
1
Patterns of Genetic Transmission
Mendelian Inheritance
There are 3 classic forms of genetic inheritance: autosomal dominant,autosomal
recessive, and X-linked. These remain the foundation of single-gene inheritance.
Autosomal Dominant Inheritance
- Autosomal dominant inheritance is determined by the presence of 1 abnormal gene
on 1 of the autosomes (chromosomes 1-22).
- Autosomal genes exist in pairs, with each parent contributing 1 copy.
- In an autosomal dominant trait, a change in 1 of the paired genes has an effect on
the phenotype.
- The disorder is transmitted in a vertical (parent-to-child) pattern and can appear in
multiple generations.
- An affected individual has a 50% (1 in 2) chance of passing on the deleterious gene
in each pregnancy and, therefore, of having a child affected by the disorder. This is
referred to as the recurrence risk for the disorder.
- Unaffected individuals (family members who do not manifest the trait) do not pass
the disorder to their children.
- Males and females are equally affected.
- Although parent-to-child transmission is a characteristic of autosomal dominant
inheritance, for many patients with an autosomal dominant disorder there is no
history of an affected family member. There are several possible reasons:
1. New mutation.
2. Incomplete penetrance, meaning that not all individuals who carry the mutation
have phenotypic manifestations.
3. Variable expression: Individuals with the same autosomal dominant mutation can
manifest the disorder to different degrees.
4. Somatic mutations: Some spontaneous genetic mutations occur not in the egg or
sperm that forms a child, but rather in a cell in the developing embryo.
5 .Germ-line mosaicism: the mutation occurs in cells that populate the germline that
produce eggs or sperm.
Examples
of
AD
disease:
achondroplasia,
hereditary
spherocytosis,
neurofibromatosis, Marfan syndrome, and osteogenesis imperfecta.
Autosomal Recessive Inheritance
-Horizontal transmission, the observation of multiple affected members of a kindred
in the same generation, but no affected family members in other generations.
- Recurrence risk of 25% for parents with a previous affected child.
- Males and females being equally affected.
- The affected individual should be homozygous for the affected gene.
- If both parents are heterozygous the chance of having affected child is 25%.

2
- If the affected person married from normal person, all the children will be
heterozygous.
- If affected person married from a heterozygous person the children will be: -
• 50% affected. It is called Pseudodominant inheritance, which refers
to the observation of apparent dominant (parent to child) transmission
of a known AR disorder. It is most likely to occur for relatively
common traits, such as sickle cell anemia.
• 50% heterozygous (carrier).
Examples of AR disease: Sickle cell disease, PKU, Tay-sachs disease, Cystic
Fibrosis, Hurler Disease, Werdnig-Hoffmann Disease, and Alkaptonuria, Albinism,
and Congenital adrenal hyperplasia.
X-Linked Recessive Inheritance
◆
Males are more commonly and more severely affected than
females.
◆
Female carriers are generally unaffected, or if affected, they are
affected more mildly than males.
◆
Female carriers have a 25% risk for having an affected son, a 25%
risk for a carrier daughter, and a 50% chance of having a child that
does not inherit the mutated X-linked gene.
◆
Affected males have carrier daughters and unaffected sons because
they pass their X chromosome to all of their daughters and their
Y chromosome to all of their sons. Male-to-male transmission
excludes X-linkage but is seen with autosomal dominant and
Y-linked inheritance.
◆
A female occasionally exhibits signs of an X-linked trait similarly to
a male. This occurs rarely owing to:
-homozygosity for an X-linked trait.
- the presence of a sex chromosome abnormality (45X0).
- nonrandom X-inactivation.
X chromosome inactivation occurs early in development and involves random and
irreversible inactivation of most genes on one X chromosome in female cells.
Examples of X-linked recessive diseases: Hemophilia A and B, Lesch Nyhan
Syndrome, Hunter`s syndrome,+ D.M.D., G6PD, and Color-blindness.
X-Linked Dominant Inheritance:
+ Female carriers typically manifest abnormal findings.
+An affected man will have only affected daughters and unaffected sons.
+ Half of the offspring of an affected woman will be affected.
+ Some X-linked dominant conditions are lethal in a high percentage
of males.

3
+ The pedigree shows only affected females and an overall ratio
of 2 : 1 females to males with an increased number of miscarriages.
Examples of X-linked dominant diseases: Incontinentia pigmenti, and vit.D resistant
rickets.
