Pathology 4 lectures dr.lameia
Cellular response to stress and harmful stimuli
Objectives:-
Simple introduction
Adaptation:- definition and its types
Cell injury and cell death:- causes, morphological example of reversible injury(cellular
swelling, fatty change) and irreversible injury (necrosis, apoptosis). Morphological
features of necrotic cells. Morphological patterns of tissue necrosis
Apoptosis:- causes, mechanism, morphology, examples
Mechanisms of cell injury
Intracellular accumulations:- fatty change, cholesterol and cholesterol ester
accumulations, protein accumulations, pigments
Pathological calcification:- dystrophic and metastatic classifications
Cellular response to stress and harmful stimuli
Each cell in the body is destined to carry a specific function which dependent on this
machinery and metabolic pathways.
This structural and the related functional specificities are genetically determined.
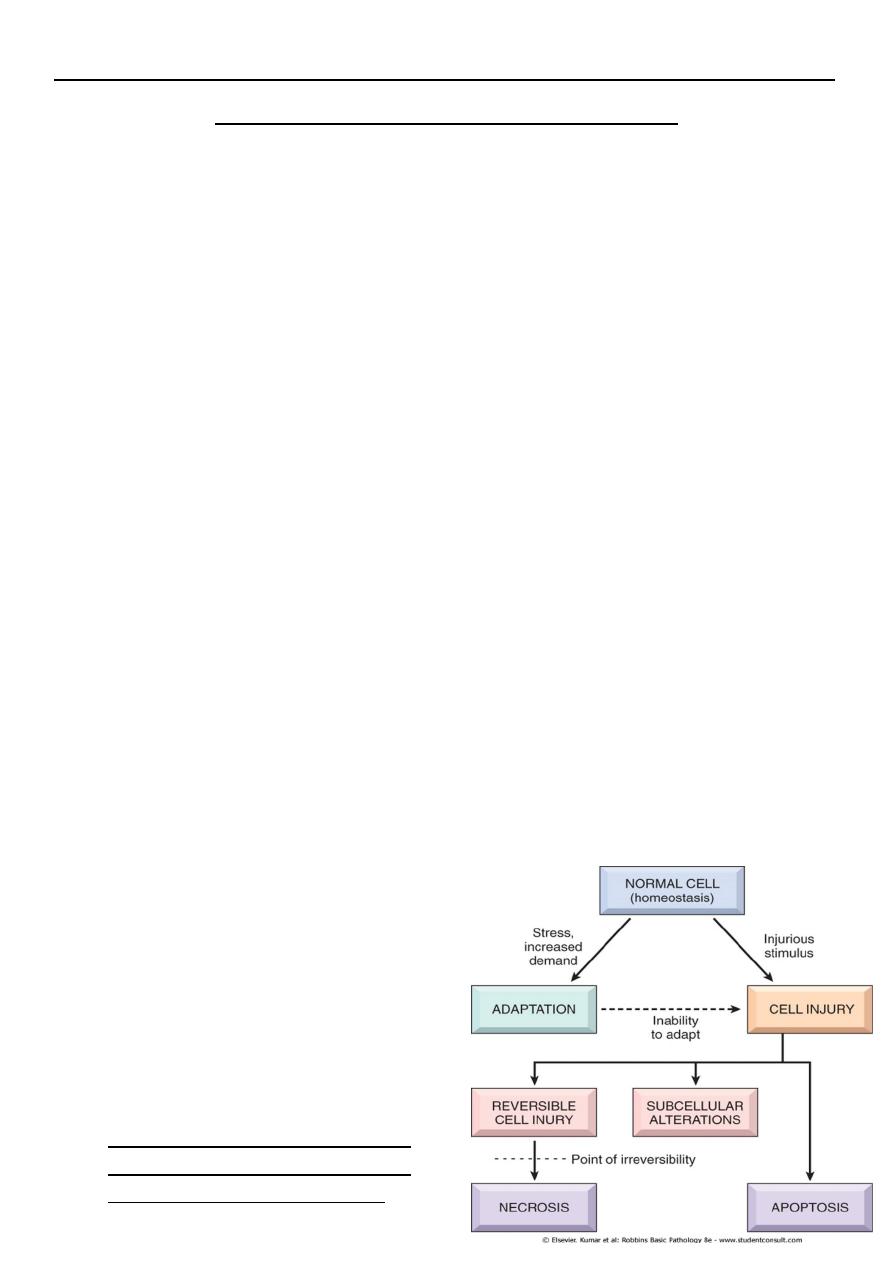
Cells are continuously adjust their structure and function to accommodate changing
demand and extra cellular stresses. However these adjustment occur in a narrow
range. This is referred to as homeostasis .
should the cells encounter physiologic or pathologic stresses or stimuli, they can
modify the homeostatic state and achieve a new steady state to counteract the
noxious effect of these external stresses and stimuli. These changed are referred to
as adaptation. The aim of these adaptations is to avoid cell injury and death.
Adaptation are reversible changes and are divided into physiologic adaptation and
pathologic adaptation.
• The injury may be reversible i.e. cells
return to normal state on the removal of
the offending agent.
• Reversible cell injury occur when the
injurious agent is mild or short lived, the
functional and morphological
(structural) changes are reversible.
• At this stage although there may be
significant structural and functional
abnormalities, the injury has not
progressed to severe damage of the cellular membranes and nucleus that are
equated with cell death. With continuing damage. The injury becomes irreversible, at
which time the cell cannot recover and it dies.
• or irreversible i.e there is no possibility of making a U-turn to normal. Irreversible
injury ultimately eventuates in cell death.
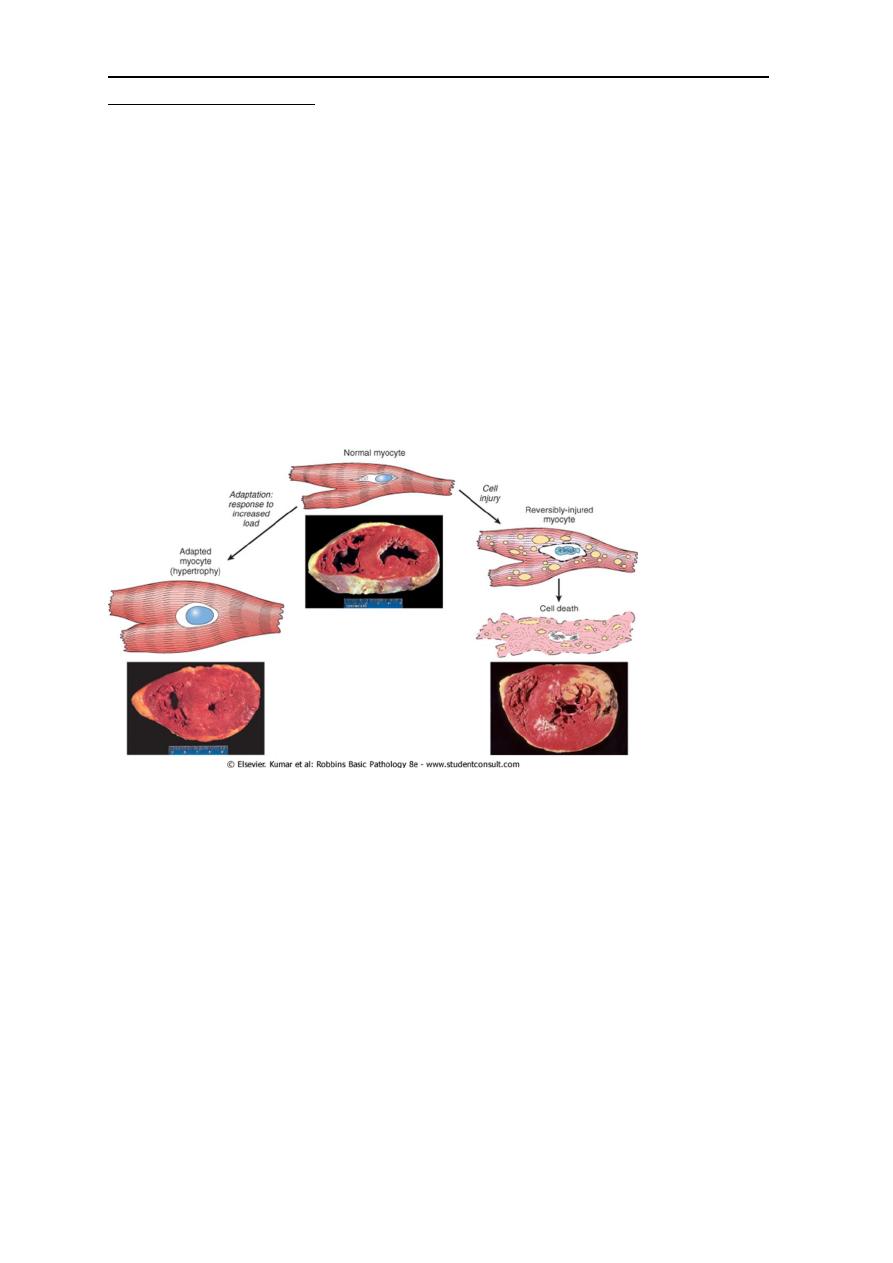
• The above mentioned possibility can be exemplified by the responses of the heart to
different types of stress. A myocardium that is subjected to persistently increased
pressure load , as in hypertension, adapts by undergoing hypertrophy as increase in
the size of individual cells and ultimately the entire heart to generate the required
higher contractile force. If the increased demand is not relieved; the muscle cell may
undergo injury. The injury may be reversible if the hypertension is mild; otherwise
irreversible injury ( cell death) occurs.
Adaptation
1. Hypertrophy
2. Hyperplasia
3. Atrophy
4. Metaplasia
Hypertrophy : refers to increase in the size of the cells resulting in increase in the size of the
relevant organ and its either physiologic or pathologic
Physiologic hypertrophy is caused either by:-
• increased in functional demand as in hypertrophy of the skeletal muscles in athletes
and mechanical workers
• by specific hormonal stimulation e.g. is the massive physiologic enlargement of the
uterus during pregnancy due to estrogen stimulated smooth muscle hypertrophy
(and hyperplasia).
Pathologic hypertrophy is exemplified by
cardiomegaly secondary to hypertension. The stimuli of hypertrophy turn in signals that
lead to induction of a number of genes, which in turn stimulate synthesis of numerous
myofillaments per cell; this lead to improve performance to house the excessive demand
imposed by the external burden. There is however a limit for the adaptation and when this
happened example in the heart, several degenerative changes occur in the myocardial
fibers that culminate in loss of the contractile myofibrils. This limitation of cardiac
hypertrophy may be related to the amount of available blood to the enlarged fibers. The
net result of these regressive changes is ventricular dilation and ultimately cardiac failure.
This mean that an adaptation can progress to dysfunction if the stress is not relieved.
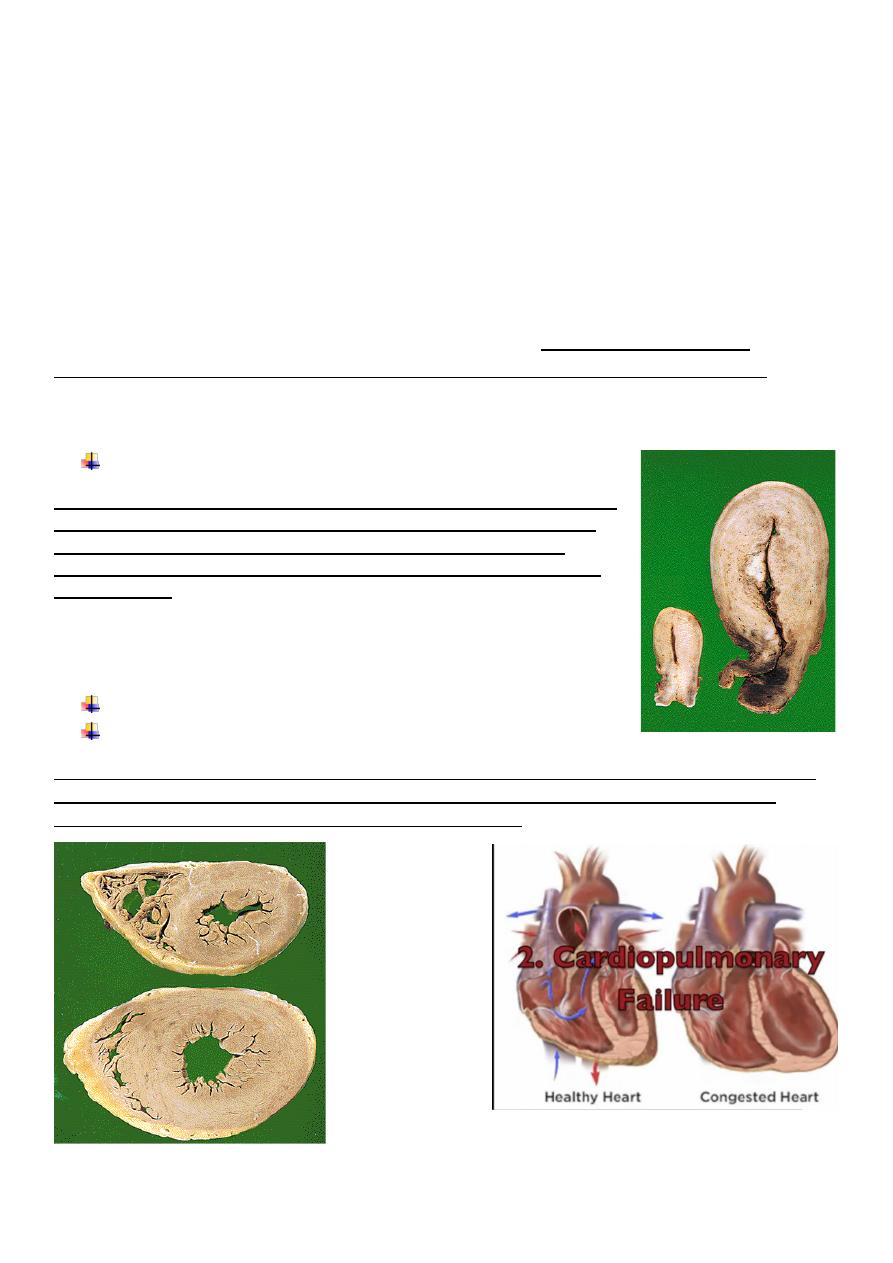
Enlargement of the uterus in pregnancy
On the left is a normal uterus showing the normal mass of smooth muscle
in its wall. On the right is a uterus from a recently pregnant women, in
which the striking increase in mass of smooth muscle is evident. At
cellular level this is due to both hyperplasia and hypertrophy of uterine
smooth muscle.
