Definitions
Pharmacology: The word pharmacology is derived from the Greek: pharmakon meaning drug or poison and logos: word or discourse. It is the study of interaction of drugs with living organisms. It covers the use of drugs in the prevention (prophylaxis) and treatment of diseases.It also includes history, source, physicochemical properties, dosage forms, methods of administration, absorption, distribution mechanism of action, biotransformation, excretion, clinical uses and adverse effects of drugs.
Clinical Pharmacology: It evaluate the pharmacological action of drug preferred route of administration and safe dosage range in human by clinical trails.
Drugs: Drugs are chemicals that alter functions of living organisms. Drugs are generally given for the diagnosis, prevention, control or cure of disease.
Pharmacy: It is the science of identification, selection, preservation, standardization,
compounding and dispensing of medical substances.
Toxicology: It is the science of poisons. It is the area of pharmacology concerned with the undesirable effects of chemicals and biologicals on cellular functions.
Poisons are substances that cause harmful, dangerous or fatal symptoms in living substances. Many drugs in larger doses may act as poisons.
Origin and sources of drugs:1. Minerals: Liquid paraffin, magnesium sulfate, magnesium trisilicate, kaolin, etc.2. Animals: Insulin, thyroid extract, and antitoxin sera, etc.3. Plants: Morphine, digoxin, atropine, castor oil, etc.4. Synthetic source: Aspirin, sulphonamides, paracetamol, zidovudine, etc.5. Micro organisms: Penicillin, streptomycin and many other antibiotics.6. Genetic engineering: Human insulin, human growth hormone etc.
Drugs formulations:1- Injections ( Ampoules, vials)2. Liquid ( SYRUP, SUSPENSION)3. Tablets, capsule4. Suppositories, pessaries5. Sprays and inhalants6. Ointments and creams7. Trans dermal patches8. Drug implants9. Micro and nanoparticles10. Targeted drug delivery








Drug Nomenclature: Every drug has at least three names - a chemical name (e.g. 6-dimethylamino-4,4-diphenyl-3-heptanone hydrochloride),- a generic name (e.g., methadone hydrochloride) and -a proprietary (or trade) name (e.g., Dolophine). Chemical and generic names are written in lower case whereas trade names are capitalized. If drug is marketed by more than one pharmaceutical company, then the same drug may have several trade names but only one official generic name.



Pharmacopoeia: An official code containing a selected list of the established drugs and medical preparations with descriptions of their source, chemical nature, physical properties and tests for their identity, purity and potency, route of administration, dynamic effects and kinetic parameters. It also included: Indications (under what circumstances is the drug used). Drug action (what clinical effect does the drug have. Adverse effects (are there clinically relevant side effects of the drug). Contraindications (are there circumstances in which the drug should not be administered to certain patient populations e.g: the elderly, those with renal insufficiency, pregnant women etc).
Such as [British Pharmacopoeia (B.P),
United States Pharmacopoeia (USP)…etc



Drugs administration:
A. Oral:Oral route is the most common route of drug administration. It is mostly used for the neutral drugs. It may be in the form of tablets, capsules, syrup, emulsions or powders.
Advantages:
It is convenient, It is the cheapest available route, It is easy to use, It is safe.
Disadvantages:
Not suitable for all drugs, Some drugs are destroyed by gastric juices. eg: insulin., Absorption is slow, so is not preferred during emergency., May cause gastric irritation, May objectionable in taste., May cause discoloration of teeth e.g. tetracyclines below 14 cause brown discoloration so are not advisable during pregnancy., First pass effect : hepatic metabolism of drug when absorbed and delivered through portal blood. Greater the first pass effect, less amounts of the drug reach the systemic circulation
B. Parenteral Routes 1-Intravenous injections:Intravenous injections might be applied to the cubital, basilic and cephalic veins.Advantages: Immediate action takes place , This route is preferred in emergency situations, This route is preferred for unconscious patients., Titration of dose is possible., Large volume of fluids might be injected by this route, Diluted irritant might be injected. , Absorption is not required.,No first pass effect takes place., Blood plasma or fluids might be injected.Disadvantages:There is no retreat. , This method is more risky. , suitable for insoluble prSepsis-Infection might occur. , Phlebitis(Inflammation of the blood vessel) might occur., Infiltration of surrounding tissues might result., This method is not suitable for oily preparations. ,This method is not eparations2-Intramuscular injection: Drugs may be injected into the arm (deltoid), thigh (vastus lateralis) or buttocks (gluteus maximus). The volume used is 3 ml.Advantages:Absorption is rapid than subcutaneous route., Oily preparations can be used. ,Slow releasing drugs can be given by this route.DisadvantagesSome time painful, Not suitable for irritant drugs., Using this route might cause nerve or vein damage.3. Subcutaneous injection: Some drugs, notably insulin, are routinely administered SC. Drug absorption is generally slower SC than IM, due to poorer vascularity. Absorption can be facilitated by heat, massage or vasodilators. It can be slowed by co-administration of vasoconstrictors, a practice commonly used to prolong the local action of local anesthetics. As above, arm > thigh.4- Intraperitoneal injection: is the injection of a substance into the peritoneum (body cavity). In general , it is rarely used in human (fearing from peritonitis), it is preferred when large amounts of replacement fluids are needed, or when low blood pressure or other problems prevent the use of a suitable blood vessel for intravenous injection5- Intradermal injection: is injection of drug in the dermis, it is one of the routes used for administration vaccins and for the test of allergy.