Y-Linked Inheritance
# This demonstrates only male-to-male transmission.
# Only males are affected.
# Most Y-linked genes are related to male sex determination and reproduction and
are associated with infertility. Therefore, it is rare to see familial transmission of a Y-
linked disorder. Example of YLD:Leri-Weil dyschondrosteosis,a rare skeletal
dysplasia that involves bilateral bowing of the forearms with dislocations of the ulna
at the wrist and generalized short stature.
Multifactorial and Polygenic Inheritance
Multifactorial inheritance refers to traits that are caused by a combination
of inherited and environmental factors.
Characteristics include the following:
◆
There is a similar rate of recurrence among all 1st-degree relatives
(parents, siblings, offspring of the affected child).
◆
The risk of recurrence is related to the incidence of the disease.
◆
Some disorders have a sex predilection, as indicated by an unequal
male : female incidence. Pyloric stenosis, for example, is more common in males,
whereas congenital dislocation of the hips is more common in females. Where there
is an altered sex ratio, the risk is higher for the relatives of an index case whose
gender is less commonly affected than relatives of an index case of the more
commonly affected gender. For example, the risk to the son of an affected female
with infantile pyloric stenosis is 18%, compared with the 5% risk for the son of an
affected male.
◆
The likelihood that both identical twins will be affected with the
same malformation is less than 100% but much greater than the
chance that both members of a nonidentical twin pair will be
affected.
◆
The risk of recurrence is increased when multiple family members are affected. A
simple example is that the risk of recurrence for unilateral cleft lip and palate is 4%
for a couple with 1 affected child and increases to 9% with 2 affected children.
◆
The risk of recurrence may be greater when the disorder is more severe. For
example, an infant who has long-segment Hirschsprungdisease has a greater chance
of having an affected sibling than the infant who has short-segment Hirschsprung
disease.
Examples include pyloric stenosis, neural tube defects, congenital heart
defects, diabetes, and cleft lip and cleft palate.

4
Cytogenetics
:
Clinical cytogenetics is the study of chromosomes: their structure, function,
inheritance, and abnormalities.
The specific indications for this study include:
1.Advanced maternal age (>35 yr)
2.Multiple abnormalities on fetal ultrasound(prenatal testing),
3.Multiple congenital anomalies,
4.Unexplained growth restriction in the fetus or postnatal problems in growth and
development,
5.Ambiguous genitalia,
6.Unexplained intellectual disability with or without associated anatomic
abnormalities,
7.Primary amenorrhea or infertility,
8.Recurrent miscarriages (≥3) or prior history of stillbirths and neonatal deaths,
9.A 1st-degree relative with a known or suspected structural chromosome
abnormality,
10.Clinical findings consistent with a known anomaly, some malignancies, and
chromosome breakage syndromes (e.g.Fanconi anemia).
Abnormalities of Chromosomal Number
A haploid cell (n) has 23 chromosomes (typically in the ovum or sperm). If a cell’s
chromosomes are an exact multiple of 23 (46, 69, 92 in humans), those cells are
referred to as euploid.
Polyploidcells are euploid cells with more than the normal diploid number of 46 (2n)
chromosomes: 3n, 4n. Polyploid conceptions are usually not viable, but the presence
of mosaicism with a karyotypically normal line can allow survival.
Abnormal cells that do not contain a multiple of haploid number of chromosomes are
termed aneuploidcells.
The most common cause of aneuploidy is nondisjunction, thefailure of
chromosomes to disjoin normally during meiosis resulting in amonosomic or trisomic
zygote.
Trisomy is characterized by the presence of 3 chromosomes, instead of the normal 2,
of any particular chromosome. Trisomy is the most common form of aneuploidy and
can occur in all cells or it may be mosaic(the presence of 2 or more cell lines in a
single individual). Most individuals with trisomy exhibit a consistent and specific
phenotype depending on the chromosome involved. The occurrence of trisomy 21
and other trisomies increases with advanced maternal age (≥35 yr).
Down Syndrome
The incidence of Down syndrome in live births is approximately 1 in 733; the
incidence at conception is more than twice that rate; the difference is accounted by
early pregnancy losses.
