
Myelodysplastic syndrome

Myelodysplastic syndrome
• Myelodysplastic syndrome (MDS) consists of a
group of clonal haematopoietic disorders which
represent steps in the progression to the
development of leukaemia.
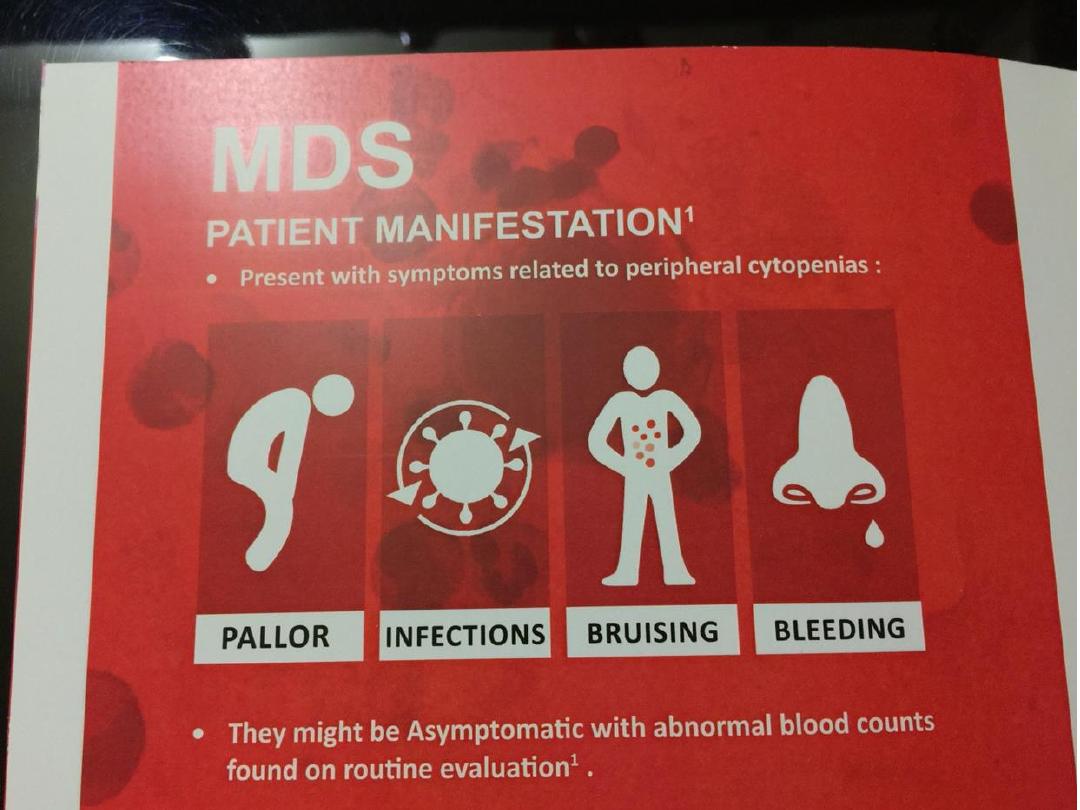
• MDS presents with consequences of bone marrow
failure (anaemia, recurrent infections or bleeding),