C. Topical application to the skin and mucus membranes: a. Eye For desired local effects. b. Intravaginal for infections or contraceptives. c. Intranasal for alleviation of local symptoms. d. Respiratory mucus membrane: inhalors e. Skin Topical drug administration for skin disorders minimizes systemic exposure. However, systemic absorption does occur and varies with the area, site, drug, and state of the skin. f-Sublingual (buccal) Certain drugs are best given beneath the tongue or retained in the cheek pouch and are absorbed from these regions into the local circulation. These vascular areas are ideal for lipid-soluble drugs that would be metabolized in the gut or liver, since the blood vessels in the mouth bypass the liver (do not undergo first pass liver metabolism), and drain directly into the systemic circulation. This route is usually reserved for nitrates and certain hormones.g. Rectal: The administration of suppositories is usually reserved for situations in which oral administration is difficult. This route is more frequently used in small children. The rectum is devoid of villi, thus absorption is often slow.h. Inhalation Volatile anesthetics, as well as many drugs which affect pulmonary function, are administered as aerosols. Other obvious examples include nicotine and tetrahydrocannabinol (THC), which are absorbed following inhalation of tobacco or marijuana smoke. The large alveolar area and blood supply lead to rapid absorption into the blood. Drugs administered via this route are not subject to first-pass liver metabolism.
D- Administration of drugs by special routes: -Intrathecal and epidural injection: drugs are given via lumbar puncture and injection into the subarachnoid space such as using life-threatening, antibiotics, antifungals and anticancer and also in spinal anesthesia to by bass blood-brain barrier -Intra-arterial injection: Used in certain special situations, notably with anticancer drugs, in an effort to deliver a high concentration of drug to a particular tissue. Typically, the injected artery leads directly to the target organ.-Intra-articular: Injected directly into a joint e.g. hydrocortisone
Pharmacodynamics: what the drug does to the body : The study of the biological and therapeutic effects of drugs. Pharmacokinetics: what the body does to the drug: Study of the absorption, distribution metabolism and excretion
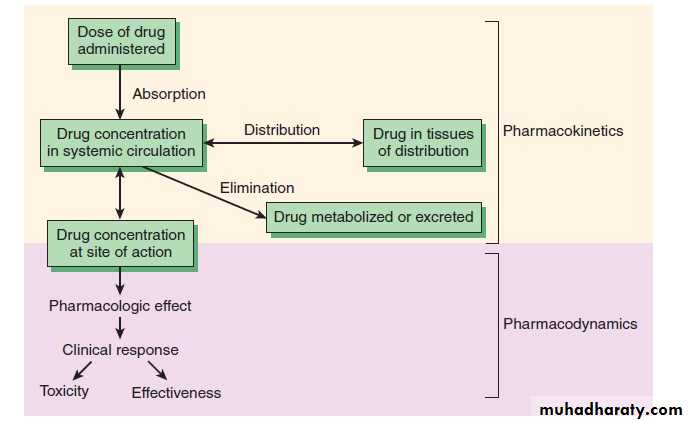
Pharmacokinetics: Absorption
Absorption is movement of the drug from its site of administration into the circulation. Not only the fraction of the administered dose that gets absorbed, but also the rate of absorption is important. Except when given i.v., the drug has to cross biological membranes; absorption is governed by the above described principles. Other factors affecting absorption are:Biological Membrane:
Biological membranes consist of a lipid bilayer separating different compartments, with protein molecules acting as enzymes, channels or carrier proteins.
Drugs have to cross the biological membranes to get absorbed.
Drugs are transported across the membranes by:
(a) Passive: Simple diffusion and filtration:(b) Specialized transport ( carrier transport):
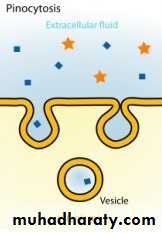
(c) Pinocytosis:
(a) Passive: Simple diffusion and filtration:
1-Simple diffusion
The drug diffuses across the membrane in the direction of its concentration gradient, the membrane playing no active role in the process. This is the most important mechanism for majority of drugs. Depend on: concentration gradient, lipid solubility and molecular weight.
Influence of pH Most drugs are weak electrolytes, i.e. their ionization is pH dependent (contrast strong electrolytes that are nearly completely ionized at acidic as well as alkaline pH). The ionization of a weak acid HA is given by the equation: I pH= pKa + log ------- …………………….. (1) I isonized, UI unionized UIpKa is the negative logarithm of acidic dissociation constant of the weak electrolyte. While for weak base UI pH= pKa + log ------- …………………….. (2) I (a) Acidic drugs, e.g. aspirin (pKa 3.5) are largely unionized at acid gastric pH and are absorbed from stomach, while bases, e.g. atropine (pKa 10) are largely ionized and are absorbed only when they reach the intestines.(b) The unionized form of acidic drugs which crosses the surface membrane of gastric mucosal cell, reverts to the ionized form within the cell (pH 7.0) and then only slowly passes to the extracellular fluid. This is called ion trapping. (c) Basic drugs attain higher concentration intracellularly (pH 7.0 vs 7.4 of plasma).(d) Acidic drugs are ionized more in alkaline urine-do not back diffuse in the kidney tubules and are excreted faster. Accordingly, basic drugs are excreted faster if urine is acidified.To enhance acidic drug excretion alkalinize urine, and to enhance alkaline drug excretion acidify urine.
2-FiltrationFiltration is passage of drugs through aqueous pores in the membrane or through paracellular spaces. This can be accelerated if hydrodynamic flow of the solvent is occurring under hydrostatic or osmotic pressure gradient, e.g. across most capillaries including glomeruli. Lipid-insoluble drugs cross biological membranes by filtration if their molecular size is smaller than the diameter of the pores. Majority of cells (intestinal mucosa, RBC, etc.) have very small pores (4 A) and drugs with MW > 100 or 200 are not able to penetrate. However, capillaries (except those in brain) have large paracellular spaces (40 A) and most drugs can filter through these. As such, diffusion of drugs across capillaries is dependent on rate of blood flow through them rather than on lipid solubility of the drug or pH of the medium.