Athletes as an example of muscular hypertrophy
Left ventricular hypertrophy
The upper specimen demonstrates the normal thickness of the Lt ventricular wall for comparison with
the greatly thickened wall in the lower specimen. The increased mass of the Lt ventricle is due to
enlargement of cardiac muscle cells as a result of hypertrophy.
Hyperplasia : refers to increase in the number of the cells. It takes place only if the cell

population is capable of replication it may occur with hypertrophy and often in response to
the same stimuli.
1. Hormonal hyperplasia exemplified by the proliferation of the glandular epithelium of
the female breast at puberty and during pregnancy. The enlargement of the gravid
uterus is due to a combination of hypertrophy and hyperplasia.
2. Compensatory hyperplasia which occurs when a portion of the tissue is removed or
diseased for example when a liver is partially resected, mitotic activity in the
remaining cells begins that eventually restores the liver to its normal weight. The
stimuli for hyperplasia in this setting are growth factors produce by the remaining
hepatocytes and non-parenchymal cells in the liver. After restoration of the liver
mass, cell proliferation is turned off by various growth inhibitors.
3. pathologic hyperplasia are caused by excessive hormonal or growth factors
stimulation.
• For example if there is persistent or excessive estrogen stimulation of the
endometrium, endometrial hyperplasia occurs a common cause of abnormal
menstrual bleeding.
• Papilloma virus cause skin warts composed of masses of hyperplastic epithelium. The
growth factor responsible may be produce by the virus or by the infected cells.
Hyperplasia
• above situation the hyperplastic process remained controlled if hormonal or growth
factor stimulation subsides the hyperplasia disappears. It is this response to normal
regulatory control mechanisms that distinguishes benign pathologic hyperplasia from
cancer. In which the growth control mechanisms become ineffective. However
pathologic hyperplasia may be a fertile soil for the development of the carcinoma.
Atrophy : refers to shrinkage in the size of the cell by the loss of the cell substance. This
situation is exactly opposite to hypertrophy. When sufficient number of cells are involved
the entire tissue or organ diminishes in size. i.e becomes atrophic
Causes of atrophy include:-
1. A decreased workload (e.g. immobilization of a limb to permit healing of a fracture)
2. Denervation as in poliomyelitis
3. Diminished blood supply e.g. decreased blood supply to a limb.
4. Inadequate nutrition as in starvation and famines
5. Loss of endocrine stimulation as in postmenopausal endometrial atrophy and
testicular atrophy
6. Aging ( senile atrophy)
Atrophy
• When there is diminished blood supply, nutrition or any trophic stimulation (whether
physiologic e.g. loss of hormonal stimulation in menopause or pathologic e.g.
denervation) so a new equilibrium state should be achieved at which survival is still
possible and this done by retreating the cell into smaller size.
• So atrophy result from decreased protein synthesis (because of reduced metabolic
activity) and increase protein degradation in cells. In many situations atrophy is also
accompanied by increased autophagy (self-eating) with resulting increase in the
number of the autophagic vacuoles. The starved cells eats its own component in an
attempt to fined nutrient and survive.
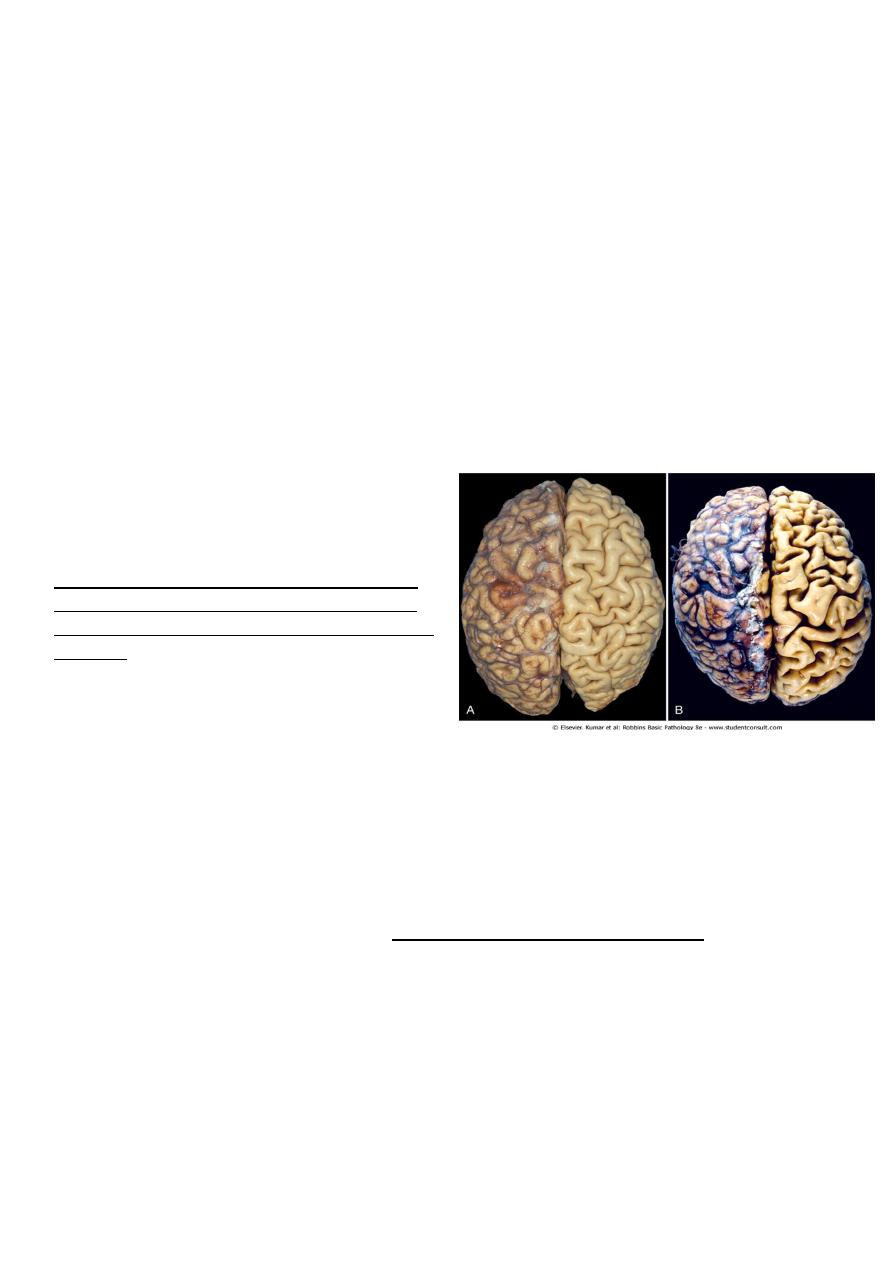
Figure 1-4 Atrophy. A, Normal brain of a young adult.
B, Atrophy of the brain in an 82-year-old male with
atherosclerotic disease. Atrophy of the brain is due
to aging and reduced blood supply. Note that loss of
brain substance narrows the gyri and widens the
sulci. The meninges have been stripped from the
right half of each specimen to reveal the surface of
the brain.
Metaplasia : refers to a reversible changes in which one adult mature cell type ( epithelia or
mesenchymal) is replaced by another adult cell type. In this type of cellular adaptation cells
sensitive to a particular stress are replaced by other cell types that are more capable of
withstanding the adverse environment.
• Metaplasia is thought to arise by genetic reprogramming of stem cells.
• The epithelial metaplasia is exemplified by
1. the squamous changes that occur in the respiratory epithelium in habitual cigarette
smokers. The normal ciliated columnar epithelial cells of the trachea and bronchi are
focally or widely replaced by stratified squamous epithelial cells. Although the
metaplastic squamous epithelium has survival advantages but important protective
mechanisms are lost, such as mucus secretion and ciliary clearance of particular
matter. Moreover the influences that induce metaplastic transformation; if
persistent, may predispose to malignant transformation of the epithelium. In fact
squamous cell carcinoma of the bronchi often coexists with squamous metaplasia. It
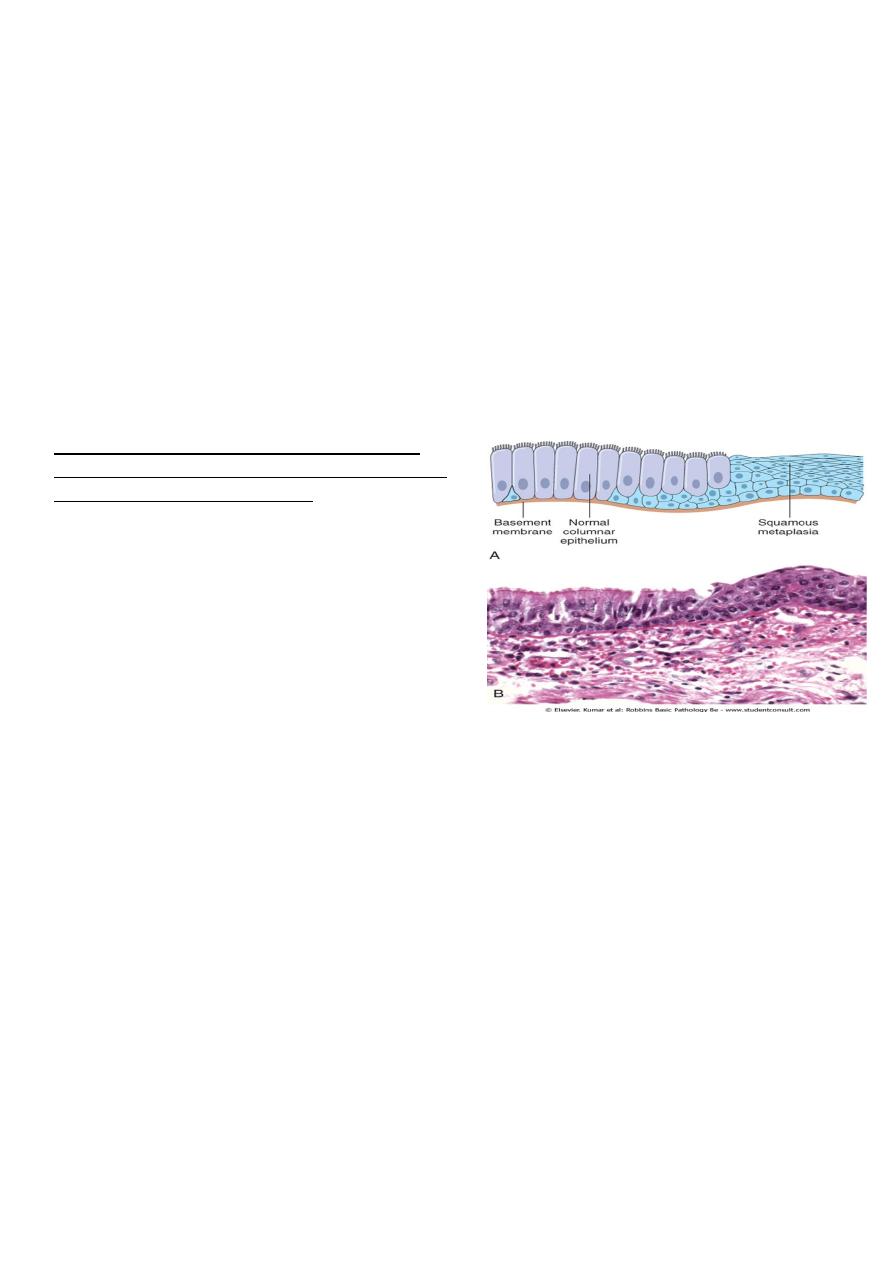
is thought that cigarette smoking initially causes sq. metaplasia and cancer arise later
in some of these altered foci.
2. S.M. metaplasia is also seen in the urinary bladder harboring shistosomal ova.
Metaplasia
• In chronic gastro-esophageal reflux disease (GERD)the normal stratified sq.
epithelium of the lower esophagous may undergo glandular metaplastic
transformation to intestinal type columnar epithelium.
• A closely related topic to metaplasia is a process of dysplasia which is a definite step
in the evolution of cancer but this process is not an adaptation.
Figure 1-5 Metaplasia of normal columnar (left) to
squamous epithelium (right) in a bronchus, shown (A)
schematically and (B) histologically.
Cell injury and cell death
Cell injury results when cells are exposed to
1. Severe stress so that they no longer able to adapt or
2. Inherently damaging agents
Reversible cell injury occur when the injurious agent is mild or short lived, the functional
and morphologic changes are reversible.
With continuing damage. The injury becomes irreversible, at which time the cell cannot
recover and it dies.
Causes of cell injury
1. Oxygen deprivation (hypoxia) insufficient supply of oxygen interferes with aerobic
oxidative respiration and is a common cause of cell injury and death. Causes of
hypoxia include
• Ischemia i.e. loss of blood supply in a tissue due to interference with arterial flow or
reduced venous drainage . this the most commonest cause of hypoxia
• Inadequate oxygenation of the blood as in pneumonia
• Reduction in the oxygen carrying capacity of the blood as in anemia or carbon
monoxide (Co) poisoning
2. Chemical agents include concentrated glucose or salt and oxygen at high partial
pressures. Various poisons cause damage by affecting their membrane permeability or the
integrity of the enzymes. Environmental toxins as pollutants, insecticides, Co ,and alcohol as
well as drug can cause cell or tissue injury.
3. Infectious agents including virus
bacteria, rickettsia, fungi and parasites
4. Immunologic reactions are primary defensive in nature but they can also result in cell
and tissue injury. Example include autoimmune disease and allergic reactions in
genetically susceptible individuals.