usually in older people (median age at diagnosis is
69 years).
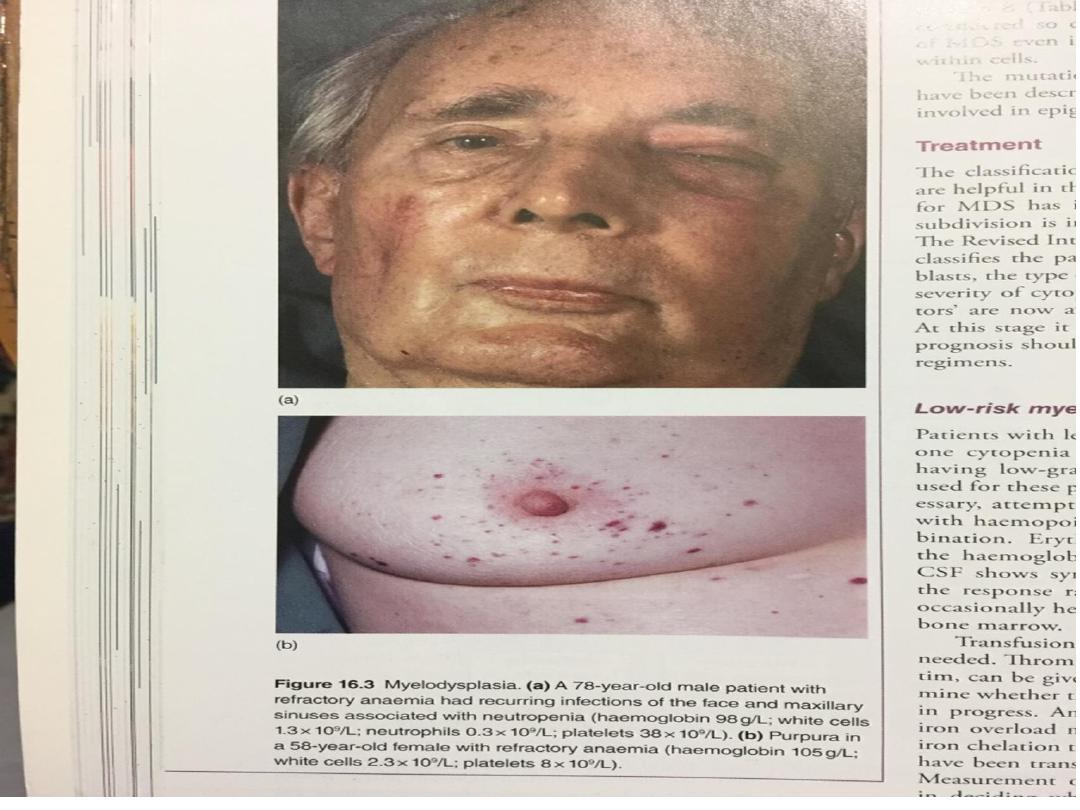
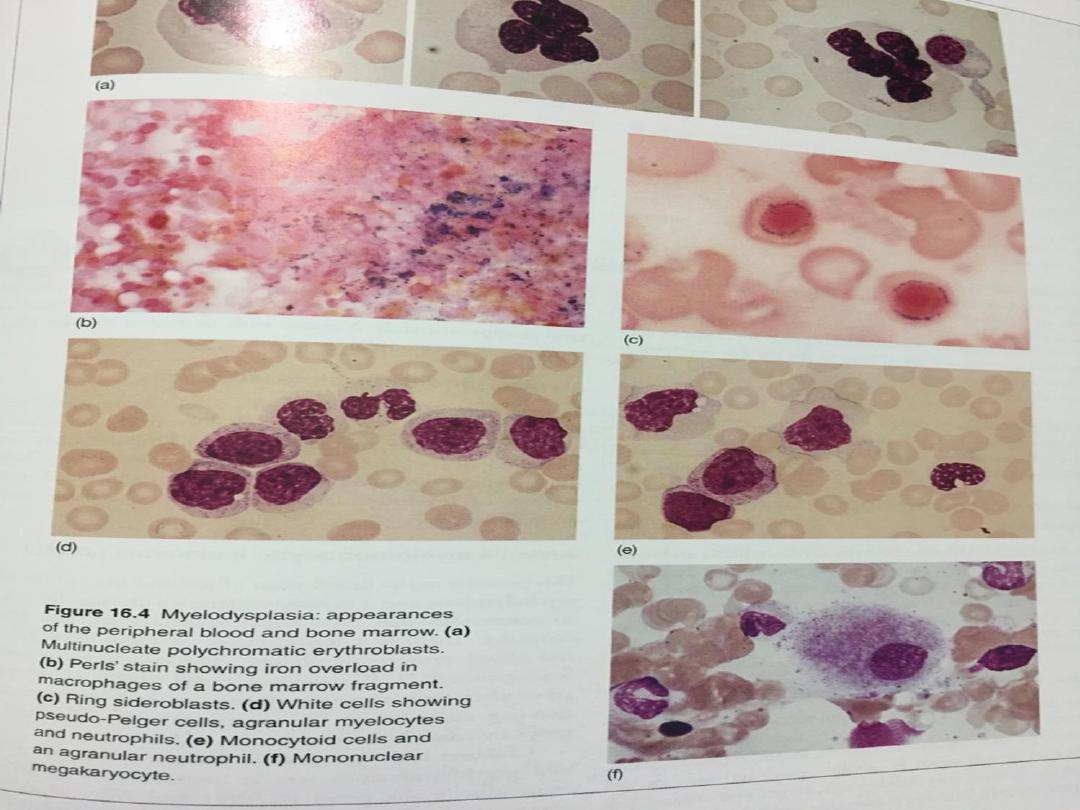
• The blood film is characterised by cytopenias and
abnormal-looking (dysplastic) blood cells, including
macrocytic red cells and hypogranular neutrophils
with nuclear hyper- or hyposegmentation.