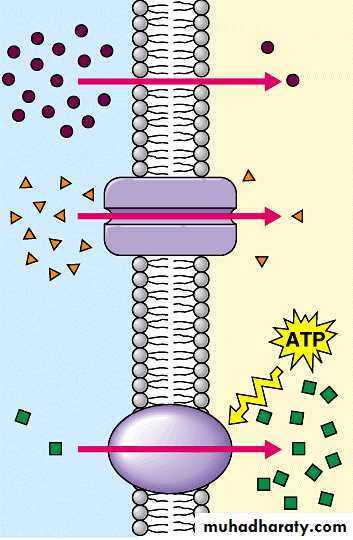
(b) Specialized transport ( carrier transport): 1. Facilitated transport The transporter, belonging to the super-family of solute carrier (SLC) transporters, operates passively without needing energy and translocates the substrate in the direction of its electrochemical gradient, i.e. from higher to lower concentration . lt mearly facilitates permeation of a poorly diffusible substrate, e.g. the entry of glucose into muscle and fat cells by GLUT 4. 2. Active transport It requires energy, is inhibited by metabolic poisons, and transports the solute against its electrochemical gradient (low to high), resulting in selective accumulation of the substance on one side of the membrane. It is saturable and follows the Michaelis-Menten kinetics. The maximal rate of transport is dependent on the density of the transporter in a particular membrane, and its rate constant (Km), i.e. the substrate concentration at which rate of transport is half maximal, is governed by its affinity for the substrate. Genetic polymorphismcan alter both the density and affinity of the transporter protein for different substrates and thus affect the pharmacokinetics of drugs. Moreover, tissue specific drug distribution can occur due to the presence of specific transporters in certain cells. (c) Pinocytosis:Large macromolecules (e.g., proteins, viruses, lipoprotein particles) require more complex mechanisms to traverse membranes, and are transported into and out of cells selectively via endocytosis and exocytosis (secretion). Interestingly, endocytosis and exocytosis are not only important for the import/export of large molecules. Often, essential small molecules that are hydrophobic or toxic (e.g., iron) travel through the bloodstream bound to proteins, which enter and exit cells via these mechanisms. Receptor-mediated endocytosis will be discussed in more detail in the Organs block.
Factors Related to Drugs:1. Lipid water solubilityLipid water solubility coefficient is the ratio of dissolution of drug in lipid as compared to water. Greater the lipid water solubility coefficient, more is the lipid solubility of the drug and greater is the absorption. Less the coefficient, less is the lipid solubility and less is the absorption.Water film exists on the membranes so part of the drugs must be water soluble to cross this water film. Drugs with benzene ring, hydrocarbon chain, steroid nucleus and halogen groups in their structures are lipid soluble. 2. Molecular sizeSmaller the molecular size of the drug, rapid is the absorption. Those with a large molecular size undergo active transport or endocytosis, while those with smaller molecular sizes utilize aqueous diffusion or lipid channels. 3. Particle sizeLarger particle size, slower will be the diffusion and absorption and vice versa. 4. Degree of IonizationDrugs are either acidic or basic and are present in ionized or unionized form. In the body, the ratio of the ionized and unionized forms depend on the pH of the medium. Acidic drugs are unionized in the acidic medium and basic drugs are unionized in the basic medium. Acidic drugs are better absorbed from the acidic compartment and basic drugs from alkaline compartment. 5. Physical FormsDrugs may exist as solids, liquids or gases. Gases are rapidly absorbed than the liquids, while the liquids are rapidly absorbed than the solids. Volatile gases used in general anesthesia are quickly absorbed through the pulmonary route. Syrup or suspension form are rapidly absorbed than the tablets or capsules.
Factors affected absorption:
6. Chemical NatureChemical nature is responsible for the selection of the route of administration of drug. Drugs that cannot be absorbed from GIT are given by the parenteral route, eg: heparin, insulin and benzyl penicillin is degraded in the GIT, so is given parenterally. 7. Dosage FormsDosage forms affect the rate and extent of absorption. A drug can be given in the form of tablets, capsules or transdermal forms. Examples nitroglycerin which when given by sublingual route, disintegrates rapidly but stays for a shorter duration. When it is given orally, it disintegrates slowly and stays for longer duration. When given by transdermal route, the drug can cover an even longer duration. IM injections may be aqueous or oily. Oily was slowly absorbed.a. Disintegration:Disintegration is the breaking up of the dosage form into smaller particles. When rapid is the disintegration, rapid will be the absorption.b. Dissolution:After disintegration, the drug dissolves in the gastric juices, which is called dissolution. It is only then that the drug can be absorbed. When these two processes occur rapidly, the rate of absorption increases. 8. FormulationWhen the drugs are formed, apart from the active form some inert substances are included. These are the diluents, excipients and the binders. Normally they are inert, but if they interact, they can change the bioavailability. 9. ConcentrationThe higher the concentration more flux occurs across the membrane. The rate is less affected than the extent of absorption.Factors Related to Body
1. Area of Absorptive Surface
Area of absorptive surface affects oral as well as other routes. Most of the drugs are given orally because of the large area of absorptive surface, so that greater absorption occurs. Similarly, when the topically acting drugs are applied on a large surface area, they are better absorbed.
2. Vascularity
More the vascularity, more is the rate and extent of absorption and vice versa. In shock, the blood flow to the peripheries is decreased, so absorption in those areas is diminished, intravenous route is preferred.
Vasoconstrictors decrease the blood supply of an area, thus are useful to restrict the local anesthesias so that they remain for a longer duration. Their wash away as well as their toxic effects are decreased in this way.
Massage in intramuscular injections improves vascular supply to enhance absorption.
3. pH
Acidic pH favors acidic drug absorption while basic pH is better for basic drugs.
4. Presence of other Substances
Foods or drugs may interact with the drugs to alter their rate of absorption. Especially for the drugs given orally, food can increase or decrease the absorption.
Antihyperlipidemic drugs like the statins are better absorbed when taken with the food.
Iron when given with milk has decreased absorption.
Vitamin C enhances the absorption of iron.