The risk of having a child with trisomy 21 is highest in women who
conceive at >35 yr of age. Even though younger women have a lower

5
risk, they represent half of all mothers with babies with Down syndrome
because of their higher overall birth rate. All women should be
offered screening for Down syndrome in their 2nd trimester by means
of 4 maternal serum tests (free β-human chorionic gonadotropin
[β-hCG], unconjugated estriol, inhibin, andα-fetoprotein). This is known as the
quad screen; it can detect up to 80% of Down syndrome pregnancies compared to
70% in the triple screen. In approximately 95% of the cases of Down syndrome there
are 3copies of chromosome 21.
v
95% of cases are due to non-disjunction (the supernumerary chromosome 21
is maternal in 97% of the cases). The incidence of this syndrome increased
with maternal age from (1/750) of birth in those 30 years old to (1/365) in
those >35 years, (1/100) in those 40 years old and (1/50) those >45 years.
v
1% due to mosiacism.
v
4% due to translocation involving chromosome 21. The majority of
translocations in Down syndrome are fusions at the centromere between
chromosomes 13, 14, 15, 21, and 22. Half of translocated cases were
inherited from translocated carrier parent. Those parents produce three types
of viable children
o Normal child phenotypically and karyotypically.
o Child phenotypically normal but translocation carrier.
o Child with Down syndrome who can not be differentiated
phenotypically from those resulted from non-disjunction.
So chromosomal study must be done on every case of Down syndrome. If a
translocation is identified, parental studies must be done. The recurrence risk for
normal women <40 years old is 1%. If the mother is translocation carrier, the risk will
be 10%. If the father is translocation carrier, the risk will be 3-5%. If the translocation
is between (21, 21), the risk will be 100%.
Clinical features
Central nervous system: Hypotonia, developmental delay, poor moro reflex,
seizures.
Craniofacial: Brachycephaly with flat occiput, flat face, upward slanted palpebral
fissures, epicanthal folds, speckled irises (brushfield spots), nystagmus, strabismus,
glaucoma, congenital or acquired cataract, three fontanels, delayed fontanel closure,
frontal sinus and midfacial hypoplasia, mild microcephaly, short hard palate, small
nose, flat nasal bridge, protruding tongue, open mouth, small dysplastic ears.
Cardiovascular(50%): Endocardial cushing defects(most characteristic), ventricular
septal defect(most common), atrial septal defect, patent ductus arteriosus, aberrant
subclavian artery, pulmonary hypertension
MusculoskeletaL: Joint hyperflexibility, short neck, redundant skin, short
metacarpals and phalanges, short 5th digit with clinodactyly, single transverse palmar

6
crease, wide gap between 1st and 2nd toes, pelvic dysplasiashort sternum, two sternal
manubrium ossification centers, slipped capital femoral epiphyses.
Gastrointestinal: Duodenal atresia,annular pancreas,tracheoesophageal fistula,
hirschsprung disease, imperforate anus, neonatal cholestasis.
Cutaneous: Cutis marmorata.
Up to 15% of children with Down syndromehave misalignment of cervical vertebra
C1, which places them at riskfor spinal cord injury with neck hyperextension or
extreme flexion.
Developmental delay is universal, social development is relatively spared, but
children with Down syndrome have considerable difficulty using expressive
language. Patient with Down syndrome had increased risk for hypothyrodism,
diabetes mellitus, obesity, AML, ALL, problems with hearing and vision, Alzheimer
disease, celiac disease, delayed tooth eruption, obstructive sleep apnea, and frequent
infections (sinusitis, nasopharyngitis, pneumonia). Most males are sterile; some
females have been able to reproduce with 50% chance of having trisomy 21
pregnancies. Most adults with Down syndrome are able to perform activities of daily
living. However, most adults with Down syndrome have difficulty with complex
financial, legal, or medical decisions. The life expectancy is approximately 50-55
years.
A, Face of a child with Down syndrome. Foot of a 1
mo child with Down syndrome
B, Karyotype of a male with trisomy 21.
Trisomy 18 (Edwards syndrome)
Incidence: 1/6000
Head and Face:small and premature appearance, tight palpebral fissures, narrow
nose and hypoplastic nasal alae, narrow bifrontal diameter, prominent occiput,
micrognathia, cleft lip or palate, microcephaly.
Chest:congenital heart disease (e.g., VSD, PDA, ASD), short sternum, small nipples.
Extrimities:limited hip abduction, clinodactyly and overlapping fingers; index over
3rd, 5th over 4th;closed fist, rocker-bottom feet, hypoplastic nails.
General: severe developmental delays and prenatal and postnatal growth restriction,
premature birth, polyhydramnios, inguinal or abdominal hernias, only 5% live >1 yr.

7
Overlapping of fingers and hypoplastic nailsRocker-botton feet
Male infant with trisomy 18
Trisomy 13 (Patau syndrome)
Incidence: 1/10000
Head and Face: scalp defects (e.g., cutis aplasia), microphthalmia, corneal
abnormalities, cleft lip and palate in 60%-80% of cases, microcephaly,
microphthalmia, sloping forehead, holoprosencephaly, capillary hemangiomas,
malformed ears, deafness.
