
1
Pediatric surgery
bO� d��«Æœ
wLOL��«
INTRODUCTION
Children are not small adults. They suffer from different disorders
their physical and psychological responses are different. Their
capacity for adaptation is greater but they must endure any
consequences of disease and its management for longer. In contrast
to adults they rarely have comorbidity from degenerative diseases
but they can suffer the unique consequences of congenital
malformations. Children must be treated within the context of their
families.
ANATOMY AND PHYSIOLOGY
anatomical differences between adults and children are important.
Infants and small children have a wider abdomen, a broader costal
margin and a shallower pelvis Thus, the edge of the liver is more
easily palpable below the costal margin and the bladder is an intra-
abdominal organ. The ribs are more horizontal and respiratory
function is more dependent on diphragmatic movement. The child
abdomen is square rather than rectangular therefore transverse
supraumbilical incisions are preferred to vertical midline ones for
laparotomy.
Children have small mouth cavity relatively to their tongue,
therefore they are obligatory nasal breather
Thermoregulation is important in children undergoing surgery.
body surface area to weight ratio decreases with age and small
children therefore Lose heat more rapidly. Babies have less
subcutaneous fat and immature peripheral vasomotor control
mechanisms. The operating theatre must be warm and the infant’s
head (which may account for up to 20% of the body surface area
compared with 9% in an adult) should be insulated. Infusions and

2
respiratory gases may need to be warmed. The central temperature
should be monitored and a warm air blanket is advisable during
lengthy operations. Infants undergoing surgery are vulnerable in
other ways to Impaired gluconeogenesis renders them more
susceptible to Hypoglycemia blood glucose must be monitored and
maintained above 2.6 mmol/L Newborns are at risk of clotting
factor deficiencies and should be given intramuscular vitamin K
before major surgery. They are less able to concentrate urine or
conserve sodium and have greater obligatory water loss to excrete
a given solute load ;therefore Fluid and sodium requirements are
relatively high. Infants are prone to gastro-oesophageal reflux and
have less well-developed protective reflexes, rendering them more
at risk of pulmonary aspiration; adequate nasogastric aspiration is
essential in those with gastrointestinal obstruction. Immaturity of
the immune system increases the risk of infection, which can
present with non specific features such as poor feeding, vomiting
and lethargy