Milk decreases the absorption of tetracyclines. Calcium salts when given with iron salts or tetracyclines interfere with their absorption
Aspirin is given with food while antibiotics are given in empty stomach. Liquid paraffin may affect drug absorption. Some acidic drugs bind with cholestyramine to from a complex which is not absorbed in GIT.
5. GI MobilityGI mobility must be optimal for absorption of oral drugs. It should be neither increased nor decreased which may affect the rate or extent of absorption.Different diseases or drugs may alter the mobility. Diarrhea causes rapid peristalsis, decreasing contact time and thus the extent of absorption is affected more. Constipation affects disintegration and dissolution so decreases motility. 6. Functional Integrity of Absorptive SurfaceFlattening and edema of mucosa decreaes absorption. Burns and wounds in the skin affects the absorption of topical drugs.Parasympathomimetic drugs can decrease drug absorption and parasympatholytic drugs can increase absorption. Prokinetic drugs prevents vomiting and accelerates gastric emptying. It increases gastric emptying increasing drug absorption. 7. Diseasesa. Diarrhea and vomiting: Decreases absorption.b. Malabsorptive syndrome : Decreases absorptionc. Achlorhydria: Decrease stomach acid secretion decrease absorption of acidic drugs. Methods for Delaying Absorption1. Vasoconstrictors: Vasoconstrictors decrease absorption.2. Formulation: Slow releasing (SR) preparations and Slowly absorbed oily preparations.
Bioavailability of drugsBioavailability is the fraction of the dose of a drug contained in any dosage form that reaches the systemic circulation in unchanged or active form administered through any routeBioavailability = AUC (oral)/ AUC (I/V) x 100 (AUC is the area under the curve)When the drug is given orally, only part of the administered dose appears in the plasma. The area under the curve (AUC) can be measured by plotting plasma concentrations of the drug versus time.Drugs injected using intravenous route of administration have 100% bioavailability, while others have much less bioavailability, because:Not all the drug adsorbed Metabolism of the drug might occur before reaching circulation
Distribution of drugs:Penetration of a drug to the sites of action through the walls of blood vessels from the administered site after absorption is called drug distribution. Drugs distribute through various body fluid compartments.1-Plasma compartment: If a drug has a very large molecular weight or bindsextensively to plasma proteins, it is trapped within the plasma (vascular)compartment.2- Extracellular fluid: If a drug has a low molecular weight but is hydrophilic,it can move through the endothelium of the capillaries into the interstitial fluid.3- Total body water: If a drug has a low molecular weight and is hydrophobic,not only can it move into the interstitium through the slit junctions, but it canalso move through the cell membranes into the intracellular fluid.4- Other sites: In pregnancy, the fetus may take up drugs and thus increase the volume of distribution.
Volume of DistributionVolume of distribution (Vd) is the ratio between the amount of drug in body (dose given) (D) and the concentration of the drug (C) measured in blood or plasma. D Vd = ------ C Factors affected volume of distribution:- Lipid: water partition coefficient of the drug- pKa value of the drug- Degree of plasma protein binding- Affinity for different tissues- Fat: lean body mass ratio, which can vary , with age, sex, obesity, etc.- Diseases like CHF, uremia, cirrhosis
Factors affected distribution of drugs:
1. Protein binding of drug:Absorbed drug may become reversibly bound to plasma proteins (mainly albumin and to a less degree globulin). The active concentration of the drug is that part which is not bound, because only this fraction is free to leave the plasma. (a) Free drug leave plasma to site of action (b) binding of drugs to plasma proteins assists absorption (c) protein binding acts as a temporary store of a drug and tends to prevent large fluctuations in concentration of unbound drug in the body fluids (d) protein binding reduces diffusion of drug into the cell and there by delays its metabolic degradation e.g. high protein bound drug like phenylbutazone is long acting. Low protein bound drug like thiopental sodium is short acting.
2. Plasma concentration of drug (PC):
It represents the drug that is bound to the plasma proteins (albumins and globulins) and the drug in free form. It is the free form of drug that is distributed to the tissues and fluids and takes part in producing pharmacological effects.
The concentration of free drug in plasma does not always remain in the same level e.g.
i) After I.V. administration plasma concentration falls sharply
ii) After oral administration plasma concentration rises and falls gradually.
iii) After sublingual administration plasma concentration rise sharply and falls gradually.
3. Clearance:
Volume of plasma cleared off the drug by metabolism and excretion per unit time.
Protein binding reduces the amount of drug available for filtration at the glomeruli and hence delays the excretion, thus the protein binding reduces the clearance.
4. Physiological barriers to distribution: There are some specialized barriers in the body due to which the drug will not be distributed uniformly in all the tissues. These barriers are:a) Blood brain barrier (BBB) Blood-brain barrier: To enter the brain, drugs must pass through the endothelial cells of the capillaries of the CNS or be actively transported. By contrast, lipid-soluble drugs readily penetrate into the CNS because they can dissolve in the membrane of the endothelial cells. Ionized or polar drugs generally fail to enter the CNS because they are unable to pass the tight junctions of the endothelial cells = blood-brain barrier (BBB).b) Placental barrier: which allows non-ionized drugs with high lipid/water partitioncoefficient by a process of simple diffusion to the foetus e.g. alcohol, morphine. 5. Affinity of drugs to certain organs: The concentration of a drug in certain tissues after a single dose may persist even when its plasma concentration is reduced to low. Thus the hepatic concentration of mepacrine is more than 200 times that of plasma level. Their concentration may reach a very high level on chronic administration. Iodine is similarly concentrated in the thyroid tissue.