Chest:congenital heart disease (e.g., VSD, PDA, and ASD) in 80% of cases, thin
posterior ribs (missing ribs).
Extrimities:overlapping of fingers and toes (clinodactyly), polydactyly, hypoplastic
nails, hyperconvex nails.
General:severe developmental delays and prenatal and postnatal growth, restriction,
renal abnormalities, arely lethality in most cases, with a median survival of 7 days;
91% die by 1 year, only 5% live >6 mo.
Facial appearance of a child with trisomy 13
8
Sex Chromosomes Anomalies:
Sex chromosome abnormalities are the most common chromosome abnormalities
seen in live born infants, children, and adults. Sex chromosome abnormalities can be
either structural or numerical and can be present in all cells or in a mosaic form.
Those affected with these abnormalities might have few or no physical or
developmental problems.
Turner syndrome (45,X):
The incidence is 1/5000 of live born females.
Cytogenic aspects: Turner syndrome is a condition characterized by complete or
partial monosomy of the X chromosome. 50% of patients have (45,X)and others are
mosiasicim (45X/46XX, 45X/46XY, 45X/46X,rX). In 75% of patients, the lost sex
chromosome is of paternal origin (whether an X or a Y).
Maternal age is not a predisposing factor. 45,X is one of the chromosome
abnormalities most often associated with spontaneous abortion (95-99% of 45,X
conceptions are miscarried).
Clinical features: The phenotype is female.
Findings in the newborns can include small size for gestational age, webbing of the
neck (redundant nuchal skin), protruding ears, and lymphedema of the hands and feet,
although many newborns are phenotypically normal.
Older children and adults have short stature (cardinal finding in all girls) and
exhibit variable dysmorphic features (increased carrying angle of elbow (cubitus
valgus), scoliosis, broad chest with widely spaced nipples (shield chest), low
posterior hairline, webbed neck).
There is gonadal dysgenesis (infertility, primary amenorrhea, lack of secondary sex
characters). Congenital heart defects occur in 40% (bicuspid aortic valve, aortic
stenosis, coarctation of aorta, mitral valve prolapse ), renal malformation in 60%
(pelvic kidney, hoarseshoe kidney, double collecting system, complete absence of
one kidney, and ureteropelvic junction obstruction), recurrent otitis media,
sensorineural hearing loss. Most patients tend to be of normal intelligence but
learning disabilities (motor and visuospatial skills) is seen in 70%. Mental retardation
is seen in 6% of affected children (45,X/46,X,r(X); the ring chromosome is unable to
undergo inactivation and leads to 2 functional X chromosomes) .
Females with 45,X/46,XY mosaicism have a 15-30% risk of developing
gonadoblastoma. There is increased incidence of acquired hypothyroidism, type 2
diabetes mellitus, inflammatory bowel disease, and celiac disease.
Treatment:
1. Short stature: rhGH alone or in combination with anabolic steroid to increase
heigh velocity.
2. Lack of secondary sex characters: Replacement with estrogen at 12-13 years.
3. If Y chromosome material is identified, laparoscopic gonadectomy is
recommended..
4. Infertility: Ovum donation and in vitro fertilization.
9
5. Psychosocial support.
Noonan syndrome shares many clinical features with Turner syndrome, although
it is an autosomal dominant disorder. Features common to Noonan syndrome include
short stature, low posterior hairline, shield chest, congenital heart disease, and a short
or webbed neck. Noonan syndrome has a different pattern of congenital heart disease
typically involving right-sided lesions (left-sided lesion in turner syndrome).