5. Genetic defect including gross congenital malformation such as those associated with
down syndrome or as subtle point mutation e.g. in sickle cell anemia.
6. Nutritional imbalances :- nutritional deficiencies remain a major cause of cell injury.
protein calories among is the most obvious example;
vitamin deficiencies are not uncommon.
Excesses of nutrition are also important cause of morbidity and mortality for example
obesity markedly increases the risk for the type 2 diabetes mellitus. Moreover diet rich in fat
are strongly implicated in the development of atherosclerosis as well as in increase the
vulnerability to cancer e.g. that of colon
7. Physical agents:- trauma extremes in the temperatures, radiation, electric shock and
sudden changes in atmospheric pressure all are associated with cell injury.

8. Aging :- cellular senescence lead to alteration in replicative and repair abilities of
individual cell and tissues. All of these changes result in a diminished ability to respond to
damage and eventually the death of cells and of the organism.
Morphologic features of cell and tissue injury
• All harmful influences exert their effects first the molecular or biochemical level.
Function may be lost long before cell death occurs, and the morphologic changes of
cell injury (or death) is delayed behind both. For example myocardial cells fail to
contract after 1-2 minutes of ischemia, although they do not die until after 20-30
minutes of ischemia. These myocytes do not appear dead by electron microscopy for 3
hours and by light microscopy for 6 -12 hours.
• The cellular derangements of reversible injury can be repaired. Persistent or excessive
injury however cause cells to passes to the point of no return i.e. into irreversible
injury and cell death.
• There are two alteration that consistently characterized irreversible injury (cell death):-
1. Mitochondrial dysfunction :- manifested as lack of oxidative phosphorylation leading
to ATP depletion
2. Profound disturbances in membrane function including lysosomal membranes that
result in enzymatic dissolution of the injured cells which is characteristic of necrosis.
Figure 1-7 The relationship between cellular
function, cell death, and the morphologic changes
of cell injury. Note that cells may rapidly become
nonfunctional after the onset of injury, although
they are still viable, with potentially reversible
damage; a longer duration of injury may
eventually lead to irreversible injury and cell
death. Note also that cell death typically precedes
ultrastructural, light microscopic, and grossly
visible morphologic changes.
Morphologic features of cell and tissue injury
in reversible cell injury the morphological changes are exemplified by:
1. Acute cellular swelling
2. fatty change
In Irreversible cell injury there are two morphologic types of cell death
1. Necrosis
2. Apoptosis
Morphologic examples of reversible injury
• Cellular swelling (hydropic change or vacuolar degeneration) is the result of failure of
energy –dependent ion pumps in the plasma membrane, leading to an inability to
maintain ionic gradients across the membranes i.e. there is influx of sodium (with
water) into the cell and departure of the potassium out. It is the first manifestation of
almost all forms of cell injury. When it affects many cells in an organ which appear
also tense. Microscopically: there are small, clear vacuoles within the cytoplasm; these
represent distended segment of the ER.
• Fatty changes is manifested by the appearance of lipid vacuoles in the cytoplasm. It is
principally encountered in cells participating in fat metabolism (e.g. hepatocytes)
Fatty change liver Sever fatty change liver
Morphologic examples of irreversible injury
• Necrosis refers to morphological changes that accompany cell death, largely resulting
from the degradative action of enzymes on lethally injured cells. Necrotic cell are
unable to maintain membrane integrity, and their contents often leak out. The enzymes
responsible for the digestion of the cell are derived either from the lysosomes of the
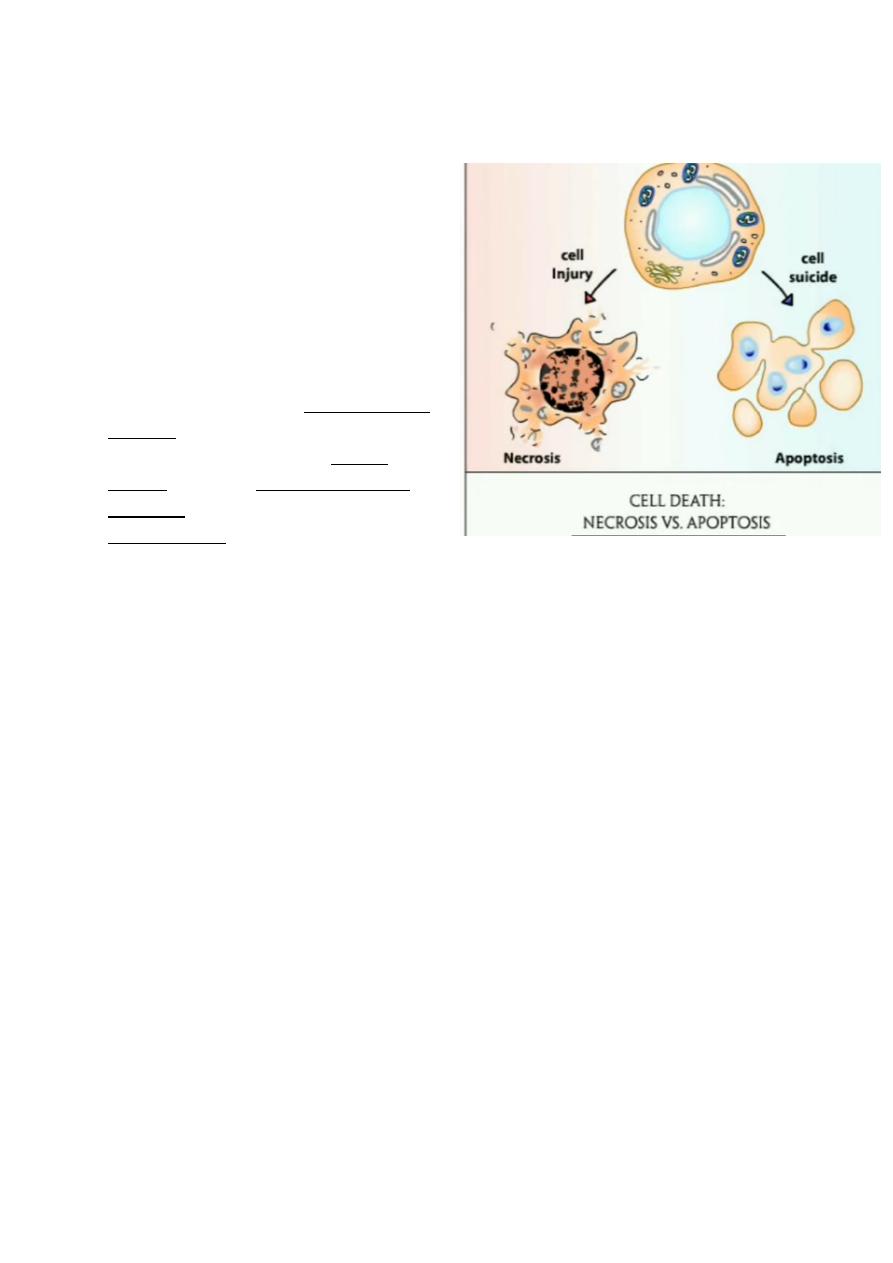
dying cells themselves or from the lysosomes of leukocytes that are recruited as part
of the inflammatory reaction to the dead cells.
• When damage to membrane is severe, enzyme leak out of lysosomes (which are
membrane bound), enter the cytoplasm
and digest the cell, resulting in necrosis.
Cellular content also leak out through
the damaged plasma membrane and
elicit inflammation.
• The leakage of intracellular proteins
through the damaged cell membrane
and ultimately into the circulation
provides a means of detecting tissue
specific necrosis using blood or serum
samples. Cardiac muscle for example
contains a unique enzyme creatin
kinase and of the contractile protein
troponin. Hepatocytes contain
transaminases. Irreversible injury and
cell death in these tissue are reflected in increase levels of such proteins and
measurement of serum levels is used clinically to assess damage to these tissues.
Apoptosis is the mode of cell death when
• The cell is deprived of growth factors or
• The cell's DNA or proteins are damaged beyond repair
This is an active, energy dependent regulated type of cell death., apoptosis servers many
normal functions and is not necessarily pathological.
Necrosis involve a large number of contagious cells (so part of tissue may involved) & it is
always a pathologic process
Morphologic features of the necrotic cells
• Cytoplasmic changes
1. The necrotic cells (e.g. as a result of oxygen deprivation) show increased eosinophilia
i.e. appear deep pink in color than normal cells. This is attributable in part to increased
binding of eosin to denaturated cytoplasmic proteins and in part to loss of the
basophilia that is normally imparted by the RNA in the cytoplasm (basophilia is the
blue staining from the hematoxilin dye)
2. The cells may have more homogenous appearance than the viable cells, mostly
because of the loss of glycogen particles
3. When enzymes have digested the cytoplasmic organelles, the cytoplasm becomes
vacuolated and appears mouth eaten.
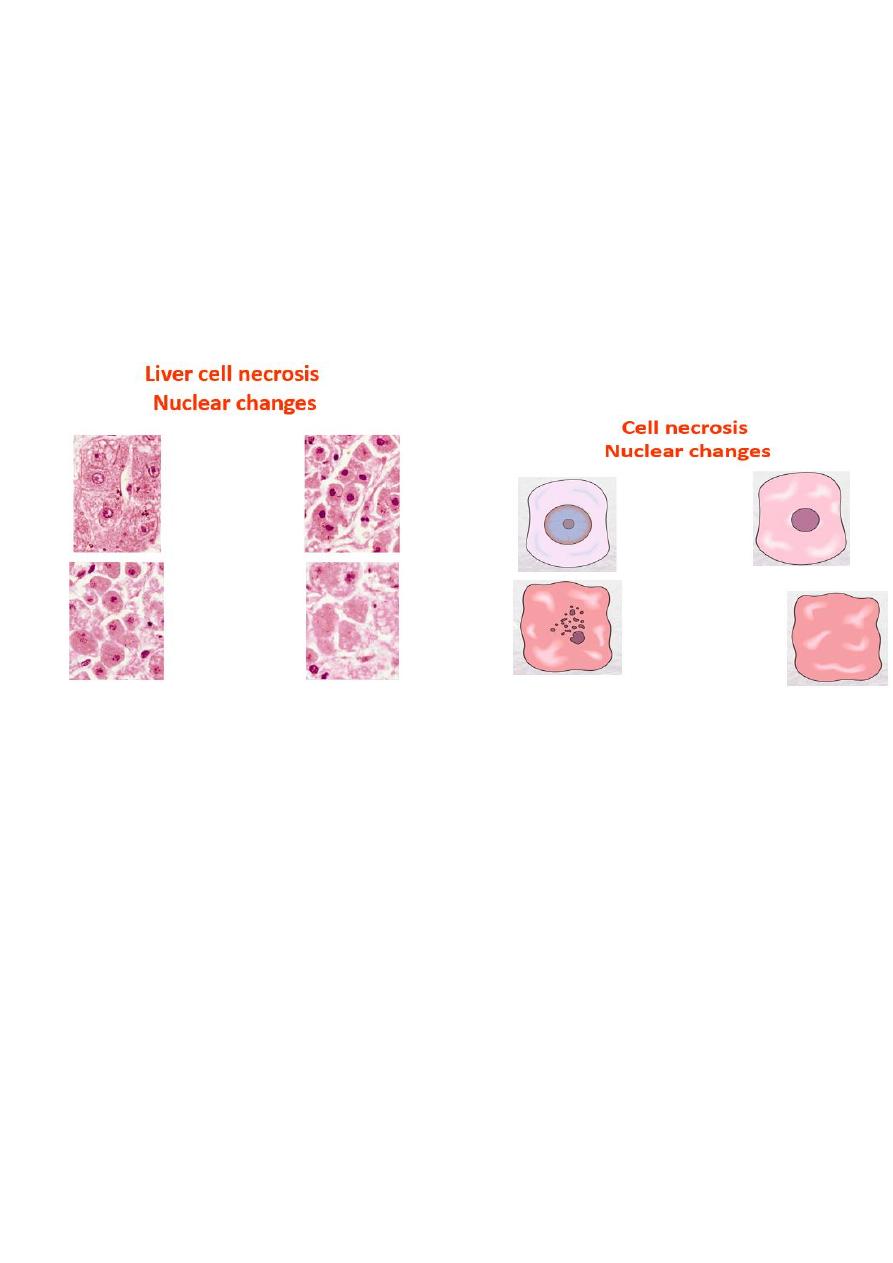
• Nuclear changes:- assume one of three patterns, all due to breakdown of DNA and
chromatin
1. Pyknosis characterized by the nuclear shrinkage and increased basophilia, the DNA
condenses into a solid shrunken mass
2. Karyorrhexis the pyknotic nucleus undergoes fragmentation onto 1 to 2 part, the
nucleus in a dead cell completely disappears.
3. Karyolysis i.e. the basophilia of the chromatin may fade, presumably secondary to
deoxyribonuclease (DNase) activity.