• The bone marrow is hypercellular, with dysplastic
changes in all three cell lines. Blast cells may be
increased but do not reach the 20% level that
indicates acute leukaemia
• Chromosome analysis frequently reveals
abnormalities, particularly of chromosome 5 or 7.
• Inevitably, MDS progresses to AML, although the
time to progression varies (from months to years)
with the subtype of MDS, being slowest in
refractory anaemia and most rapid in refractory
anaemia with excess of blasts..




• An international prognostic scoring system (IPSS)
predicts clinical outcome based upon karyotype and
cytopenias in blood, as well as percentage of bone
marrow blasts
• . In low-risk patients, median survival is 5.7 years
and time for 25% of patients to develop AML is 9.4
years; equivalent figures in high-risk patients are 0.4
and 0.2 years, respectively

WHO classification of myelodysplastic syndromes:
• Refractory anaemia (RA) Blasts < 5% Erythroid
dysplasia only
• Refractory anaemia with sideroblasts (RARS)
Blasts < 5% , Ringed sideroblasts > 15%
• Refractory cytopenias with multilineage dysplasia
(RCMD) Blasts < 5% 2–3 lineage dysplasia
• Refractory anaemia with excess blasts (RAEB)
Blasts 5–20% 2–3 lineage dysplasia
• Myelodysplastic syndrome with 5q− Myelodysplastic
syndrome associated with a del (5q) cytogenetic
abnormality Blasts < 5% Often normal or increased
blood platelet count
• Myelodysplastic syndrome Unclassified None of the
above or inadequate material

Management
• For the vast majority of patients who are elderly, the
disease is incurable, and supportive care with red cell
and platelet transfusions is the mainstay of treatment.
• A trial of erythropoietin and granulocyte–colony-
stimulating factor (G–CSF) is recommended in some
patients with early disease to improve haemoglobin
and white cell counts.
• younger patients with higher-risk disease, allogeneic
HSCT may afford a cure. Transplantation should be
preceded by intensive chemotherapy in those with
more advanced disease., More recently, the
hypomethylating agent azacytidine has improved
survival by a median of 9 months for high-risk patients