1
Pediatric surgery
Lec.
د.اﯾﺴﺮ ﺣﻤﯿﺪ اﻟﺘﻤﯿﻤﻲ
Esophageal Atresia/Tracheo-Esophageal Fistula
(EA/TEF)
It is a rare congenital birth defect which affects approximately 1 in 4,000
babies. With EA/TEF, a baby is unable to swallow, and may also have
trouble breathing. Esophageal atresia (EA) is a birth defect in which the
esophagus, closed off at some point along its length. Esophageal Atresia
almost always occurs in conjunction with tracheoesophageal fistula (TEF), a
condition in which the esophagus is improperly attached to the trachea, It is
believed that these defects occur around the fourth week of pregnancy when
the digestive tract is forming. There is no known cause for the defects.
During fetal development, the esophagus and trachea arise from the same
original tissue, forming into two side-by-side passageways, the esophagus
leading from the throat to the stomach and digestive tract, and the trachea
leading from the larynx to the lungs and respiratory system. Normally, the
two tubes form separately (differentiate); however, in the case of EA/TEF,
they do not differentiate, which results in various malformed configurations.
There are five configurations:
•
Type I (87 %): Esophageal atresia with tracheoesophageal fistula, in
which the upper segment of the esophagus ends in a blind pouch (EA)
and the lower segment of the esophagus is attached to the trachea
(TEF).
•
Type II (8% of cases): Esophageal atresia in which both segments of
the esophagus end in blind pouches. Neither segment is attached to the
trachea.
•
Type III (also called Type H) (4%): Tracheoesophageal fistula in
which there is no esophageal atresia because the esophagus is
continuous to the stomach. Fistula is present between the esophagus
and the trachea.
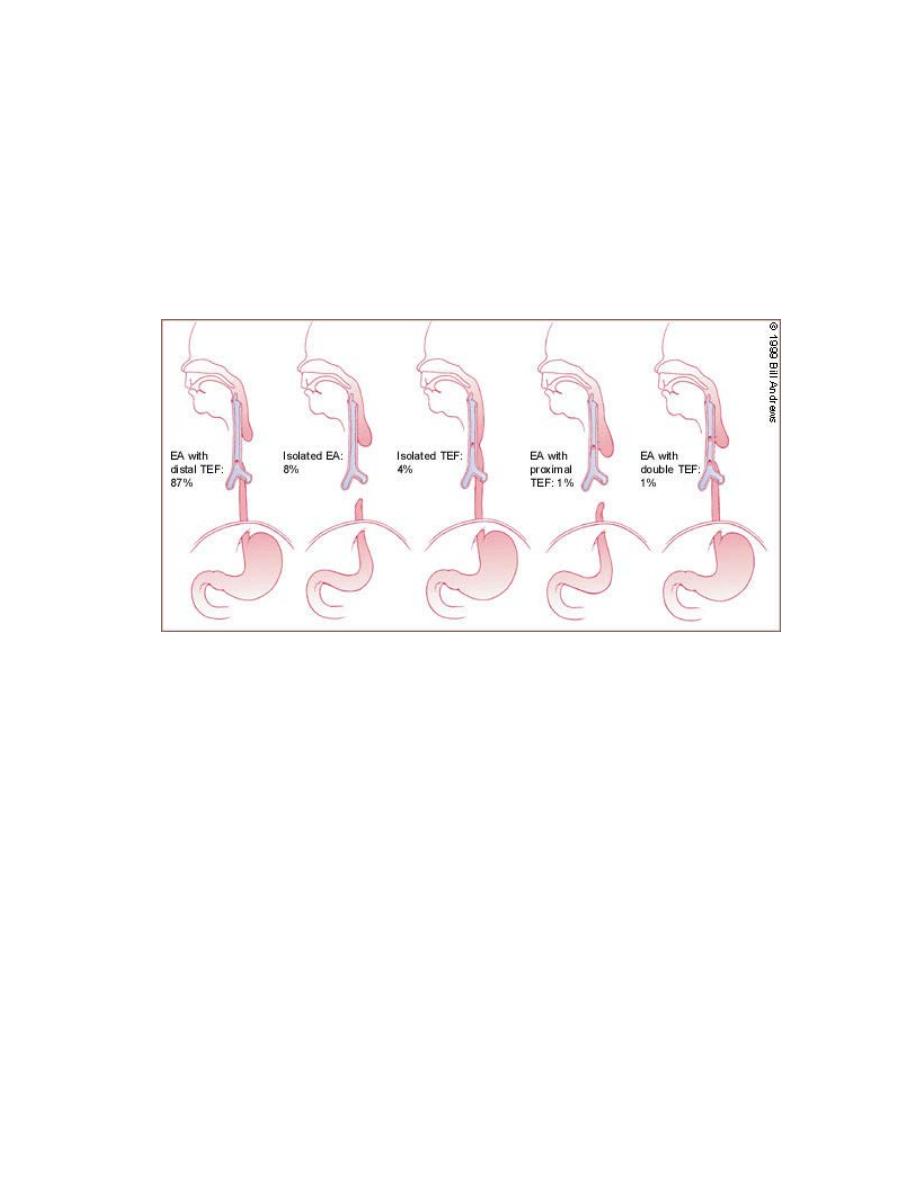
2
•
Type IV (1%): Esophageal atresia with tracheoesophageal fistula in
which the upper segment of the esophagus forms a fistula to the
trachea. The lower segment of the esophagus ends in a blind pouch.
This condition is very rare.
•
Type V (1%): Esophageal atresia with tracheoesophageal fistula, in
which both segments of the esophagus are attached to the trachea.
This is the rarest form of EA/TEF.
Figure 1 (Tracheoesophageal anomalies)
When the esophagus ends in a pouch instead of emptying into the stomach,
food, liquids, and saliva cannot pass through. The combination of ea with tef
compromises digestion, nutrition, and respiration (breathing), creating a life-
threatening condition that requires immediate medical attention. All babies
with ea/tef require surgical repair to correct the condition and allow proper
nutrition and swallowing.
Symptoms
The cause of esophageal atresia, like that of most birth defects, is unknown.
An infant born with ea/tef may initially appear to swallow normally.
However, the first signs of ea/tef may be the presence of tiny, white, frothy
bubbles of mucus in the infant’s mouth and sometimes in the nose as well.
When these bubbles are suctioned away, they reappear. This symptom
occurs when the blind pouch begins to fill with mucus and saliva that would
normally pass through the esophagus into the stomach. Instead these
secretions back up into the mouth and nasal area, causing the baby to drool

3
excessively. Aspiration pneumonia, an infection of the respiratory system
caused by inhalation of the contents of the digestive tract, may also
develope, that present with coughing, chocking and cyanosis
Diagnosis
1-prenatal diagnosis
About 40% of pregnant ladies with esophageal atresia have a history of
polyhydrominion in addition to small fetal stomach by ultrasound
examination .
2-postnatal diagnosis
When a physician suspects esophageal atresia after being presented with the
typical symptoms, diagnosis usually begins with gently passing a catheter
through the nose and into the esophagus. Esophageal atresia is indicated if
the catheter stops at the blind pouch, indicating that it has hit an obstruction.
If EA is present, the catheter will typically stop at 4 to 5 inches (10–12 cm)
from the nostrils.
As EA and TEF associated with many other anomalies that can be
summarized by VACTRAL which refer to :vertebral ,anorectal
,cardiothoracic ,respiratory and limbs anomalies ,so search for these
anomalies take part in the diagnosis.
Treatment
Infants with ea, with or without tef, are unlikely to survive without surgery
to reconnect the esophagus. The procedure is done as soon as possible;
however, prematurity, the presence of other birth defects, or complications
of aspiration pneumonia may delay surgery. Once diagnosed, the baby may
be fed intravenously until surgery is performed. Mucus and saliva will also
be continuously removed via a catheter. Healthy infants who have no
complications, such as heart or lung problems or other types of intestinal
malformations, can usually have surgery within the first 24 hours of life.
Surgery techniques used to treat the five types of ea/tef defects are similar.
Surgery is conducted while the infant is under general anesthesia; a tube is
placed through the mouth to continuously suction the esophageal pouch
during the procedure. An intravenous line is established to allow fluids to be
administered as needed during surgery. Usually, the infant is placed on a
ventilator, with a tube placed down the airway for at least the length of the
surgery.
During surgical operation we are going to connect the two end of the
Esophageal and ligation of fistula