Six types of necrosis
1. Liquefactive
2. coagulative
3. gangrenous
4. fibrinoid
5. fat
6. caseous
morphological patterns of tissue necrosis
There are several morphological patterns of tissue necrosis, which may
provide clues about the underlying cause
Coagulative necrosis is:-
• a form of tissue necrosis. Coagulative necrosis is characteristic of infarcts (area of
ischemic necrosis) in all solid organ except brain. Grossly the affected tissue have a
firm texture
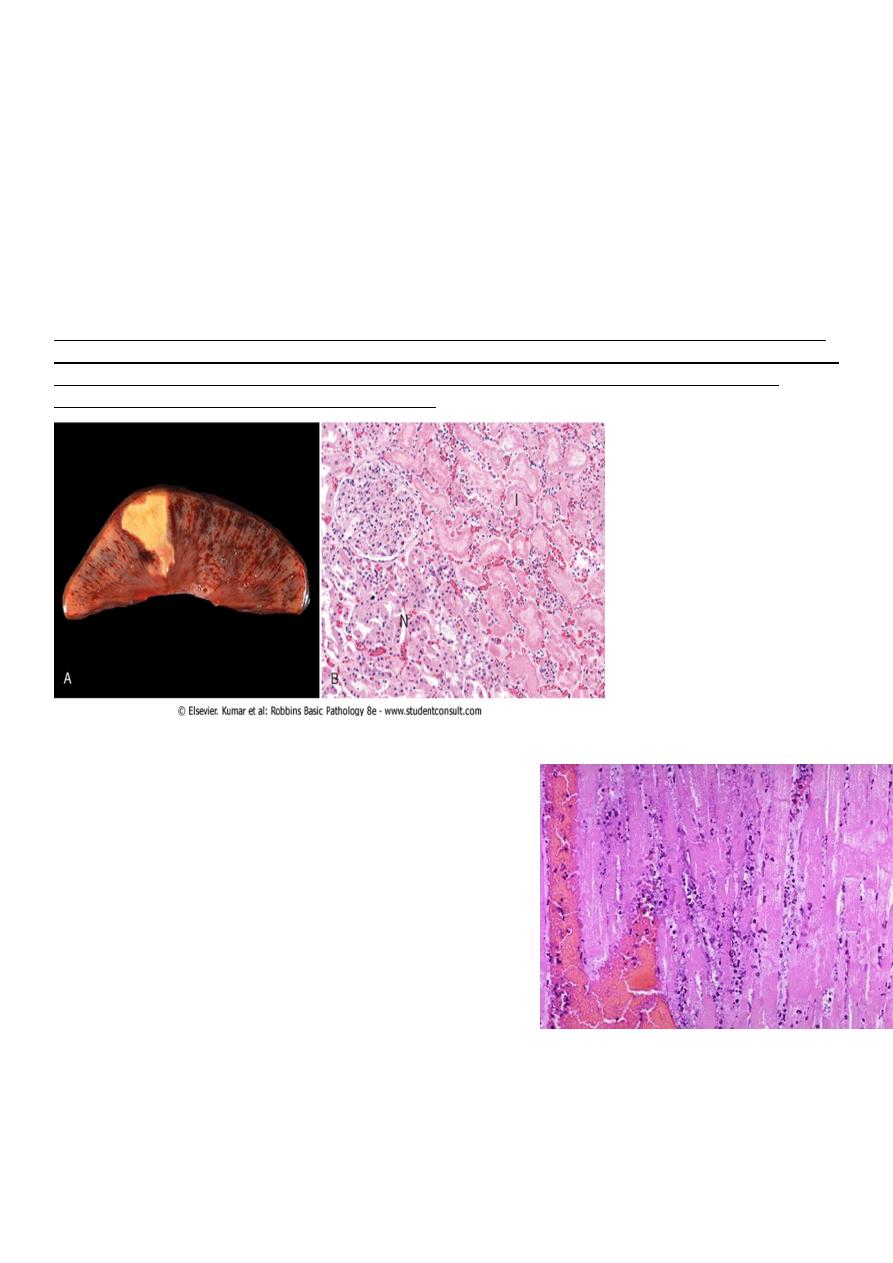
• in which the final structural details are lost but the basic tissue architecture is
preserved. This appears to be due to denaturation not only of structural proteins but
also of enzymes which blocks proteolysis of the dead cells. These changes are
reflected microscopically as homogeneously eosinophilic cells that are devoid of
nuclei.
• Ultimately the necrotic cells are removed by phagocytosis of the cellular debris by
infiltrating leukocytes and by digestion by the action of the lysosomal enzymes of the
leukocytes.
Coagulative necrosis. A, A wedge-shaped kidney infarct (yellow) with preservation of the outlines. B,
Microscopic view of the edge of the infarct, with normal kidney (N) and necrotic cells in the infarct (I).
The necrotic cells show preserved outlines with loss of nuclei, and an inflammatory infiltrate is
present (difficult to discern at this magnification).
Coagulative necrosis
myocardial cells
This myocardial infarction is about 3 to 4 days old.
There is an extensive acute inflammatory cell infiltrate
and the myocardial fibers are so necrotic that the
outlines of them are only barely visible.
The cytoplasm is rather homogeneous, deeply
eosinophilic, devoid of cross striation and there are no
nuclei.
Liquefactive necrosis :-
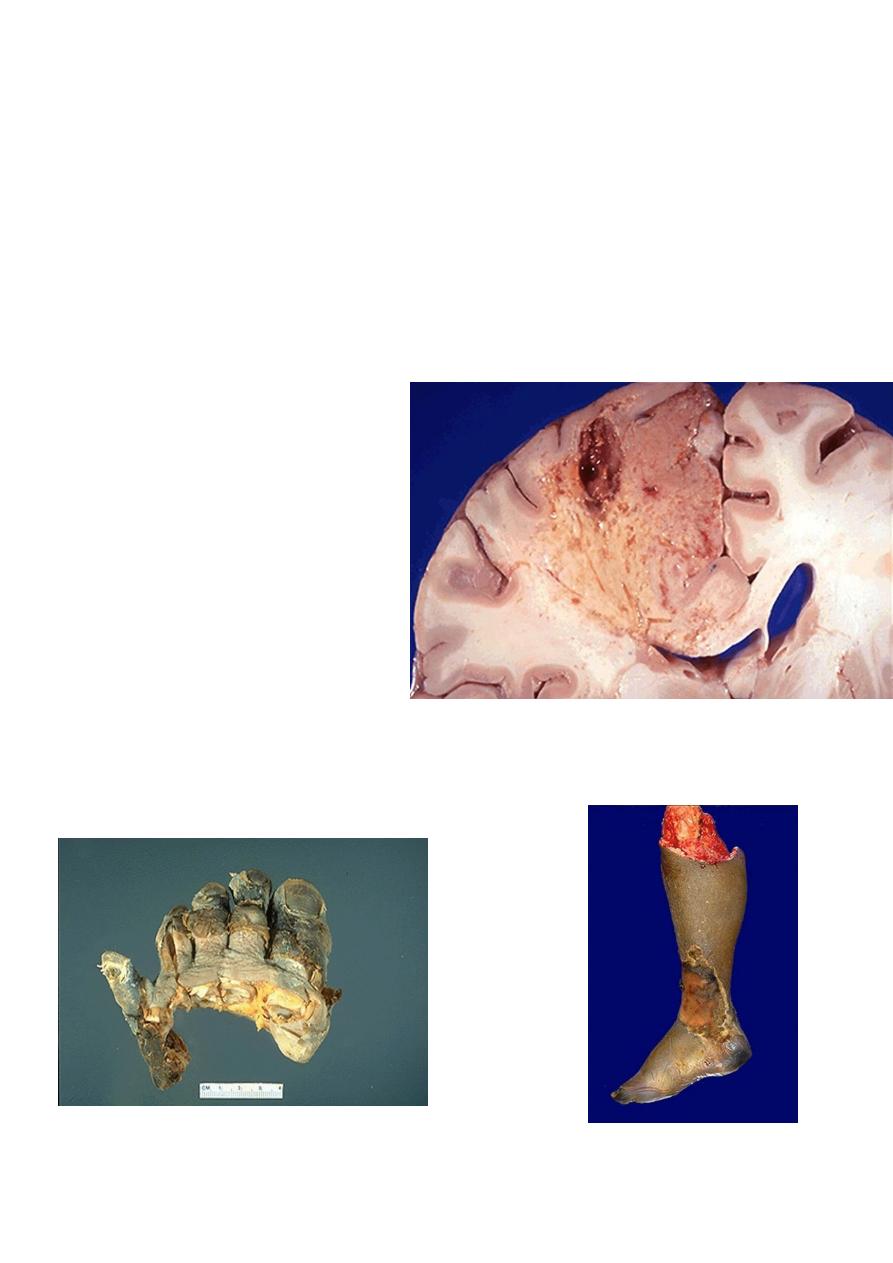
in this type of necrosis there is complete digestion of the dead cells resulting in
transformation of the affected tissue into a liquid viscous mass enclosed within a cystic
cavity. Liquefaction necrosis is seen in two situations
1. Focal bacterial infection( or occasionally fungal) infections. This is because microbes
stimulate the accumulation of inflammatory cells and the enzymes of the leukocytes
digest (liquefy) the tissue. The process is usually due to pyogenic bacterial –
associates acute suppurative inflammation ( abscess); the liquefied material is
frequently creamy yellow and is called pus
2. Ischemic destruction of the brain tissue : for obscure reasons hypoxic death of cells
within the central nervous system often evokes liquefactive necrosis.
• Gangrenous necrosis:- it is not a distinctive pattern of cell death, however the term is
still commonly used in clinical
practice. It is usually applied to a
limb, generally the lower leg that
has lost its blood supply and has
undergone coagulative necrosis
involving multiple tissue layers
(dry gangrene) when bacterial
infection is superimposed,
coagulative necrosis is modified
by the liquefactive action of the
bacteria and the attracted
leukocytes (wet gangrene)
Liquefactive necrosis brain
Gangrene-lower limb
Left: this is gangrene, or necrosis of the toes that were involved in a frostbite injury. This is an
example of "dry" gangrene in which there is mainly coagulative necrosis from the anoxic injury.
Right: this is gangrene of the lower extremity. In this case the term "wet" gangrene is more applicable
because of the liquefactive component from superimposed infection in addition to the coagulative
necrosis from loss of blood supply. This patient had diabetes mellitus.
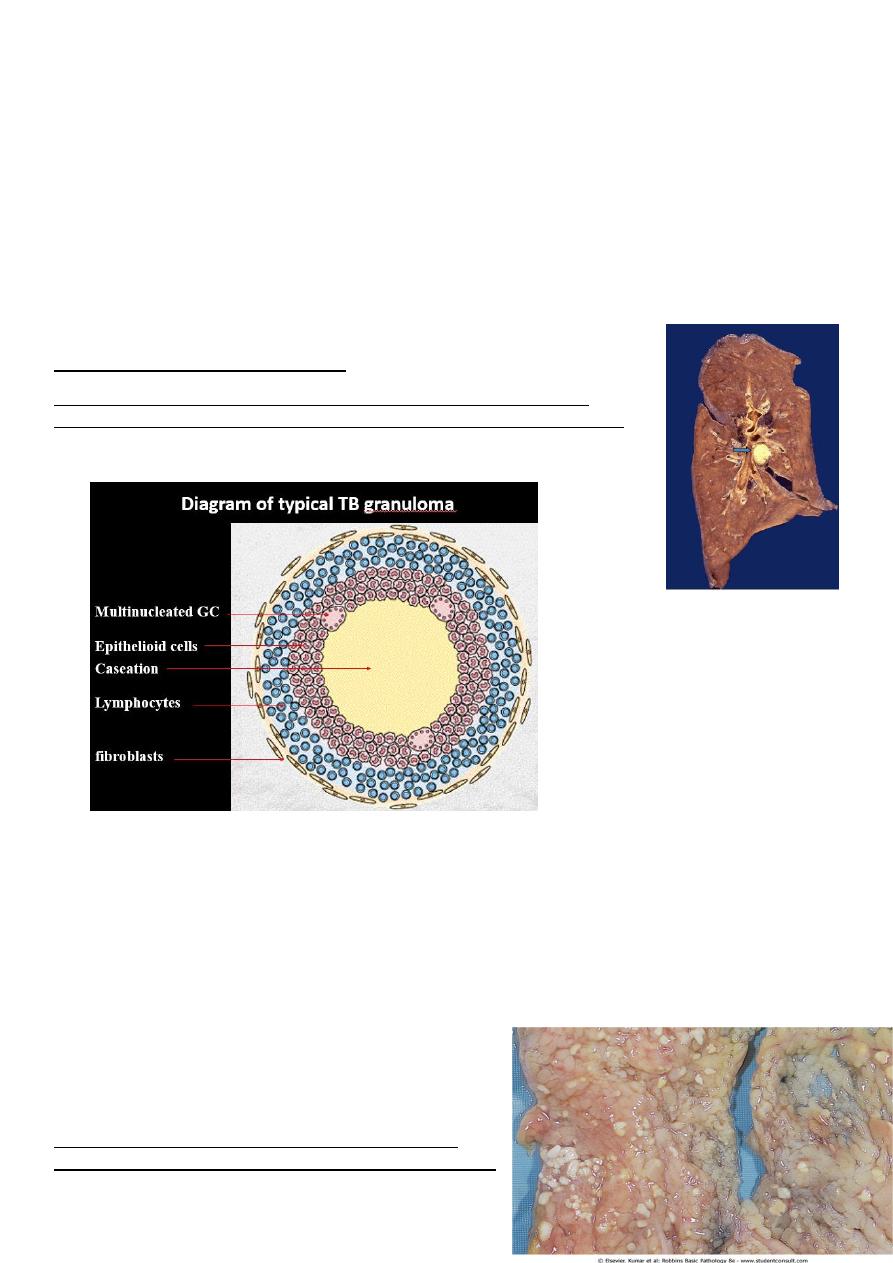
Caseous necrosis :-
is encountered most often in the foci of the tuberculous infection. The term caseous is ( a
cheese like) is derived from friable yellow white appearance of the area of the necrosis. The
tissue architecture is completely lost and cellular outlines cannot be discerned. Caseous
necrosis is often enclosed within a distinctive inflammatory border; this appearance is
characteristics of granulomatous inflammation.
Caseous necrosis-pulmonary hilar LN
This is the gross appearance of caseous necrosis in a hilar lymph node
infected with tuberculosis. The node has a cheesy tan to white appearance.
Fat necrosis typically
seen in acute pancreatitis and results from release of activated pancreatic lipases
into the substance of the pancreas and the peritoneal cavity. Pancreatic enzymes
that have leaked out of acinar cells liquefy the membranes of fat cells releasing
fatty acids that combined with calcium to produce grossly visible chalky white
areas (fat saponification).this typical appearance enables the surgeon to identify the
lesions. Microscopically the foci of necrosis contain shadowy outlines of necrotic
fat cells with bluish calcium deposits surrounded by an inflammatory reaction.
Another example of fat necrosis is seen in female breasts; at least some of these
cases are preceded by a history of trauma
(traumatic fat necrosis)
Fat necrosis in acute pancreatitis. The areas of white
chalky deposits represent foci of fat necrosis with calcium
soap formation (saponification) at sites of lipid breakdown in the mesentery.