Half-Life (t1/2) defined as the time it takes for blood level of a drug to fall to one-half (50%) of the level measured at some prior time Half life of a drug is directly proportional to the volume of the distribution and inversely proportional to the clearance. Half life = 0.693 x Vd/ total body clearance
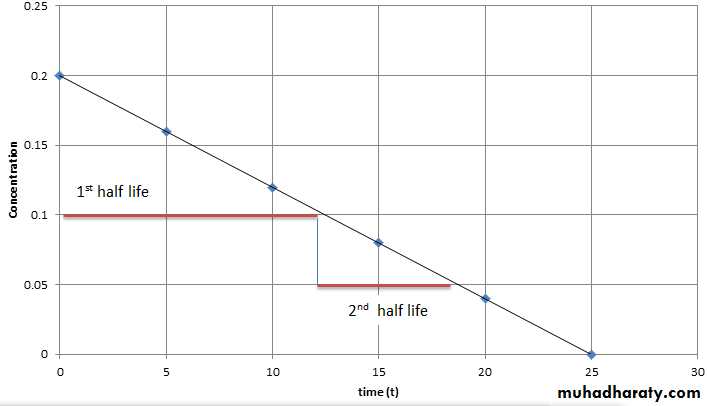
Steady State Concentration A steady-state plasma concentration of a drug occurs when the rate of drug elimination is equal to the rate of administration. Any change in drug dose and interval will change the steady state level
Drug Metabolism (Biotransformation): Sites:Liver is the main organ. Others include GIT, lungs, kidneys, skin, adrenals and blood (plasma).Types of Biotransformation:1. Enzymatic : a. Microsomal and b. Non-microsomal 2. Non-enzymaticNon Enzymatic Elimination:Spontaneous, non-catalyzed and non-enzymatic type of biotransformation for highly active, unstable compounds taking place at physiological pH. Very few drugs undergo non-enzymatic elimination. Enzymatic Elimination:Biotransformation taking place due to different enzymes present in the body/cells is known as enzymatic elimination.
Enzymatic Elimination:Biotransformation taking place due to different enzymes present in the body/cells is known as enzymatic elimination. Non-Microsomal Biotransformation:The type of biotransformation in which the enzymes taking part are soluble and present within the mitochondria. eg: Xanthine oxidase converting hypoxanthine into xanthine. Monoamine oxidase involved in non-microsomal metabolism of catecholamines and noradrenaline. Alcohol dehydrogenase responsible for metabolism of ethanol into acetaldehyde Microsomal Biotransformation:Enzymes responsible are present within the lipophilic membranes of endoplasmic reticulum. Enzymes isolated from ER possess enzymatic activity termed as microsomal mixed function oxidase system.Components:Cytochrome P450 (ferric, ferrous forms)NADPH (flavoprotein)
Biochemical Reactions:Phase I reactionsPhase II reactionsPhase I reactions:Phase I reactions are non-synthetic chemical reactions occurring mainly within the ER. The parent drug is converted into more soluble agents by introduction or unmasking of functional component.Phase I reactions include: -Oxidation(Hydroxylation, Dealkylation, O-Dealkylation, N-oxidation, Sulfoxidation, Deamination, Desulfuration)- Reduction ( eg: Chloramphenicol).- Hydrolysis ( eg: Esters: and Amides). Consequences of Phase I reactions:1. Active drug may be converted into inactive metabolite ( mainly). 2. Active drug may be converted into more active metabolite. Eg: morphine is converted into more active metabolite.3. Prodrug may be converted into active metabolite4. Active drug may be converted into toxic metabolite e.g. halothane used in general anesthesia, is converted into trifluoroacetylated compound or trifluoroacetic acid, leading to hepatic toxicity.
Phase II reactions ( conjugation Reaction):Phase II is a conjugation reaction followed the phase I reaction, the products of phase I were conjugated with endogenous substrates like glucuronic acid, sulphuric acid, amino acid or acetic acid, yielding more excretable drug conjugates which are excreted by the kidneys. Phase II reactions lead to :-Usually inactivation of drug- Production of water soluble metabolites, which is the main aim of biotransformation.- Usually detoxification reaction , with some exception ( producing of toxic conjugates eg: methanol is converted into formaldehyde, which is toxic)
Factors Affecting BiotransformationBiotransformation is significantly affected by a number of factors, these include:1. Enzyme Induction:When drugs given over prolonged period of time, upregulation of enzymes takes place. The rate of metabolism increases as enzyme induction takes place. The drugs which bring about these changes are known as enzyme inducers. Some examples include anticonvulsants like phenytoin, carbomycin, chronic alcoholism. various sedatives, hypnotics and tranquilizers. Consequencs of Microsomal Enzyme InductionDecreased intensity and duration of action of drugs e.g. failure of contraceptivesIncreased intensity of action of drugs activated by metabolism. E.g. acute paracetamol toxicity is due to one of its metabolites.If drug induces its own metabolism e.g. cicobarbitone it develops tolerance so effects are not produced.Precipitation of acute intermittent porphyria. Enzyme induction might increase porphyrin synthesis.Intermittent use of an inducer might interfere adjustment of dose of another drug e.g. oral anti coagulants, oral hypoglycemic, antiepileptics and antihypertensives.Auto induction: The phenomenon in which a drug induces metabolism of other drugs as well as its own. eg. carbamazepine-antiepileptic.
2. Enzyme InhibitionThe process in which drug metabolizing capacity of cytochrome is decreased is known as enzyme inhibition. The rate of metabolism is decreased. Drugs bringing about these changes are known as enzyme inhibitors. Examples include ketoconazole- antifungal drug, cimetidine and verapamil- calcium channel blocker.3. Presystemic Metabolism/First pass effect/Route of AdministrationDrugs following first pass metabolism have decreased bioavailability. Most of the drugs are metabolized within the liver. Changing the route of administration might change the first pass metabolism.Propanolol is 80% metabolized before reaching systemic circulation.4. Genetic Variations:Inter individual variations might occur, as drugs behave differently in different individuals due to genetic variations resulting in absent or malformed enzymes due to absent or malformed genes. eg: fast acetylators and slow acetylatorst. 5. Environmental factors:Cigarette smokers, Chronic alcoholism and pesticides might lead to enzyme In hot and humid climate biotransformation is decreased and vice versa. At high altitude, decreased biotransformation occurs due to decreased oxygen leading to decreased oxidation of drugs.
