and pseudohypoparathyroidism are treated with oral calcium
salts and vitamin D analogues,
either 1α-hydroxycholecalciferol (alfacalcidol) or 1,25-
dihydroxycholecalciferol (calcitriol). This therapy needs careful monitoring because of the
risks of iatrogenic hypercalcaemia, hypercalciuria and nephrocalcinosis. Recombinant PTH
is available as subcutaneous injection therapy for osteoporosis and, although not currently
licensed, has been used in hypoparathyroidism (but not in pseudohypoparathyroidism). It is
much more expensive than calcium and vitamin D analogue therapy but has the advantage
that it is less likely to cause hypercalciuria. There is no specific treatment for AHO other
than to try to maintain calcium levels within the reference range using active vitamin D
metabolites.