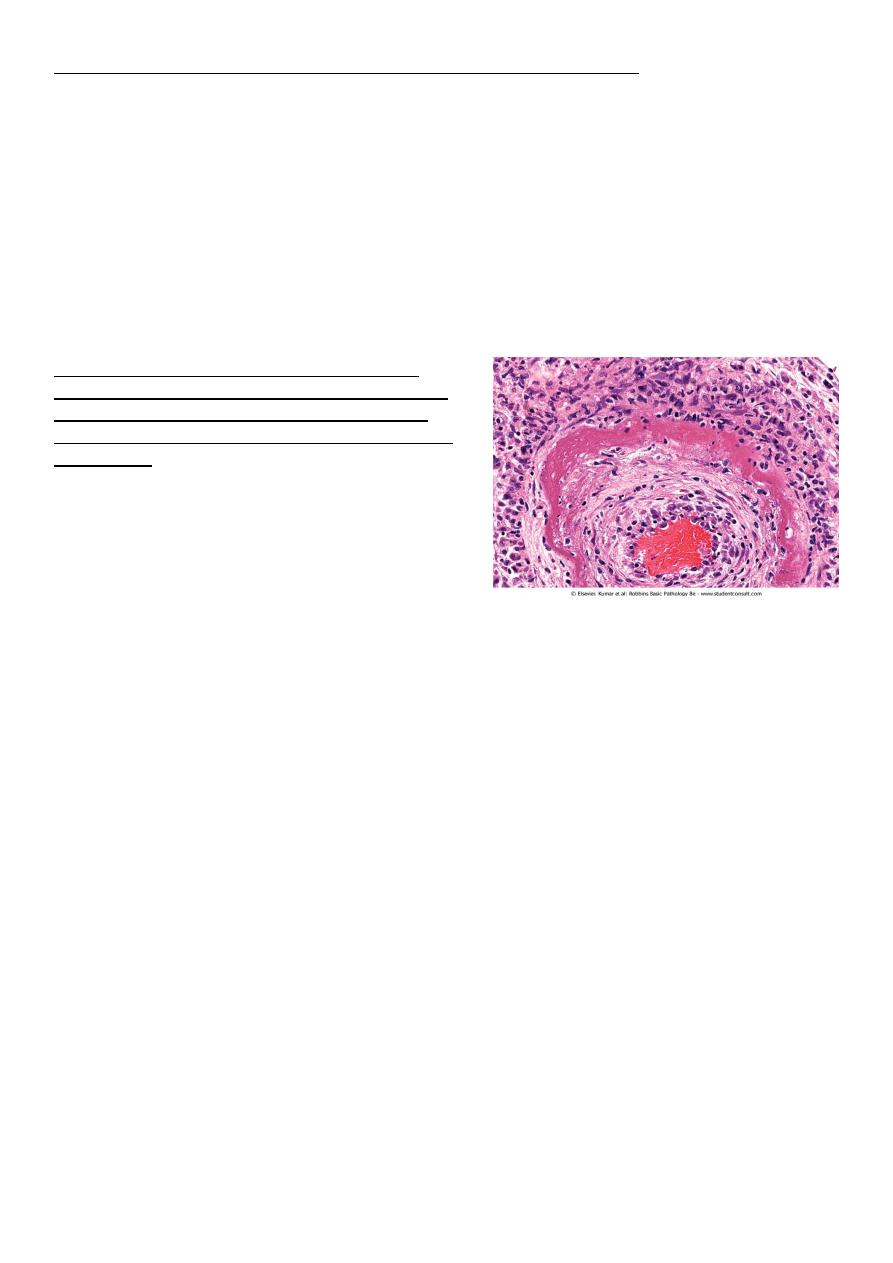
• Fibrinoid necrosis. typically seen immune reactions involving blood vessels. Deposits
of immune complexes, together with fibrin that has leaked out of vessels result in a
bright pink and amorphous appearance in H&E stains. This type is exemplified by the
necrosis seen in polyarteritis nodosa.
Fibrinoid necrosis in an artery in a patient with
polyarteritis nodosa. The wall of the artery shows a
circumferential bright pink area of necrosis with
protein deposition and inflammation (dark nuclei of
neutrophils).
Mechanisms of cell injury
• The outcome of the interaction between the injurious agent & the cell depend on:-
1) The type of injury, its duration and its severity.
Thus low doses of toxins or a brief duration of ischemia may lead to reversible cell
injury while large toxin doses or longer ischemic intervals may result in irreversible
injury and cell death.
2)
The type, adaptability and genetic makeup of the injured cell The same injury has
vastly outcomes depending on the cell type.
Thus striated skeletal muscle in the leg resist complete ischemia for 2-3- hours
without irreversible injury, whereas cardiac muscle dies after only 20-30 minutes.
The nutritional or hormonal status can also be important; clearly a glycogen filled
hepatocytes will tolerate ischemia much better than one that has just burden its last
glucose molecules.
Genetically determined diversity in metabolic pathways can also be important. For
instance, when exposed for the same dose of a toxin. Individual who inherit variants in
genes encoding cytochrom p-450 may cataboliz the toxin at different rates leading to
different outcomes.
Mechanisms of cell injury
The most important targets of injurious stimuli are:-
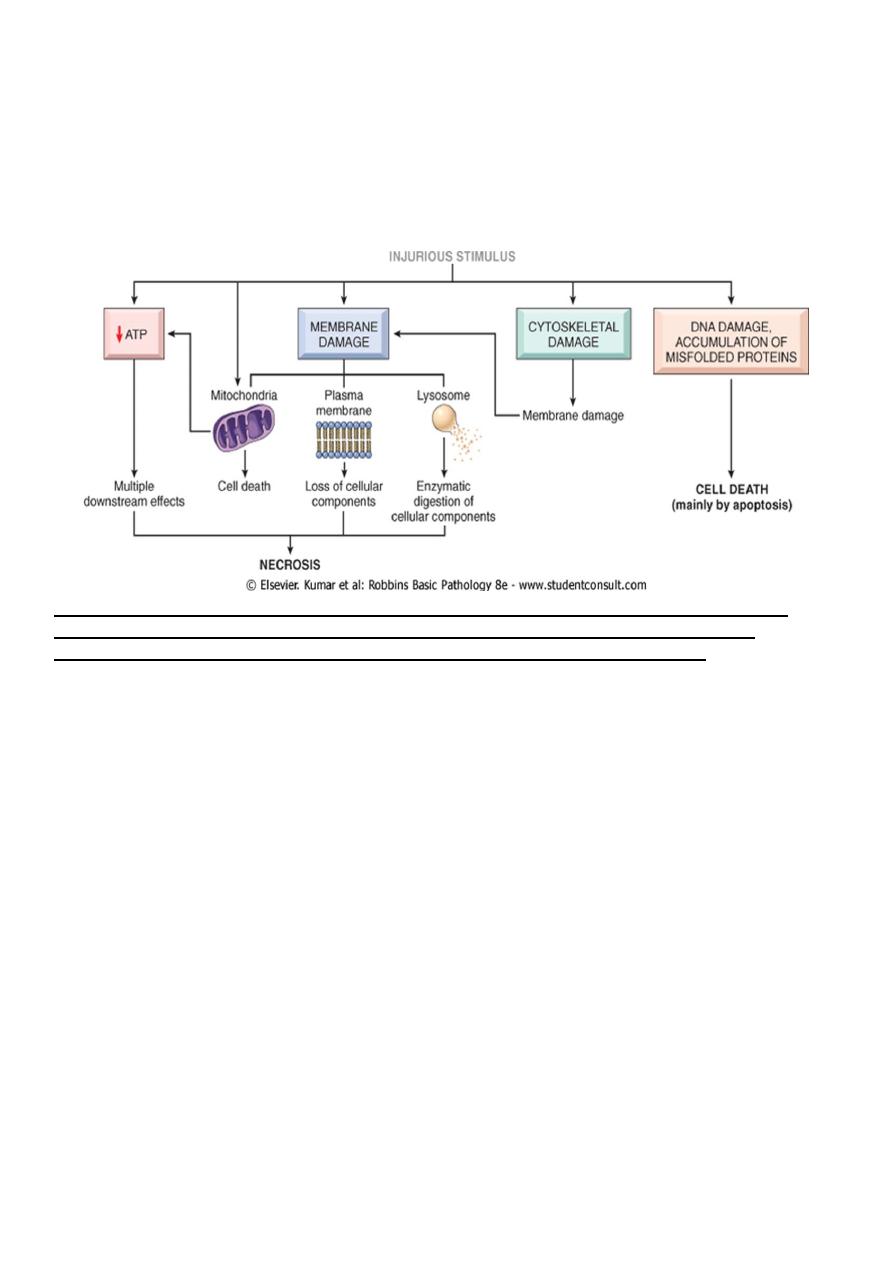
1. Mitochondria (the site of ATP generation).
2. Cell membrane which influence the ionic and osmotic homeostasis of the cell.
3. Protein synthesis (ribosome).
4. The cytoskeleton (microtubules and various filaments).
5. The genetic apparatus of the cell (nuclear DNA)
The principal cellular and biochemical sites of damage in cell injury. Note that loss of adenosine
triphosphate (ATP) results first in reversible injury (not shown) and culminates in necrosis.
Mitochondrial damage may lead to reversible injury and death necrosis or apoptosis.
ATP depletion
ATP the energy fuel of cells, is produced mainly by the oxidative phosphorylation of ATP
of the mitochondria. In addition the glycolytic pathway can generate ATP in the absence of
oxygen using glucose derived either from the circulation or from the hydrolysis of
intracellular glycogen (anerobic glycolysis).
• The major causes of ATP depletion:-
1. Reduce supply of oxygen and nutrients.
2. Mitochondrial damage.
3. The action of some toxins (e.g. cyadine)
High energy phosphate in the form of ATP is required for virtually all synthetic
and degrdatative and processes within the cell, include membrane transport,
protein synthesis, phospholipids turnover etc. depletion of ATP to less than 5% -10% of
normal levels has widespread effect on many cellular systems
ATP depletion
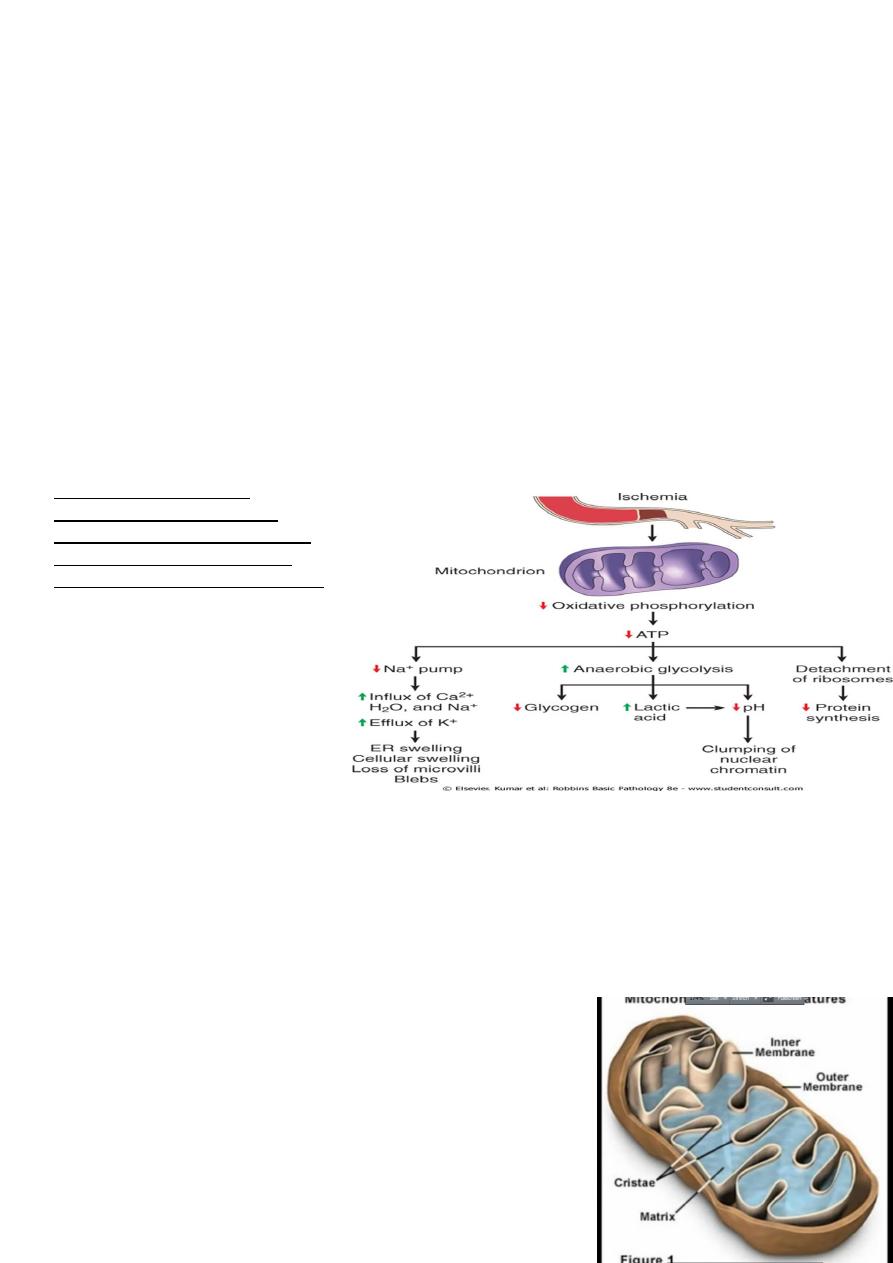
The activity of plasma membrane energy dependent sodium pump is reduce, resulting
in intracellular accumulation of sodium and efflux of potassium. The net gain of solute
is accompanied by iso- osmotic gain of water, causing cell swelling.
There is a compensatory increase in anaerobic glycolysis in an attempt to maintain the
cell energy sources. As a consequence, intracellular glycogen stores are rapidly
depleted and lactic acid accumulates, leading to decrease activity of many cellular
enzymes (due to decrease pH level).
Failure of the Ca pump lead to influx of Ca with damaging effects on numerous
cellular components, describe below.