6. AgeExtreme age groups (infants and geriatrics) associated with slow metabolic process. Chloramphenicol (antimicrobial drug) when administered in infant, does not have great efficacy. Toxic effects in the form of grey baby syndrome might occur. The baby may be cyanosed, hypothermic, flaccid and grey in color. Shock and even death might occur if toxic levels get accumulated.Diazepam (sedative hypnotic) may result in floppy baby syndrome in which flaccidity of the baby is seen.In elderly, most metabolic processes were slow down because of decreased liver functions and decreased blood flow through the liver. The drug doses should be decreased in the elderly.7. SexMale have a higher BMR as compared to the females, thus can metabolize drugs more efficiently, e.g. salicylates. Females, during pregnancy, have an increased rate of metabolism. Thus, the drug dose has to be increased. After the pregnancy is over, the dosage is decreased back to normal levels. Example includes phenytoin, whose dose has to be increased during pregnancy (specially second and third trimester).8. Drug-Drug Interaction9. Nutrition 10. Pathological Conditions ( hepatic , cardiovascular, hypothyroidism)11. Circadian rhythm
Elimination of drugThe irreversible shift of a drug from one part of the body to other part of system is known as elimination. It is from the more perfused parts to the lesser perfused parts. Elimination may be by:BiotransformationExcretionElimination Kinetics First-order kinetics (the most common type) - a constant fraction of drug is eliminated per unit time - the amount of drug eliminated is based on the concentration of drug present - this relationship is linear and predictable Zero-order kinetics (less common, associated with toxicities) - non-linear kinetics - a constant amount (number of molecules) of drug is eliminated per unit time - clearance slows as drug concentration rises - some drugs can follow first order kinetics until elimination is saturated (usually at large doses) and the clearance decreases - some drugs follow non-linear kinetics at therapeutic levels e.g. phenytoin
Excretion Excretion is the process of removing a drug and its metabolites from the body. This usually happens in the kidneys via urine produced in them. Other possible routes include bile, saliva, sweat, tears and faeces. Most drugs are insufficiently polar (and, therefore, water soluble) to be excreted directly. Instead they need to metabolise to produce more polar, water-soluble molecules. Excretory system The excretory system is made up from the two kidneys, ureters, bladder and urethra, together with the branches of the two renal arteries and veins. Blood passes into the kidney’s nephron (kidney tubule) where three processes can happen: Glomerular filtration: small drug and metabolite molecules and those not bound to plasma protein are filtered from the blood. Large molecules or those bound to plasma protein are poorly excreted by glomerular filtration. Tubular secretion: most drugs enter the kidney tubule by tubule secretion rather than glomerular filtration. The process involves active transport against a concentration gradient and, therefore, requires energy and carriers to transport basic drugs such as dopamine and histamine, and carriers for acidic drugs such as frusemide and penicillin. Tubule reabsorption: Some drugs and metabolites are absorbed back into the bloodstream.This does not require energy. It is passive transport.
Elimination by LiverLiver is the major site for metabolism. It converts lipophilic compounds into hydrophilic compounds by phase I and II reactions, which makes the drugs more excretable. In membranes of canaliculi, transporters for active secretion of drugs or metabolites are present as well. Elimination by LungsLungs constitute the most different route of drug elimination. This is the only route by which lipophilic drugs are excreted because they are absorbed through the alveolar membrane. Examples include general anesthetics, which are gases pumped through the endotracheal tubes and diffuse across the alveolar membrane. When stop their administration, pure oxygen is supplied. The body acts as a reservoir and transport occurs in reverse. Thus lipophilic compounds are lost through the lungs. Alcohol breadth is another example which can be tested by alcohol breath test, by which alcohol in the excreted air is measured.IntestinesDrugs are mostly absorbed in the small intestine. Anthracene purgatives, which act mainly on the large bowel, are partly excreted in to that area from the blood stream after absorption from small intestine. Heavy metals are also excreted through the intestine and can produce intestinal ulcerations.
Minor SourcesBreast milk is important because many drugs are excreted in it. Some effects of the drugs may be transferred to the baby, which may prove harmful. It is important to know which drugs are not to be used during breast feeding. Milk being slightly acidic than plasma, weak bases get ionized and have equal or higher concentration in milk than in plasma. Non electrolytes like ethanol and urea readily enter the milk independent of pH. 70% of plasma concentration of tetracyclines may enter milk, prolonged usage of which might cause permanent staining of teeth and weak bones in the baby. Ampicillin may lead to diarrhea and allergic sensitizations. Chloramphenicol might lead to aplastic anemia in baby, bone marrow suppression and grey syndrome. Morphine, opoids and smoking may cause lethargic baby.If the mother is taking drugs, she should lactate the baby a few hours after taking drugs or most preferably half an hour before intake. Sweat glands.
Clearance of drugUnit volume of blood which is cleared off a drug per unit time is known as clearance.Clearance is not a measure of how much drug is being eliminated; it is only a measure of how much plasma is cleared of it per minute. Units are ml/min, sometimes ml/min/kg body weight are used. Rate of elimination = Quantity or volume of urine measured (ml/min) X conc. substances in urine (mg/ml)Rate of elimination = ml/min x mg/ml = mg/min Clearance= rate of elimination of substance in urine/ concentration of the drug in the blood.Clearance = mg/min / mg/ml = ml/minTotal clearance = CL kidney + CL liver + CL…..
Pharmacodynamics:
The mechanisms of action of drugsNon receptor mechanism: Some of drugs act by non receptor mechanisms:
-Enzymes stimulation or inhibition ( MAO- inhibitors , ACE-inhibitors)
-Physical effect ( osmotic diuretic, bulk laxatives)
-Chemical effects ( Antacids)
-Local effects ( counter irritants)
B- Receptors mechanism: Most of the drugs act by occupying specific receptors, which is a macromolecular component ( usually proteins) located on the l membrane, cytoplasm or nucleus of the cell.