HAEMATOPOIETIC STEM CELL TRANSPLANTATION
• Transplantation of haematopoietic stem cells (HSCT)
has offered the only hope of ‘cure’ in a variety of
haematological and non-haematological disorders .
• As standard treatment improves, the indications for
HSCT are being refined and extended, although its use
remains most common in haematological malignancies.
The type of HSCT is defined according to the donor and
source of stem cells:
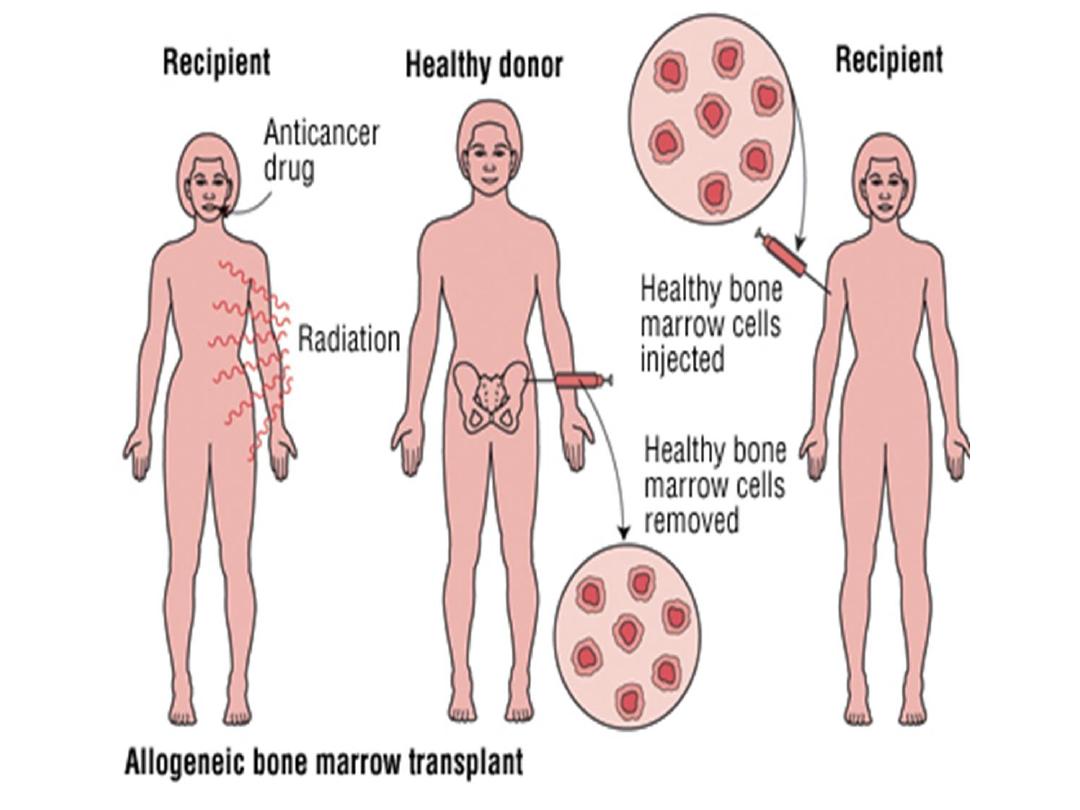
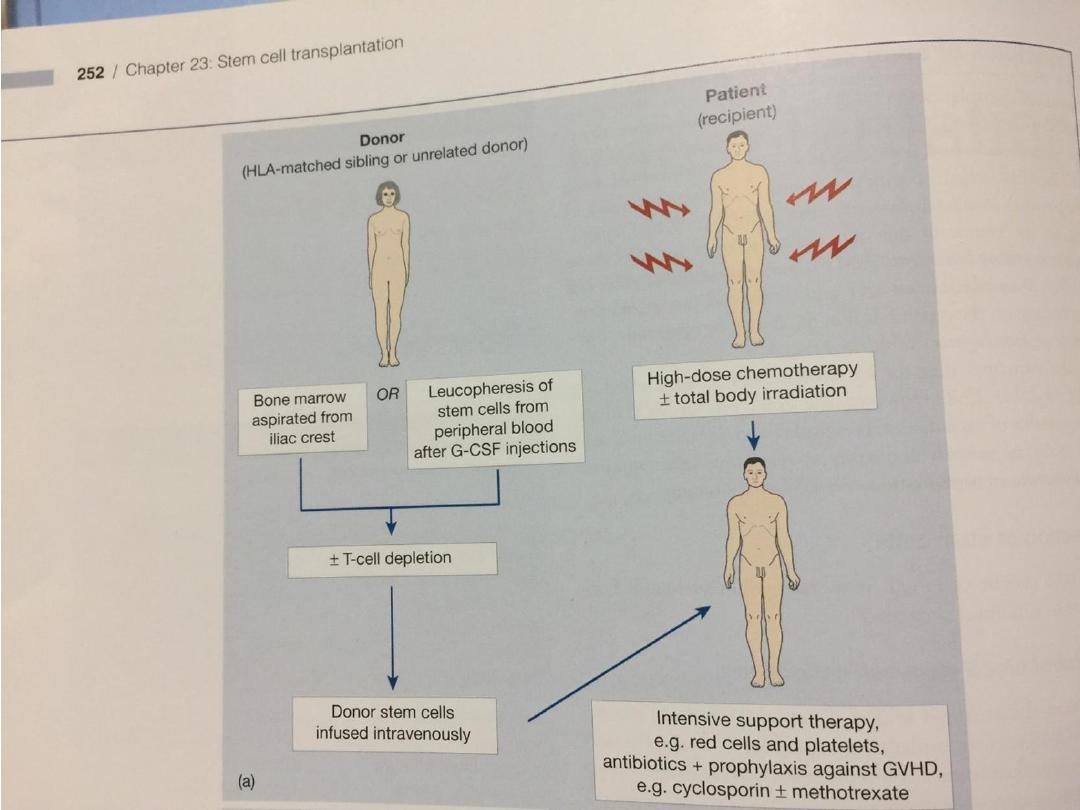
• In allogeneic HSCT, the stem cells come from a donor –
either related (usually an HLA-identical sibling) or a
closely HLA-matched volunteer unrelated donor (VUD).
• In an autologous transplant, the stem cells are
harvested from the patient and stored in the vapour
phase of liquid nitrogen until required. Stem cells can
be harvested from the bone marrow or from the blood


Allogeneic HSCT
• Healthy bone marrow or blood stem cells from a donor
are infused intravenously into the recipient, who has
been suitably ‘conditioned’.
• The conditioning treatment (chemotherapy with or
without radiotherapy) destroys malignant cells and
immunosuppresses the recipient, as well as ablating
the recipient’s haematopoietic tissues (myeloablation).
• The infused donor cells ‘home’ to the marrow, engraft
and produce enough erythrocytes, granulocytes and
platelets for the patient’s needs after about 3–4 weeks.
• During this period of aplasia, patients are at risk of
infection and bleeding, and require intensive
supportive care It may take several years to regain
normal immunological function and patients remain at
risk from opportunistic infections, in particular in the
first year.