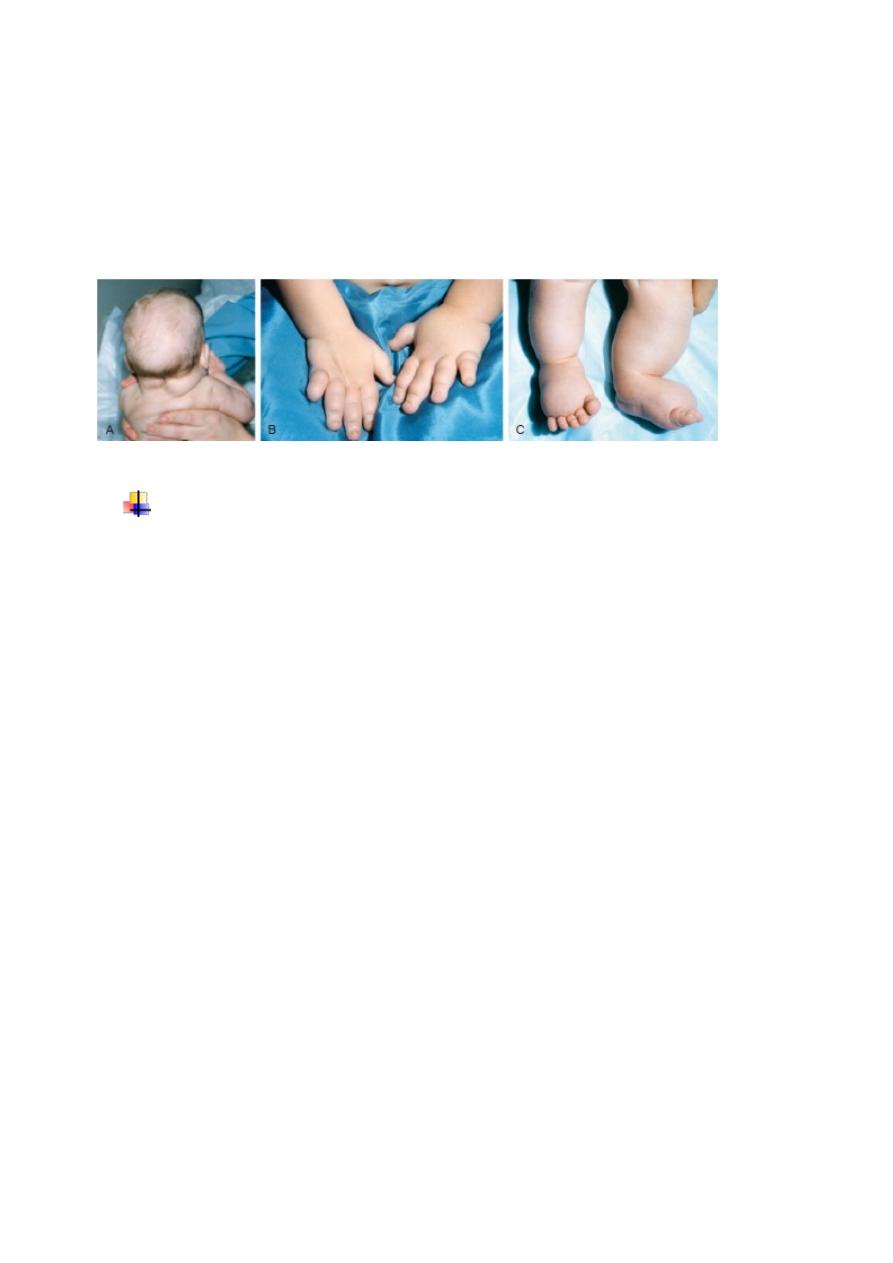
A, Redundant nuchal skin. B, Puffiness of the hands. C, Puffiness of the feet (in
Turner syndrome)
Klinefelter syndrome (males with more than 1 X chromosome):
The incidence is 1/575-1000 live born males.
Cytogenic aspects: 80% of children with Klinefelter syndrome have a male
karyotype with an extra chromosome X (47,XXY); the remaining 20% have multiple
sex chromosome aneuploidies (48,XXXY; 49,XXXXY), or mosaicism
(46,XY/47,XXY). The extra X chromosome is maternal in origin in 54% and paternal
in origin in 46% of patients.
Clinical features: Phenotypically is male with tall stature and have a specific
tendency to have long legs (out of proportion to the arms). Patients develop
secondary sex characters late and have sparser facial hair, 50% develop
gynecomastia. Usually had azospermia, small testes and phallus and infertile (patients
with 46,XY/47,XXY have a better prognosis for testicular function). Sperm counts
are higher in early puberty and decline with age. Their intelligence shows variability
and ranges from above to below average (The greater the aneuploidy, the more severe
the mental impairment and dysmorphism. Each additional X chromosome reduces the
IQ by 10-15 points, when comparing these persons with their normal siblings.).
Patient can show behavioral or psychiatric disorders.
Treatment:
1. Delayed secondary sexual characteristics: Replacement therapy with long
acting testosterone (enanthate ester), should be started at (11-12) years of age.
2. Gynecomastia:
• Medical treatment: may be treated with aromatase inhibitors (which will also
increase endogenous testosterone levels).
• Surgical treatment: indicate if: breast development is excessive causing
significant psychologic distress or fails to regress in 18-24 mo after therapy.

10
3. Infertility: Adults can father children using testicular sperm extraction (TSE)
followed by intracytoplasmic sperm injection. Because sperm counts actually
decrease with time, sperm banking is an option for older adolescents.
4. Learning difficulties and psychosocial disabilities: counseling and psychiatric
services should be provided as needed.