Potency: It is the amount of drug required to produce a certain response
Efficacy: Maximal response that can be elicited by a drug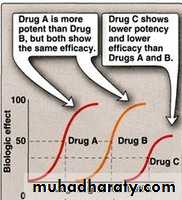
Agonist: An agent which activates a receptor to produce an effect similar to a that of the physiological signal molecule, e.g. Muscarine and Nicotine) Inverse agonist: an agent which activates receptors to produce an effect in the opposite direction to that of the agonist, e.g. DMCM Partial agonist: An agent which activates a receptor to produce submaximal effect but antagonizes the action of a full agonist, e.g. pentazocineAntagonist: an agent which prevents the action of an agonist on a receptor or the subsequent response, but does not have an effect of its own, e.g. atropine and muscarine
Antagonist could be:Physiologic (Functional) Antagonists Physiologic antagonists represent another type of antagonism in which the antagonist does not interact directly with the actions of the agonist at its molecular target. The agonist and antagonist each act on different molecular targets, but the responses elicited by these interactions are diametrically opposed and negate each other. Epinephrine and histamine are good examples of physiologic antagonists.Pharmacokinetic Antagonists One drug attenuates the action of another drug by decreasing its concentration at the site of action. This may occur through changes in absorption, distribution, metabolism, or excretion. An example is activated charcoal used in acute treatment of poisonings. Ingestion of activated charcoal binds drug in the intestine and reduces or prevents its absorption. Pharmacologic Antagonists The majority of antagonists used as drug therapy are pharmacologic antagonists that act by directly interfering with an agonist’s ability to activate its molecular target. The antagonist prevents agonist binding or agonist activation of the receptor and inhibits the biologic effects generated by the agonist.
The interaction between antagonist and agonist can take several forms, including competitive and noncompetitive antagonism.Competitive Antagonists: Compete with agonist for receptor binding => Agonist appears less potent, but can still achieve 100% effect (but at higher concentrations)Non-competitive Antagonists: Bind to receptor at different site and either prevent agonist binding or the agonist effect => maximal achievable response reduced Inverse Agonists: Not the same as antagonists! Inverse agonists trigger a negative response (= reduce baseline) (e.g. diazepam = full agonist = anticonvulsant BUT inverse agonists of benzodiazepin receptor are anticovulsants)
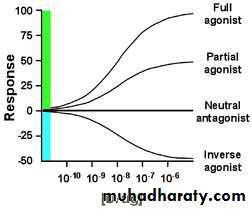
Dose Response relationshipThe exact relationship between the dose and the response depends on the biological object under observation and the drug employed.- If the concentration of the drug too low to produce the pharmacological effect, it means (no response) ie: sub-therapeutic dose.The lowest concentration of a drug that elicits a response is minimal dose --The largest concentration after which further increase in concentration will not change the response is the maximal dose.
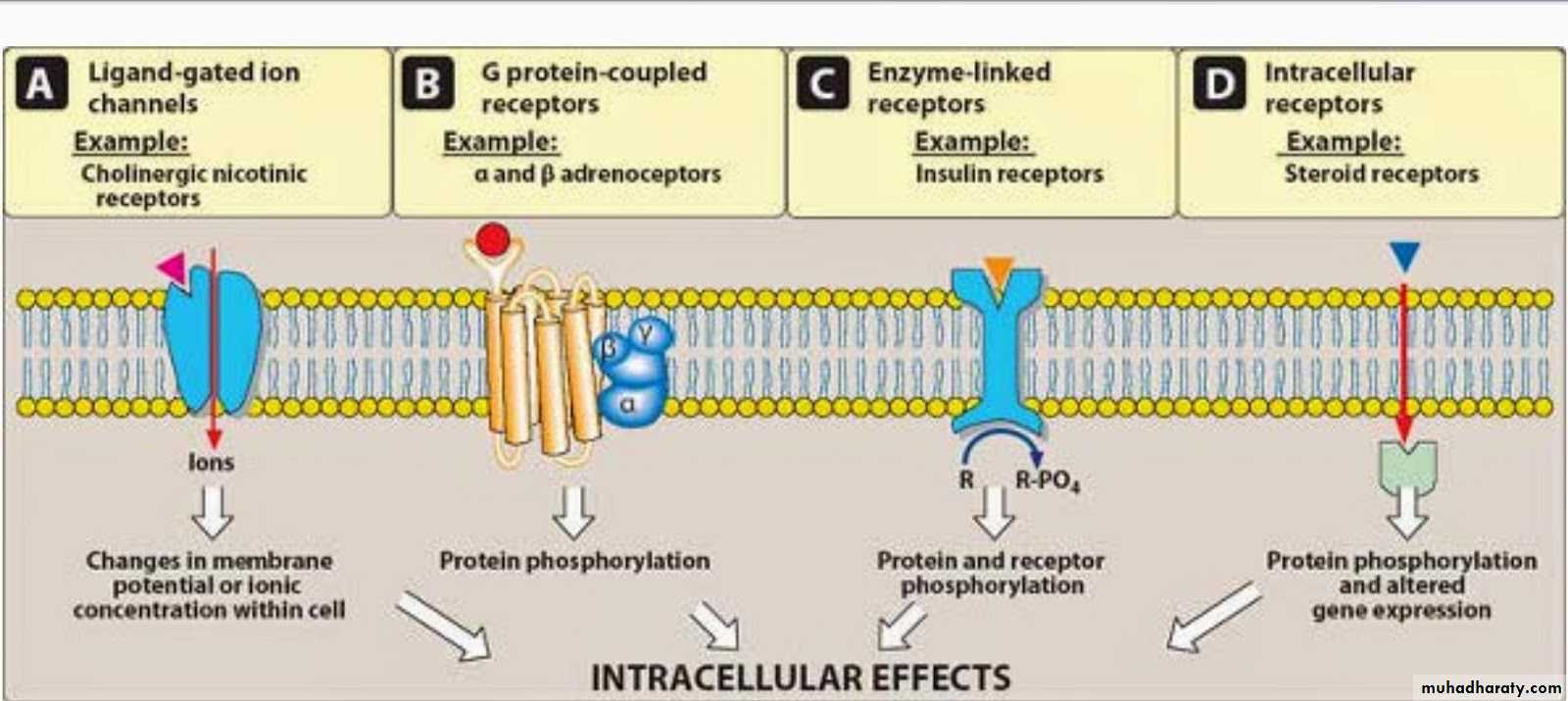
Major Receptor families: A) Ligand-gated ion channel B) G protein-coupled receptor C) Enzyme-linked receptors D) Intracellular receptorsA) Ligand-gated ion channel The most rapid cellular responses to receptor activation are mediated via ligand-gated ion channels. These kind of transmembrane receptors composed of multiple peptide subunits. The ligand binding causes conformational changes of receptor and opening of ion channel. Eg: nicotinic receptorsB) G protein-coupled receptor: It comprised of a single peptide that has seven membrane-spanning r egions.- These receptors are linked to a G Protein (Gs and others) having three subunits, alpha (α) subunit (binds guanosine triphosphate GTP) and a beta-gamma (βY) subunit.- Binding of appropriate ligand to extracellular region of the receptor activates the G Protein, so that GTP replaces guanosine diphosphate GDP on the alpha subunit.-Dissociation of G Protein occurs, and both the alpha-GTP subunit and the βY subunit interact with other cellular effectors (an enzyme or an ion channel). - Effectors then change the concentration of the 2 nd messenger that are responsible for further actions within the cell.-Stimulation of these receptors results in responses last several seconds to minutes