Structural disruption of the protein synthetic apparatus manifested as detachment of
ribosomes from the rough endoplasmic reticulum (RER) and polysomes into
monosomes, with a consequent reduction in protein synthesis
Ultimately, there is irreversible damage to the mitochonerial and lysosomal
membranes and the cells undergoes necrosis.
The initial functional and
morphologic consequences of
decreased intracellular adenosine
triphosphate (ATP) during cell
injury. ER, Endoplasmic reticulum.
Mitochondrial damage:-
Mitochondria are supplies ATP, but they are also critical players in the cell injury and death.
Mitochondria can be damaged by increase of cytosolic Ca, reactive oxygen species, and
oxygen deprivation, and so they are sensitive to virtually all types of injurious stimuli,
including hypoxia
1. The formation of a channel in the mitochondrial membrane, called the permeability
transition pore. The opening of this channel leads to the loss of mitochondrial
membrane potential and PH changes, resulting in failure
of oxidative phosphorylation and progressive depletion
of ATP, culminating in necrosis of the cells.
2. Increase permeability of the mitochondrial membrane
may result in leakage of cytochrome c ( the major
protein involved in electron transport) that are capable
for activating apoptotic pathways. Thus cytochrome c
play a key dual role in cell survival and death in its
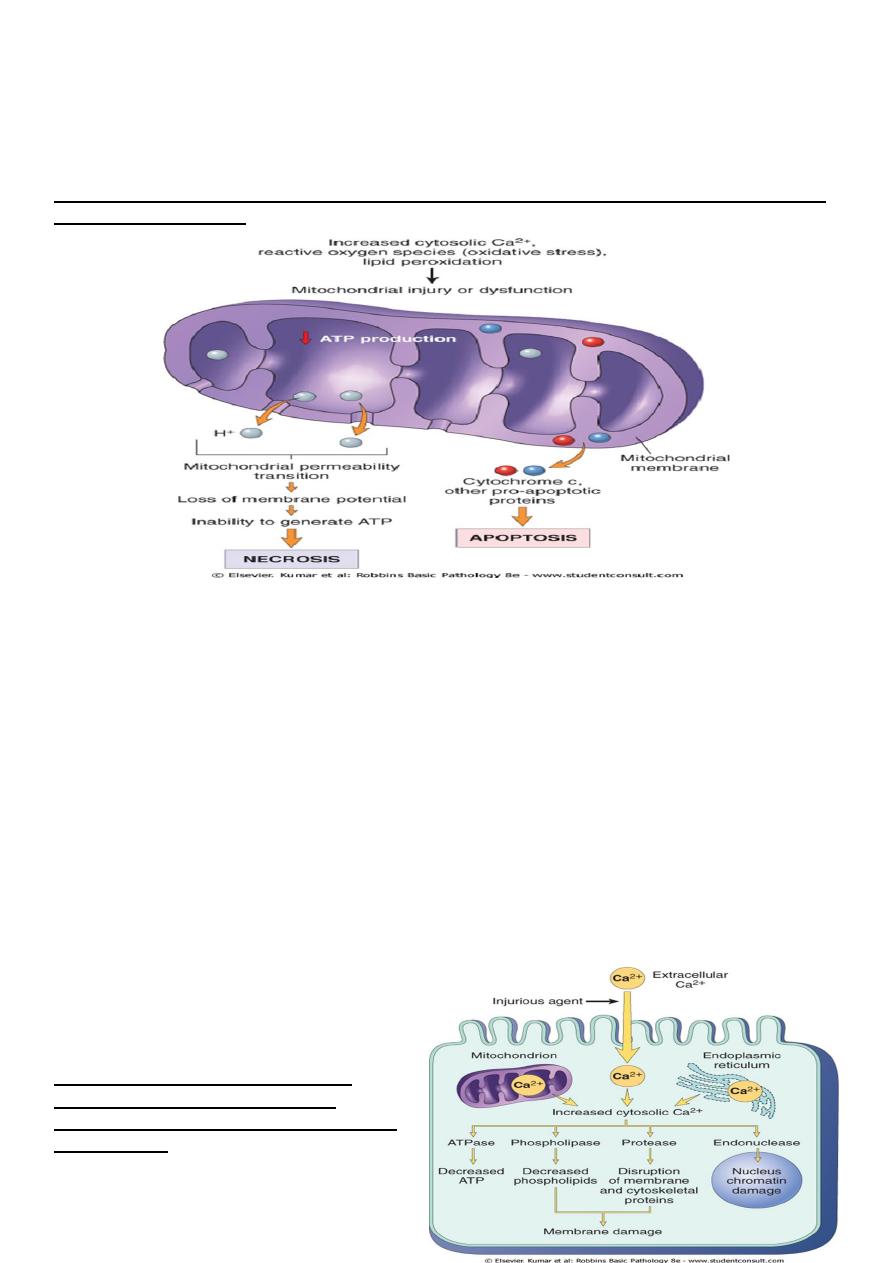
normal location inside the mitochondria, it's essential for energy generation and the
life of the cell, but when mitochoneria are damaged so severely that cytochrome c
leaks out; its signal to die by apoptosis.
Consequences of mitochondrial dysfunction, culminating in cell death by necrosis or apoptosis. ATP,
Adenosine triphosphate.
Influx of calcium:-
Cytoplasmic free calcium is normally maintained by ATP –dependent calcium pump(
transporter) at concentration that are 10,000 times lower than the concentration of
extracellular calcium or intracellular mitochondrial and ER calcium. Ischemia and certain
toxins causes an increase in cytoplasmic calcium concentration , initially because of release
of Ca from intracellular stores, and later resulting from increased influx across the plasma
membrane.of Ca from intracellular stores, and later resulting from increased influx across the
plasma membrane
.
Increased cytosolic calcium:-
1. Will activate a number of enzymes including phospholipase (which cause membrane
damage), protease (which breakdown both membrane and cytoskeletal proteins),
endonuclease which responsible for DNA and chromatin fragmentation), and ATPase
(worsen ATP depletion),
2. Induction of apoptosis by direct
activation of caspases.
Sources and consequences of increased
cytosolic calcium in cell injury. ATP,
Adenosine triphosphate; ATPase, adenosine
triphosphatase
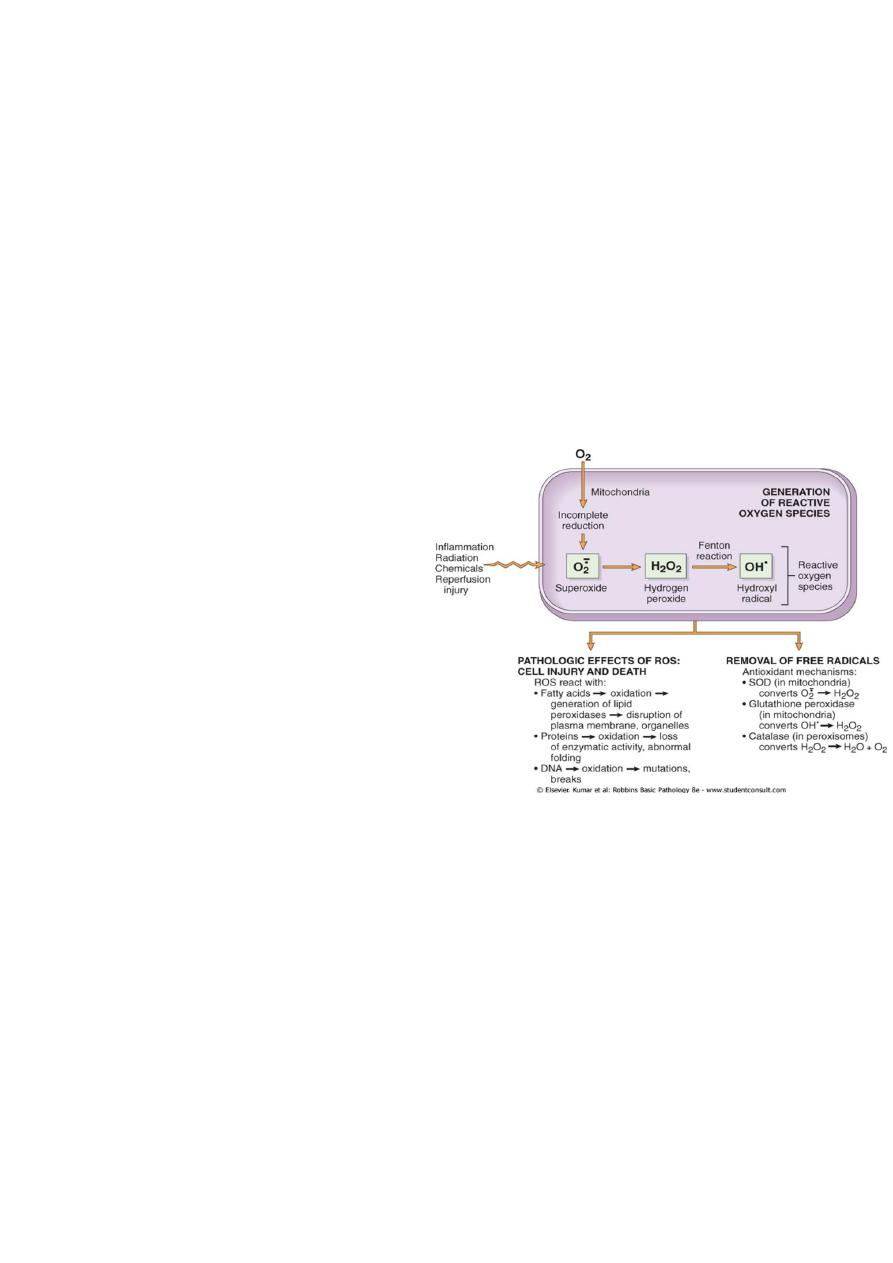
Accumulation of oxygen derived free radicals (oxidative stress)
• These are designated as reactive oxygen species (ROS) & these are units with a single
unpaired electron in their outer orbit. When generated in cells they avidly attack the
nucleic acids, cellular protein and lipids. The molecules that react with free radicals
are in turn converted into free radicals, thus propagating the chain of damage.ROS are
produced normally in cells during mitochoderial respiration and energy generation, but
they are degraded and removed by cellular defense systems. When their production
increase or the defense systems are ineffective, the result is excess of these free
radicals, leading to a condition called
oxidative stress.
Cell injury in many circumstances
involves damage by free radicals; these
include
1. Reperfusion injury.
2. Chemical and radiation injury.
3. Toxicity from oxygen and other
gases.
4. Cellular aging.
5. Inflammatory cells mediated tissue
injury
Defects in membrane permeability:-
Biochemical mechanisms contribute to membrane damage include:-
1. Decrease phopholipid synthesis due to fall in ATP levels. This affect all cellular
membrane including mitochondrial, which worsen the loss of ATP.
2. Degredation of membrane phospholipids due to activation of intracellular
phospholipase through increased levels of intracellular Ca.
3. Injury to cell membranes by oxygen free radicals (ROS) by lipid peroxidation.
4. Damaged to the cytoskeleton through activation of proteases by increased cytoplasmic
Ca.
5. They detergent effect of free fatty acids on membranes. These products result from
phospholipid degradation
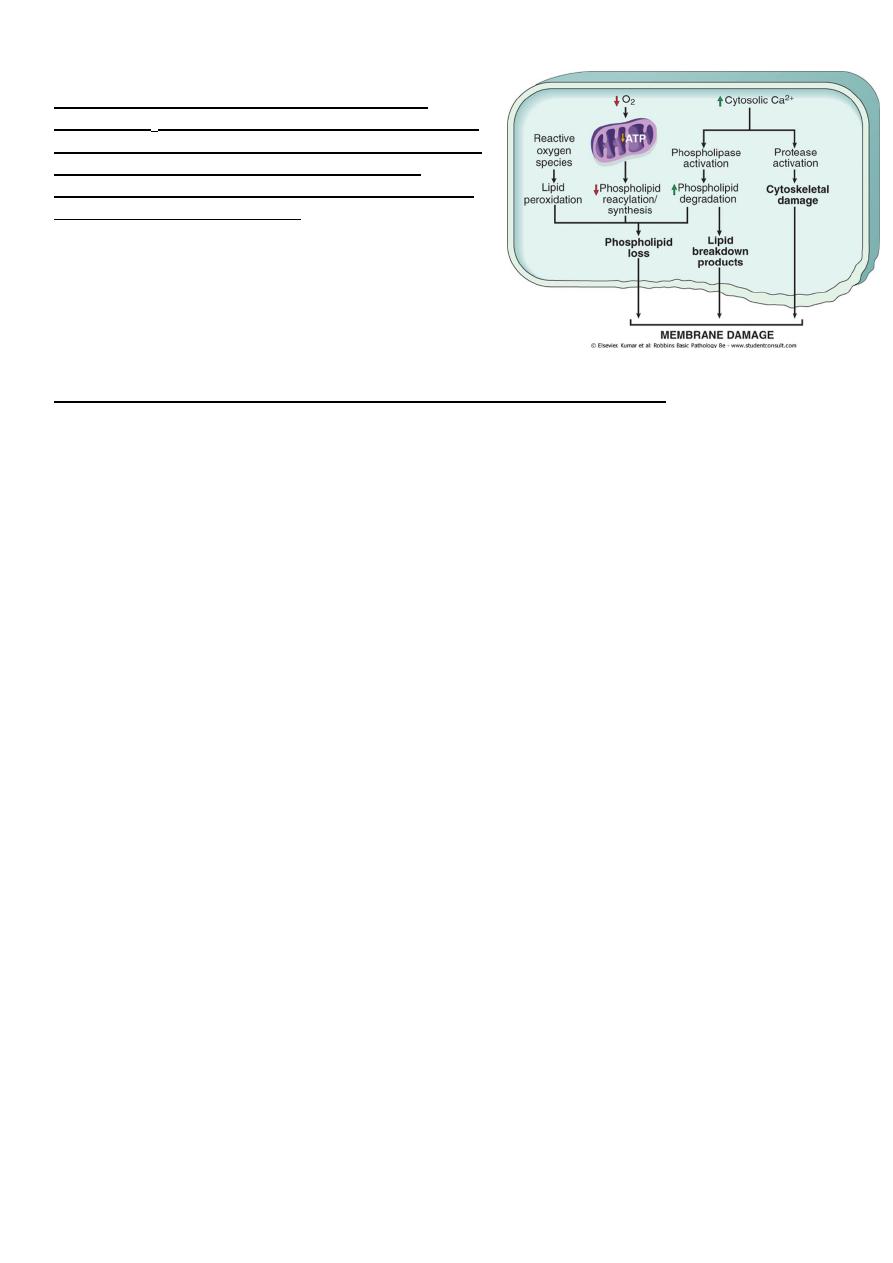
Mechanisms of membrane damage in cell injury.