Indications for allogeneic HSCT
• Neoplastic disorders affecting stem cell
compartments (e.g. leukaemias)
• Failure of haematopoiesis (e.g. aplastic anaemia)


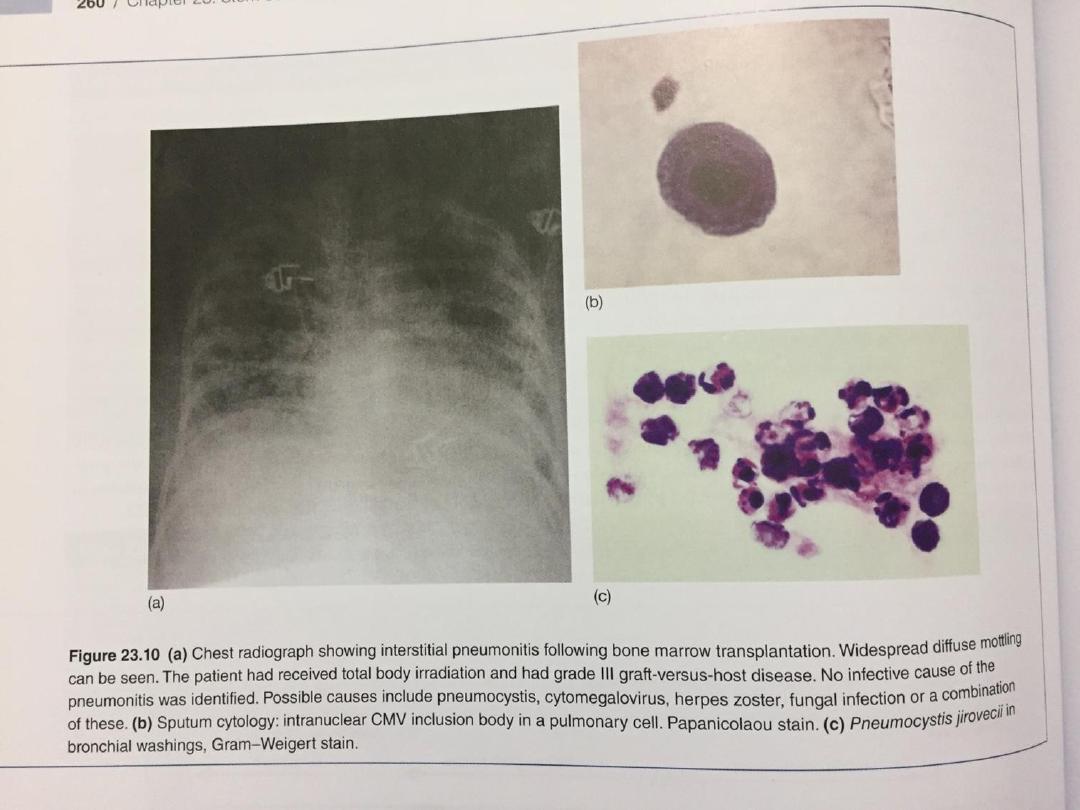
Complications of allogeneic HSCT
Early
• Anaemia
• Infections
• Bleeding
• Acute GVHD
• Mucositis – pain,
nausea, diarrhoea
• Liver veno-occlusive
disease
Late
• Chronic GVHD
• Infertility
• Cataracts
• Secondary malignancy