C) Enzyme-linked receptors: These receptors have cytosolic enzyme activity as an integral component of their structure or function. - Binding of a ligand to an extracellular domain activates or inhibits this cytosolic enzyme activity. - Duration of responses to stimulation of these receptors is in order of "minutes to hours". Intracellular (cytoplasm or nucleus) receptors: Those receptors are not associated with cell membrane. Ligands are mostly lipid soluble and passively pass cell membrane. Ligand binding activates receptor the complex then translocates to nucleus and bind to specific DNA sequences mostly located in gene promoter region. This kind of signal tranduction is slow, but duration of response can last long. Eg: receptors for glucocorticoides and gonadal steroids.
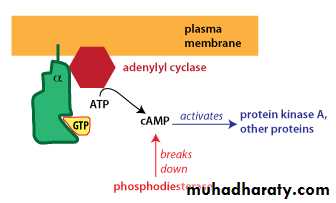
Different G-alpha proteins activate different second messenger pathways1-There are several different classes of trimeric G-proteins that are defined by their different G-alpha subunits. One type of G-alpha activates the enzyme adenylyl cyclase, which catalyzes the formation of the second messenger cyclic AMP (cAMP). Because an activated adenylyl cyclase can generate many molecules of cAMP, this is a means to amplify the signal. cAMP can have several effects, but a major effect is to bind to and activate protein kinase A (PKA; also known as cAMP-dependent kinase). PKA then phosphorylates target proteins in the cell. cAMP is rapidly broken down byphosphodiesterases, limiting the length of the signal.
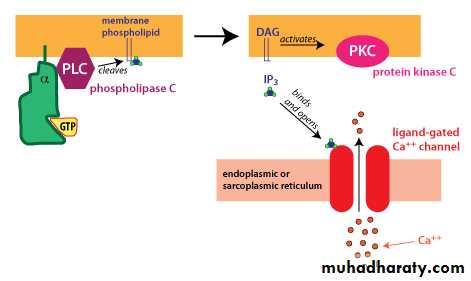
2-Another type of G-alpha activates the enzyme phospholipase C. Phospholipase C cleaves PIP2, a membrane phospholipid, to generate two second messengers, IP3 and diacylglycerol (DAG). IP3 is water soluble, diffusing through the cytosol to bind to and open a ligand-gated Ca++ channel in the endoplasmic reticulum (or sarcoplasmic reticulum in muscle cells). Thus, stimulation of a receptor linked to this G-alpha is a way to increase Ca++ inside the cytosol. Ca++ in the cytosol exerts its effects by binding to Ca++-binding proteins such ascalmodulin. DAG is lipid soluble and stays in the membrane. It activates protein kinase C (PKC), which, like PKA phosphorylates particular target proteins.
There are two types of responses:1. Graded dose effect: As the dose administered to a single subject or tissue increases, the pharmacological response also increases in graded fashion up to ceiling effect. - It is used for characterization of the action of drugs. The concentration that is required to produce 50 % of the maximum effect is termed as EC50 or ED50. 2. Quantal dose effect ( All or none): It is all or none response, the sensitive objects give response to small doses of a drug while some will be resistant and need very large doses. The quantal doseeffect curve is often characterized by stating the median effective dose and the median lethal dose.
Median lethal dose or LD50: This is the dose (mg/kg), which would be expected to kill one half of a population of the same species and strain. Median effective dose or ED50: This is the dose (mg/kg), which produces a desiredresponse in 50 per cent of test population. Therapeutic index: It is an approximate assessment of the safety of the drug. It is the ratio of the median lethal dose and the median effective dose. Also called as therapeutic window or safety. LD50 Therapeutic index (T I) = ------- ED50 The larger the therapeutic index, the safer is the drug. Penicillin has a very high therapeutic index, while it is much smaller for the digitalis preparation.
Drug toleranceTolerance defined as a state of progressively decreased responsiveness to a drug as a result of which a larger dose of the drug is needed to achieve the effect originally obtained by a smaller doseThe sensitivity of the target cells is governed by genetic factors and adaptive changes by the body. Adaptive changes occur in response to the repeated exposure to a particular drug. The result is usually a loss of sensitivity to the drug. This decreased response is called tolerance.