Decreased O
2
and increased cytosolic Ca
2+
are typically
seen in ischemia but may accompany other forms of cell
injury. Reactive oxygen species, which are often
produced on reperfusion of ischemic tissues, also cause
membrane damage (not shown).
The most important site of membrane damage during cell injury are
1. Mitochondrial membrane damage: damage to mitochondrial membranes result in
decreased production of ATP , culminating in necrosis, alternatively release of
proteins triggers apoptotic death.
2. Plasma membrane damage:- lead to loss osmotic balance and influx of fluid and ions
as well as loss of cellular contents.
3. Injury to lysosomal membranes results in leakage of their enzymes into the
cytoplasm and activation of acid hydrolases in the acidic intracellular PH of the
injured (e.g. ischemic) cell. Lysosomes contain RNases, DNases, proteases and
components and the cells die b necrosis.
Damage to DNA& proteins
Cells have mechanisms that repair damage to DNA, but if this damage is too severe to
be corrected (e.g. after radiation injury or oxidative stress), the cell initiates its suicidal
program and die by apoptosis , a similar reaction is triggered by improperly folded
(configured) protein (see unfolded protein response) which may be the result of inherited
mutations or through free radicals. These mechanisms of cell injury typically cause
apoptosis.
Examples of cell injury and necrosis:-
1. Ischemic and hypoxic injury.
2. Reperfusion injury
Subcellular response to cell injury
Certain agents and stresses induce distinctive alterations involving only subcellular
organelles. Although some of these alterations occur in acute lethal injury, other are seen in
chronic forms of cell injury and still others are adaptive responses

• E.g. Abnormalities of the cytoskeleton may be manifested as an abnormal appearance
and function of cells (e.g. Mallory bodies, which represent intracellular accumulations
of fibrillar material in alcoholic liver disease).
• Autophagy :-refers to lysosomal digestion of the cells own components it is a survival
mechanism whenever there is nutrient deprivation ; the starved cell lived by eating its
own components. In this process, intracellular organelles are first sequestrated from
the cytoplasm in an autophagic vacuoles.
• Induction (hypertrophy of smooth ER):- the smooth ER is involve in the metabolism
of various chemicals and cells exposed to these chemicals show hypertrophy of the ER
as an adaptive response that may have important functional consequences.
Apoptosis:-
This form of cell death is a regulated suicide program in which the relevant cells activated
enzymes capable of degrading the thier own nuclear DNA and other cytoplasmic proteins
Fragments of the apoptotic cells then phagocyte without elicit an inflammatory reaction in
the host. Thus apoptosis differs from necrosis ; the latter is characterized by loss of
membrane integrity, leakage of cellular contents and frequently a host reaction.
Causes of apoptosis:-
Apoptosis in physiologic situations:
1. Death by apoptosis is a normal phenomenon that serves to eliminate cells that are no
longer need it is important in the following physiologic situations.
2. During emberyogenesis (organogenesis).
3. hormone deprivation as in endomaterial cells breakdown during the menstrual cycle.
4. In proliferating cells such as intestinal crypts epithelia ( to maintain a constant
number).
5. In self-reactive lymphocytes ( to prevent self tissue destruction).
6. Cytotoxic T lymphocytes- induce cell death ( a defense mechanism against viruses and
tumors that serves to kill and eliminate virus infected and neoplastic cells.
Apoptosis in pathologic condition:-
Apoptosis eliminates cells that are genetically altered or injured beyond repair without
eliciting a host reaction , thus keeping in the damaged as restricted as possible
Mechanism of apoptosis
The basic process can be understood as four separable but overlapping components:
1. Signaling.
2. Control and integration.

3. Execution.
4. Removal of the dead cells.
1) Signaling:- apoptosis may be triggered by a variety of signals ranging
from intrinsic activation of programmed cell death pathways (e.g during
embryogenesis).
withdrawal of growth factors or hormones.
release of granzymes by cytotoxic T cells.
or selected injurious agents radiation, toxins or free radicals (which damage DNA and
activate P53 pathways).
specific receptor –ligand interactions.
The TNF receptor is belong this superfamily of plasma membrane molecules (also FAS
surface molecule is belong this family ). These plasma membrane receptors share an
intracellular (death domain) adapter protein , when this protein is oligomerized lead to
activation of initiator caspases and a cascade of enzyme activation culminating in cell death
2) Control and integration: this is accomplished by specific proteins that connect the
original death signals to the final execution program. There are two broad pathways in this
stage
Direct transmission of death signals by specific adaptor proteins to the execution
mechanism
Regulation of the mitochondrial permeability by members of the BCL-2 family of
proteins. As we know (free radicals, increase of ca,) can result into formation of
mitochondrial transition pore with loss of the mitochondrial potential and more
depletion of ATP and also increase the permeability of the outer mitochondrial
membrane resulting in releasing of the cytochrome c which binds to certain pro-
apoptotic cytosolic proteins triggering the execution caspase culminating in cell death.
The BCL-2 and BCL-X (found in the mitochondrial membrane) suppress apoptosis
by preventing increased mitochondrial permeability, (act as inhibitors) while BAX and BAD
act as promotors (promote the programmed cell death).
3) Execution :- characterized by specific biochemical events result in synthesis and or
activation of number of cytosolic catabolizing enzymes culminating in morphological change
of apoptosis
4) Removal of the dead cells. The apoptotic cells and their fragments (apoptotic bodies)
express a new ligands on their surfaces that enhancing the phagocytosis and removing these
fragments without releasing of proinflammatory mediators (so inflammation is abscent).

Morphology
• In H&E-stained tissue sections, apoptotic cells may appear as round or oval masses
with intensely eosinophilic cytoplasm. Nuclei show various stages of chromatin
condensation, aggregation and karyorrhexis. The cells rapidly shrink, form
cytoplasmic buds, and fragment into apoptotic bodies composed of membrane-bound
vesicles of cytosol and
organelles. Because these
fragments are quickly extruded
and phagocytosed without
eliciting an inflammatory
response, even substantial
apoptosis may be histologically
undetectable.
Apoptosis of a liver cell in viral hepatitis.
The cell is reduced in size and contains

brightly eosinophilic cytoplasm and a condensed nucleus.
Example of apoptosis
• Growth Factor Deprivation
Hormone-sensitive cells deprived of the relevant hormone, lymphocytes that are not
stimulated by antigens and cytokines, and neurons deprived of nerve growth factor die by
apoptosis. In all these situations, apoptosis is triggered by the mitochondrial pathway and is
attributable to activation of pro-apoptotic members of the Bcl-2 family and decreased
synthesis of Bcl-2 and Bcl-x
L
.
• DNA Damage
Exposure of cells to radiation or chemotherapeutic agents induces DNA damage, and
if this is too severe to be repaired it triggers apoptotic death. When DNA is damaged, the p53
protein accumulates in cells. It first arrests the cell cycle (at the G
1
phase) to allow time for
repair. However, if the damage is too great to be repaired successfully, p53 triggers
apoptosis, mainly by activating sensors that ultimately activate BAX and BAD, and by
stimulating synthesis of pro-apoptotic members of the Bcl-2 family. When p53 is mutated or
absent (as it is in certain cancers), it is incapable of inducing apoptosis, so that cells with
damaged DNA are allowed to survive. In such cells, the DNA damage may result in
mutations or translocations that lead to neoplastic transformation.
Example of apoptosis
Cytotoxic T Lymphocyte-Mediated Apoptosis
Cytotoxic T lymphocytes (CTLs) recognize foreign antigens presented on the surface
of infected host cells and tumor cells. Upon activation, CTL granule proteases called
granzymes enter the target cells. Granzymes are able to activate cellular caspases. In this
way, the CTL kills target cells by directly inducing the effector phase of apoptosis, without
engaging mitochondria or death receptors. CTLs also express FasL on their surface and may
kill target cells by ligation of Fas receptors.
Intracellular accumulations
Cells may accumulate abnormal amount of various substances; these may be harmless or
associated with injury. The location of these substances are either cytoplasmic within
organelles ( typically lysosomes) or in the nucleus.

Fatty change (steatosis):-
This refer to an abnormal accumulation of triglycerides within paranchymal cells. It is most
often seen in the liver ,since this is the major organ involved in fat metabolism, but it may
also occur in the heart.
Causes of fatty change include
1. Toxin including alcohol
2. Diabetes mellitus
3. Obesity
4. Protein malnutrition
5. Anoxia
Fatty change (steatosis)
Alcohol abuse and diabetes associated with obesity are the most common cause of fatty
change in the liver ( fatty liver) in industrialized nations
Free fatty acids from adipose tissue or ingested food are normally transported into
hepatocytes, where they are esterified to triglycerides converted into cholesterol or
phospholipids or oxidized to ketone bodies. Triglycerides from the hepatocytes required the
formation of complexes with apoprotiens to form lipoproteins which are able to enter the
circulation.
Excess accumulation of triglycerides may result from defect at any step from fatty acid entry
to lipoprotein exit.
Hepatotoxins e.g.(alcohol) alter mitochondrial and SER function and thus inhibit fatty
acid oxidation;
CCL4 and protein malnutrition decreases the synthesis of apoprotiens
anoxia inhibits fatty acid oxidation
starvation increase fatty acid mobilization from peripheral stores.
The significance of fatty change depends on the cause and severity of the accumulation.
When mild it may have no effect. More severe fatty change may transiently impair cellular
function, but the change is reversible. In the severe form, fatty change may precede cell
death.
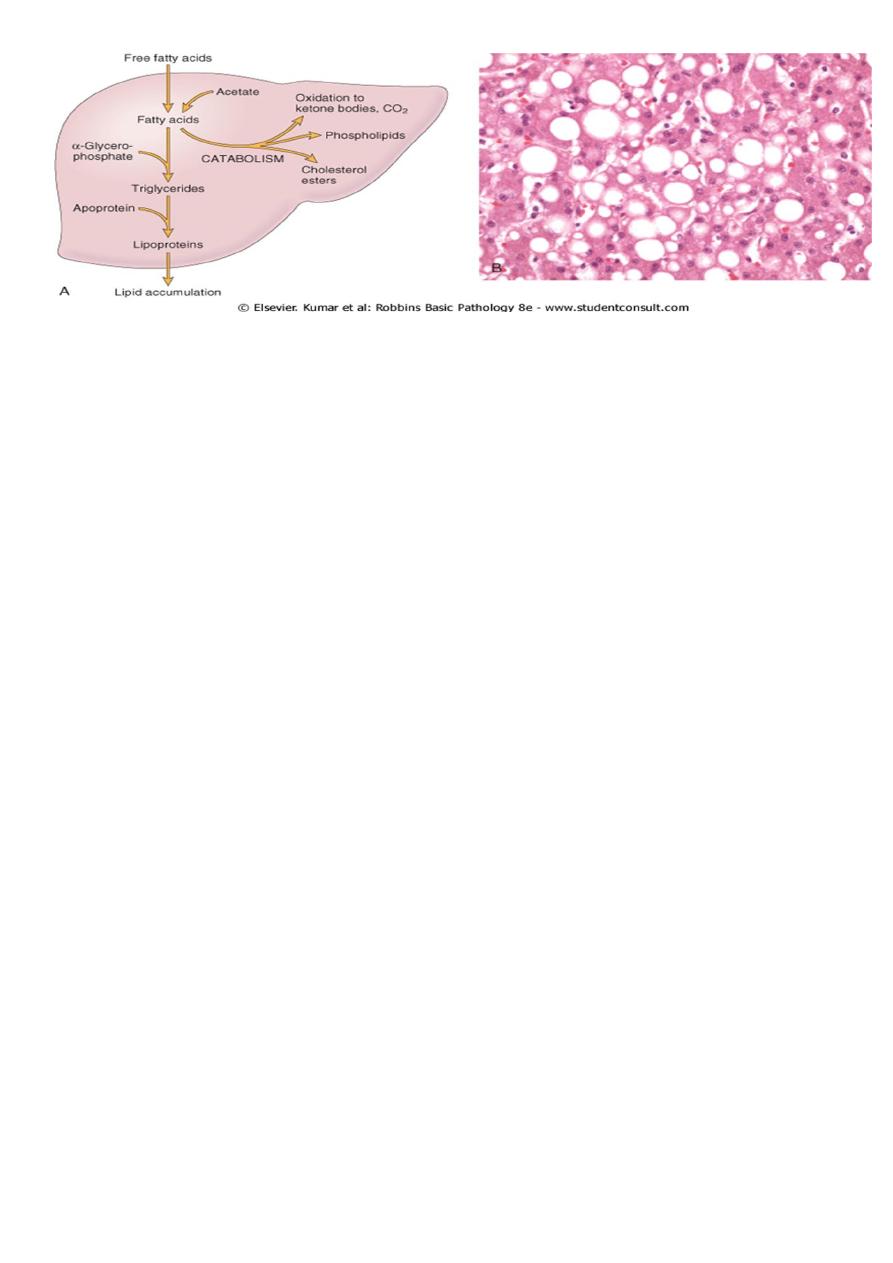
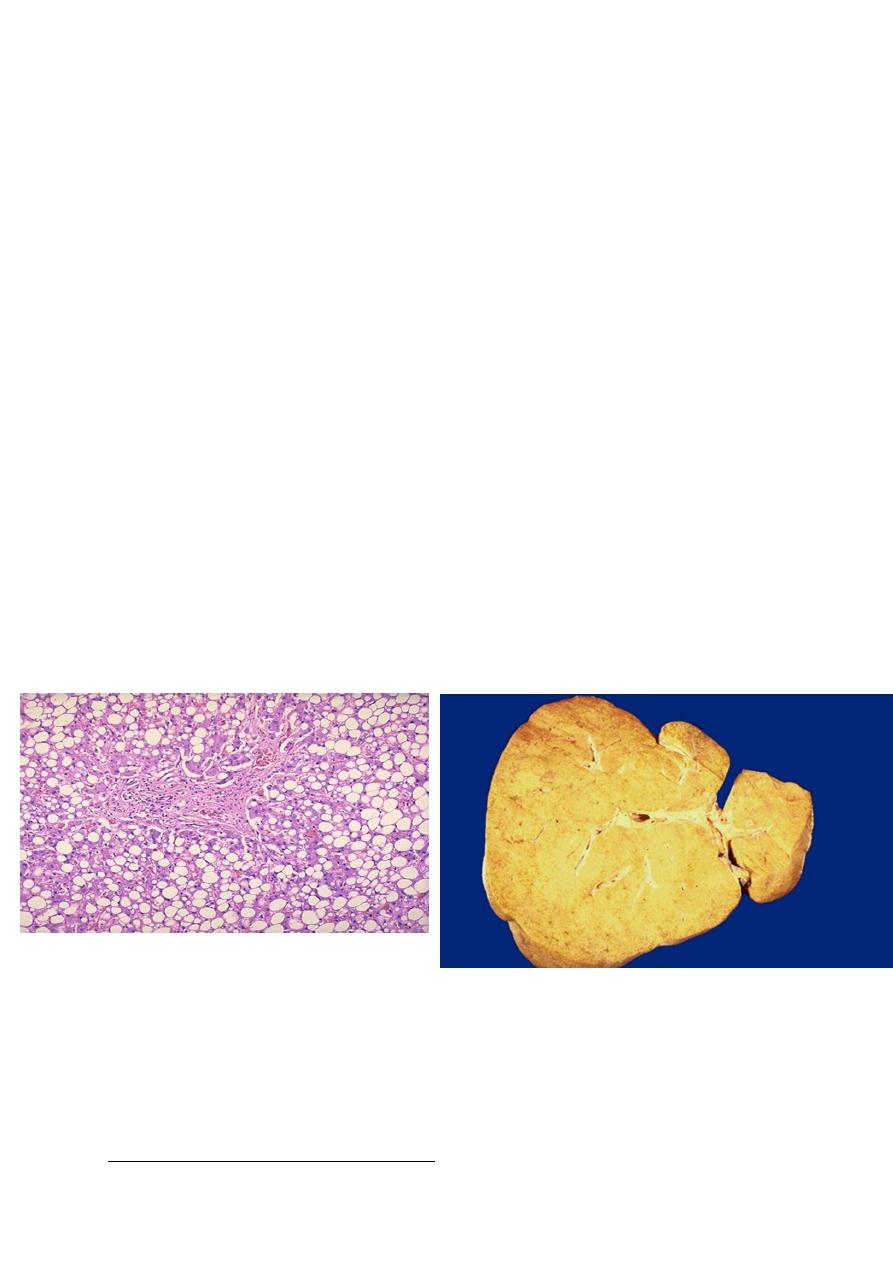
Gross features:-
• Fatty changes is most commonly seen in the liver and heart.