The chance of GVHD is:
• Around 30% to 40% when the donor and recipient
are related Around 60% to 80% when the donor and
recipient are not related
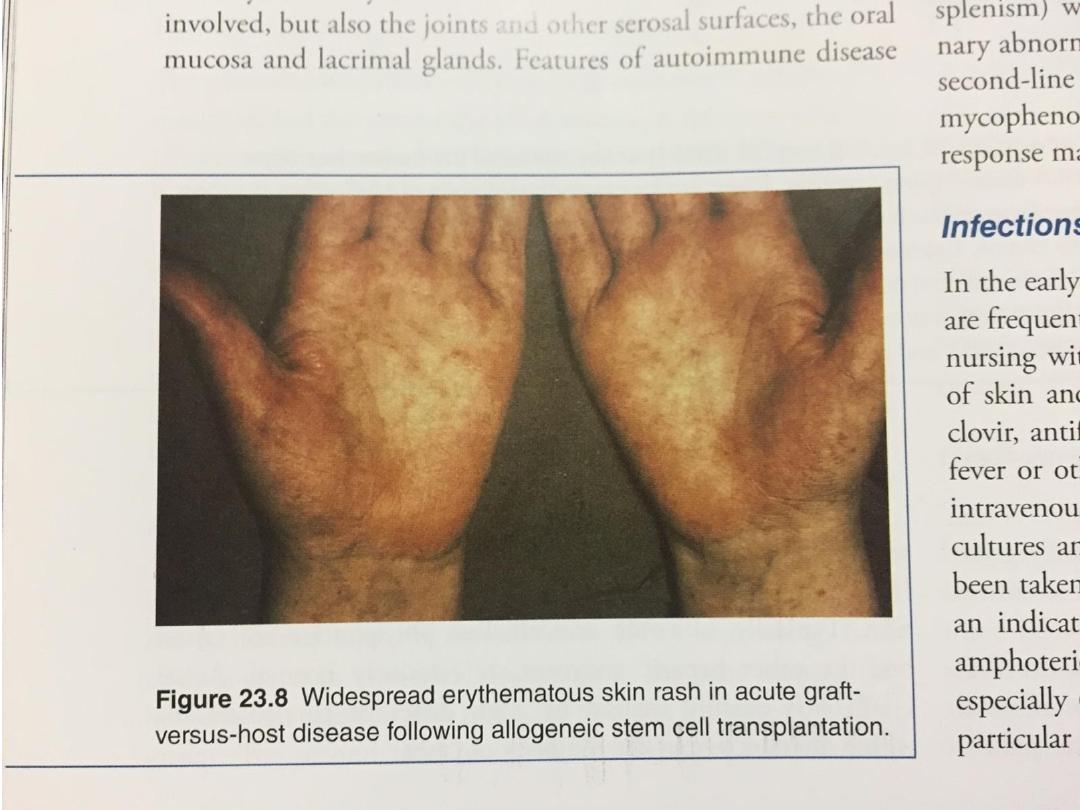
Graft-versus-host disease
• Acute GVHD usually happens within the first 6
months after a transplant. Common acute
symptoms include:
• Abdominal pain or cramps, nausea, vomiting, and
diarrhea , Jaundice ,skin rash

• Chronic GVHD usually starts more than 3 months
after a transplant, and can last a lifetime. Chronic
symptoms may include:
1. Dry eyes or vision changes
2. Dry mouth, white patches inside the mouth, and
sensitivity to spicy foods
3. Fatigue, muscle weakness, and chronic pain
4. Joint pain or stiffness
5. Skin rash with raised, discolored areas, as well as
skin tightening or thickening
6. Shortness of breath due to lung damage
7. Vaginal dryness
8. Weight loss

Donor lymphocyte infusion
• Donor leukocyte infusion is the infusion in
which lymphocytes from the original stem cell donor
are infused, after the transplant, to augment an anti-
tumor immune response or ensure that the donor stem
cells remain engrafted.
• These donated white blood cells contain cells of
the immune system that can recognize and destroy
cancer cells.
• The goal of this therapy is to induce a remission of the
patient's cancer by a process called the graft-versus-
tumor effect (GVT).
• The donor T-cells can attack and control the growth of
residual cancer cells providing the GVT effect. It is
hoped that the donor leukocyte infusion will cause GVT
and lead to a remission of the patients cancer.

• Complications of DLI include acute and
chronic graft-versus-host disease and bone
marrow aplasia, resulting
in immunosuppression and susceptibility
to opportunistic infections.

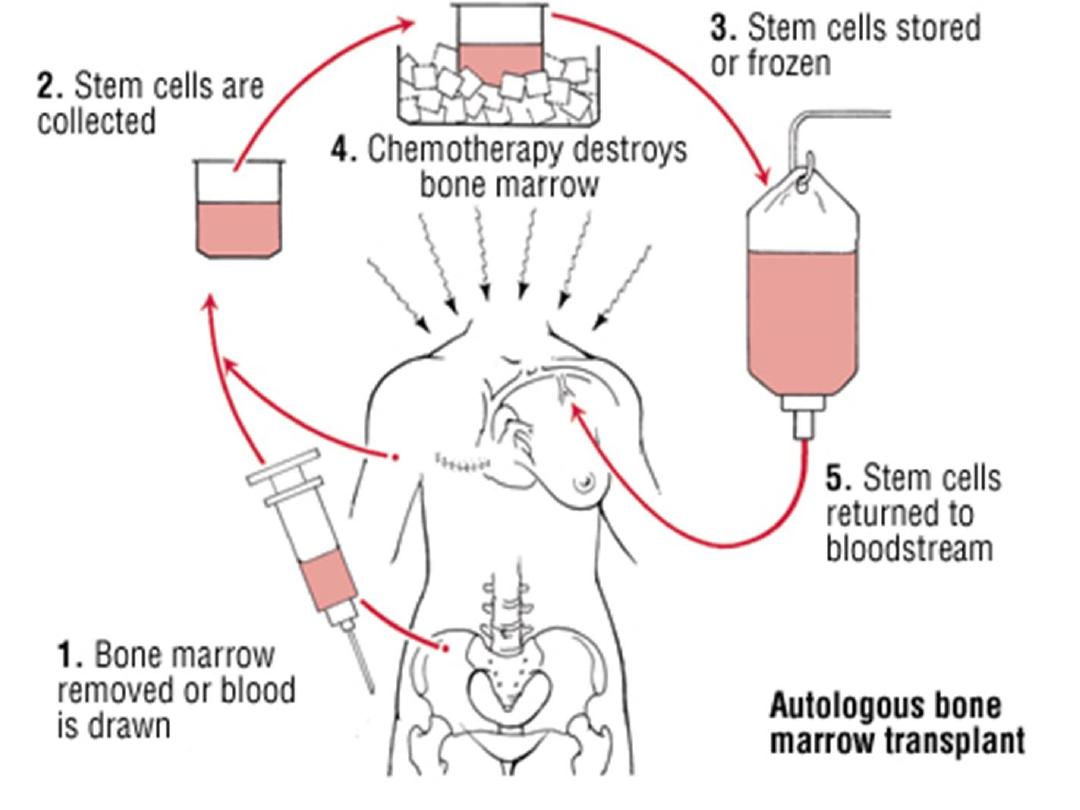
Autologous HSCT
• This procedure can also be used in haematological
malignancies. The patient’s own stem cells from
blood or marrow are first harvested and frozen.
• After conditioning myeloablative therapy, the
autologous stem cells are reinfused into the blood
stream in order to rescue the patient from the
marrow damage and aplasia caused by
chemotherapy..
• Autologous HSCT may be used for disorders which
do not primarily involve the haematopoietic tissues,
or in patients in whom very good remissions have
been achieved
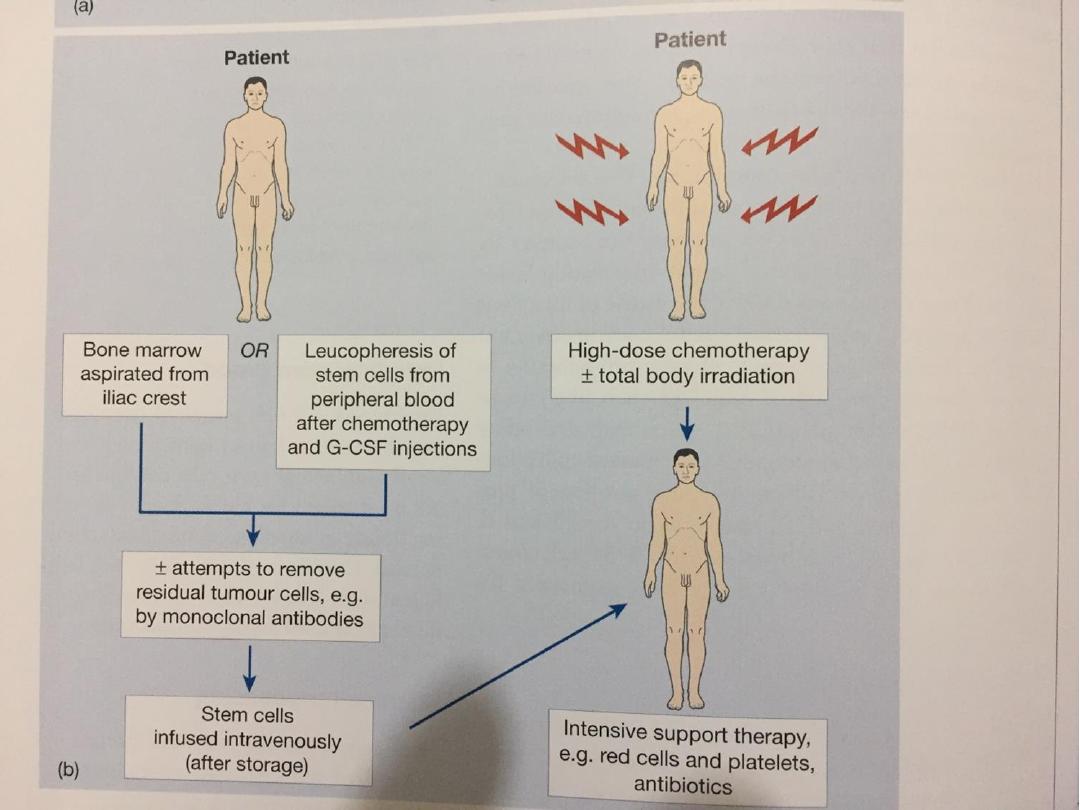