• In the liver mild fatty change may not affect the gross appearance
• With increasing the accumulation, the organ enlarged and become progressively
yellow, until in extreme cases it may weight 5 Kg (3 times the normal weight) and
appear bright yellow, soft and greasy
Microscopic features:-
• Early fatty changes is seen by light microscopy as small fat vacuoles in the cytoplasm
around the nucleus
• In later stages, the vacuoles coalesce to create cleared spaces that displace the nucleus
to the cell periphery.
Cholesterol and cholesterol esters accumulations:-
Cellular cholesterol metabolism is tightly regulated to ensure normal cell membrane
synthesis without significant intracellular accumulation. However, phagocytic cell may
become overloaded with lipid ( triglycerides, cholesterol and cholesteryl esters) in several
different pathologic processes
• Macrophage in contact with lipid debris of necrotic cells or abnormal (e.g. oxidized)
form of lipoproteins may become stuffed with phagocytosed lipid. These macrophages
may be filled with minutes, membrane bound vacuoles of lipid, imparting a foamy
appearance to their cytoplasm (foam cells).
• In atherosclerosis, smooth muscle cells and macrophages are filled with lipid vacuoles
composed of cholesterol and cholesterol esters; these give atherosclerotic plaques their
characteristic yellow color and contribute to the pathogenesis of lesion
Protein accumulations:-
• Morphologically visible protein accumulations may occur because excesses are
presented to the cells or because the cells synthesize excessive amounts. In the kidney
for example; trace amount of albumin filtered through the glomerulus are normally
reabsorbed by pinocytosis in the proximal convoluted tubules. However in disorders
with heavy protein leakage across the glomerular filter (e.g. nephrotic syndrome),
there is a much larger reabsorption of protein. Pinocytic vesicles containing this
protein fuse with lysosomes, resulting in the histologic appearance of pink, hyaline
cytoplasmic droplets.
• Another example is the accumulation of intracellular proteins are also seen in certain
type of cell injury. For example, the Mallory body (or alcoholic hyaline) is an
eosinophilic cytoplasmic inclusion in liver cells that is highly characteristic of alcohol
liver disease. Such inclusions are composed predominantly of aggregated
intermediate filaments that presumably resist degradation.
Pigments:-
are colored substances that exogenous, coming from outside the body, or endogenous
synthesized within the body itself.
• Exogenous pigments; the most common of these is carbon (an example is coal dust), a
ubiquitous air pollutant of urban life. When inhaled, it is phagocytosed by alveolar
macrophages and transported through lymphatic channels to the regional
tracheobronchial lymph nodes. Aggregates of the pigment blacken the draining lymph
nodes and pulmonary parenchyma (anthracosis) heavy accumulations may induce
fibroblastic reaction that can result in a serious lung diseases called coal workers
pneumoconiosis
• Endogenous pigments; include lipofuscin, melanin and certain derivatives of
hemoglobin material that accumulates in a variety of tissues (particularly the heart,
liver and the brain) as a function of age or atrophy.
• Lipofuscin represents complexes of the lipid and protein that derived from lipids
peroxidation of subcellular membranes. It is not injurious to the cell but is important

as a marker of past free radical injury. The brown pigment, when present in large
amounts, imparts an appearance to the tissue that is called brown atrophy
• Melanin:- is an endogenous, brown-black pigment produced in melanocytes following
the tyrosinase- catalyzed oxidation of tyrosine to dihydroxyphenylalanine. It is
synthesized exclusively by melanocytes located in the epidermis and acts as a screen
against harmful ultraviolet radiation. Although melanocytes are the only source of
melanin, adjacent basal keratinocytes in the skin can accumulate the pigment (e.g. in
freckles), as can dermal macrophages.
Pigmented nevi
Above: these are benign nevi. Small brown flat to slightly
raised nevi are quite common. They are usually less than
a centimeter in diameter.
Below: in this nevus, there are nevus cells in nests in the
lower epidermis. They show brownish melanin pigment.
Hemosiderin: is a hemoglobin-derived granular pigment that is golden yellow to brown and
accumulates in tissues when there is a local or systemic excess of iron. Hemosiderin pigment
represent large aggregates of iron, readily visualized by light microscopy; the iron can be
identified by the Prussian blue histochemical reaction. Local excesses of iron and
consequently of hemosiderin, result from hemorrahage. The best example is the common
bruise.
Whenever there is systemic overload of iron, hemosiderin is deposited in many organs and
tissues including their macrophages. This condition is called hemosiderosis . with
progressive accumulation, paranchymal cells throughout the body (but principally the liver,
pancreas, heart, and endocrine organs) become (bronzed) with accumulating pigments.
Hemosiderosis occurs in the setting of
• Increased absorption of dietary iron
• Impaired utilization of iron
• Hemolytic anemia and
• Transfusion (the transfused red cells constitute an exogenous load of iron)
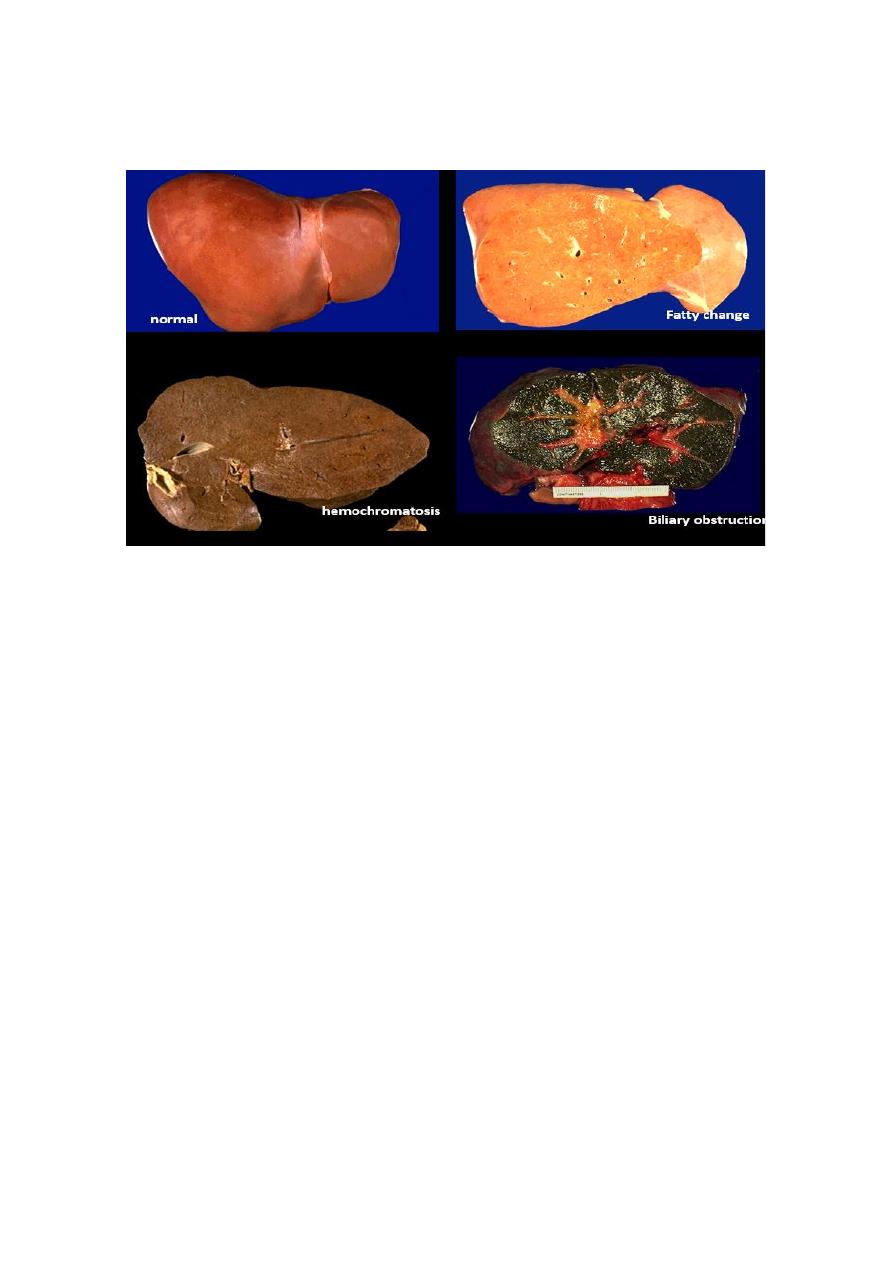
In most of instances of systemic hemosidrosis, the iron pigment does not damage he
parenchymal cells or impair organ function despite an impressive accumulation. However
more extensive accumulations of iron are seen in hereditary hemochromatosis, with tissue
injury including liver fibrosis, heart failure, and diabetes mellitus.
Pathological calcification:-
• Pathologic calcification is a common process in a wide variety of diseases states; it
implies the abnormal deposition of calcium salts.
• When the deposition occurs in dead or dying tissues, it is called dystrophic
calcification; it occur in the absence of calcium metabolic derangements (i.e. with
normal serum levels of calcium).
• In contrast, the deposition of calcium salts in normal tissues is known as metastatic
calcification and almost always reflects some derangement in calcium metabolism
(hypercalcemia). It should be noted that while hypercalcemia is not a prerequisting for
dystrophic calcification, it can exacerbate it.
Dystrophic calcification:- is encountered in
• Areas of necrosis ( of any type as coagulative, caseous, etc.)
• Advanced atherosclerosis ( as in the aorta and coronaries)
• Aging or damaged heart valves resulting in severely impaired valve motion.
Dystrophic calcification of the aortic valves is an important cause of aortic stenosis in
the elderly.
• Regardless of the site, calcium salts are grossly seen as fine white granules or clumps,
often felt as gritty deposits.

•
• microscopically:- calcification appear as intracellular and/or extracellular basophilic
(bluish) deposits. In time, metaplastic bone may be formed in the focus of
calcification.
Dystrophic calcification
Metastatic calcification:-
This is seen in cases of hypercalcemia of any cause. The four major causes of hypercalcemia
are:-
• Increased secretion of parathyroid hormone (primary parathyroid tumors or production
of parathyroid hormone-related protein by other malignant tumors)
• Destruction of bone (effect of accelerated turnover as in Paget disease, immobilization
or tumors due to increase bone catabolism associated with multiple myeloma,
leukemia or diffuse skeletal metastases
• Vitamin D related disorders including vitamin D intoxication and sarcoidosis (in
which macrophages activate a vitamin D precursor)
• Renal failure, in which phosphate retention leads to secondary hyperparathyroidism.
• Metastatic calcification can occur widely throughout the body but principally affects
the interstitial tissues of the vessels, kidneys, lungs and gastric mucosa. The calcium
deposits morphologically resemble those described in dystrophic calcification.
Although they do not generally cause clinical dysfunction, extensive calcifications in
the lungs may produce remarkable radiographs and respiratory deficits and massive
deposits in the kidney (nephrocalcinosis) can cause renal damage.
Thanks for you
like a phoenix rising from ashes