• The preferred source of stem cells for autologous
transplants is peripheral blood. These stem cells
engraft more quickly, marrow recovery occurring
within 2–3 weeks. There is no risk of GVHD and no
immunosuppression is required.
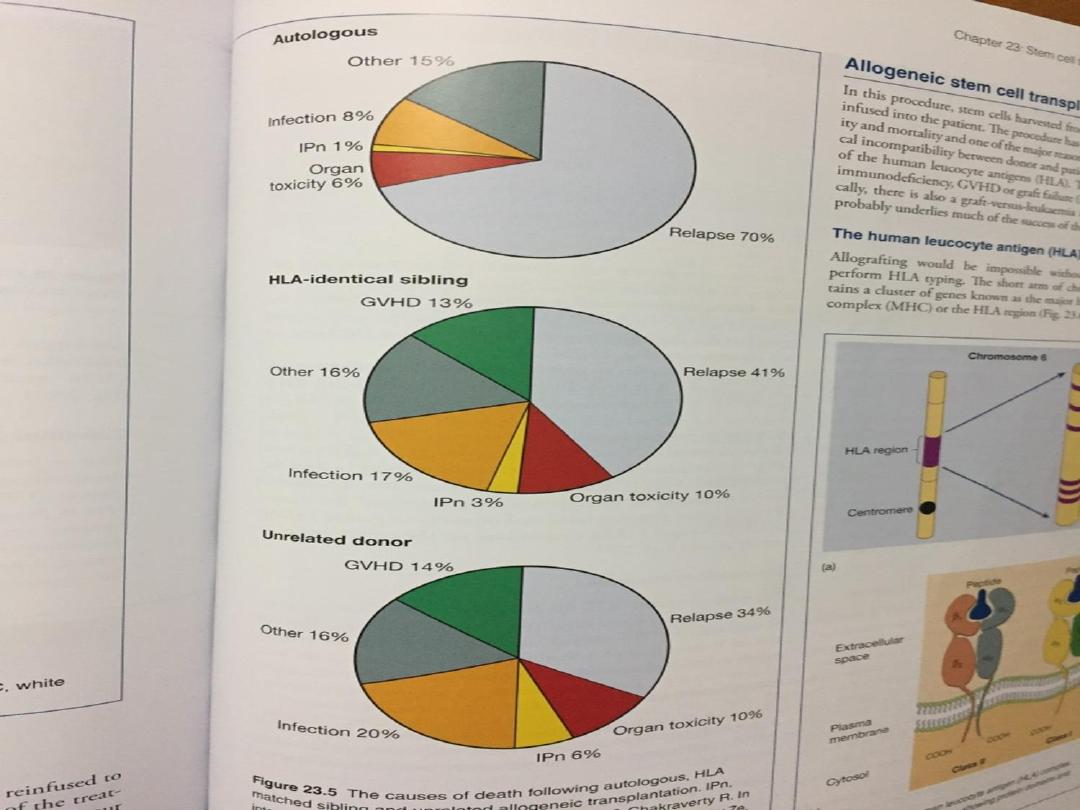
• Thus autologous stem cell transplantation carries a
lower procedure related mortality rate than
allogeneic HSCT at around 5%, but there is a higher
rate of recurrence of malignancy.
• Whether the stem cells should be treated (purged)
in an attempt to remove any residual malignant
cells remains controversial.


Cord blood stem cells
• Some people may have a stem cell transplant using
stem cells from umbilical cord blood. There are cord
blood banks which store blood taken from the umbilical
cord.
• After the baby is born and the umbilical cord has been
cut, a doctor takes blood from the umbilical cord and
placenta. The blood bank may then give the donated
stem cells to a person whose blood cells closely match
the donated cells.
• These transplants are mostly used for children because
of the lower volume of cells collected.
• It may be possible for adults to have a stem cell
transplant from 2 different umbilical cords (double cord
transplant).

