
1
Leukemia:
Objective: To discuss the types ,the etiology , clinical presentations and management of
childhood leukemia.
The leukemias may be defined as a group of malignant diseases in which genetic abnormalities
in a hematopoietic cell result in unregulated clonal proliferation of cells. The increase growth of
these abnormal cells due to increased rate of proliferation and a decreased rate of spontaneous
apoptosis which affect the growth of normal cellular elements.
The leukemias are the most common malignant neoplasms in childhood, accounting for
approximately 31% of all malignancies that occur in children younger than 15 yr of age.
Acute lymphoblastic leukemia (ALL) accounts for approximately 77%of cases of childhood
leukemia, acute myelogenous leukemia (AML)for approximately 11%, chronic myelogenous
leukemia (CML) for 2-3%, and juvenile myelomonocytic leukemia (JMML) for 1-2%.
The remaining cases consist of a variety of acute and chronic leukemias that do not fit classic
definitions for ALL, AML, CML, or JMML.
Acute Lymphoblastic Leukemia (ALL):
Childhood ALL was the first disseminated cancer shown to be curable with treatment .It is a
heterogeneous group of malignancies with a number of distinctive genetic abnormalities that
result in varying clinical behaviors and responses to therapy.
EPIDEMIOLOGY:
ALL has a peak incidence at 2-3 yr of age and occurs more in boys than in girls at all ages.
The disease is more common in children with certain chromosomal abnormalities, such as Down
syndrome, Bloom syndrome, ataxia-telangiectasia, and Fanconi anemia.
The risk is >70% if ALL is diagnosed in the first twin during the 1st yr of life and the twins
shared the same (monochorionic) placenta. If the first twin develops ALL by 5-7 yr of age, the
risk to the second twin is at least twice that of the general population, regardless of zygosity.
ETIOLOGY:
The etiology of ALL is unknown, although several genetic and environmental factors are
associated with childhood leukemia .
Most cases of ALL are thought to be caused by postconception somatic mutations in
lymphoid cells. The long lag period before the onset of the disease in some children, reported to
be as long as 14 yr, supports the concept that additional genetic modifications are required for
disease expression.
Exposure to medical diagnostic radiation both in utero and in childhood is associated with an
increased incidence of ALL.
Geographic clusters of cases have raised the concern that environmental factors can increase the
incidence of ALL.
There is an association between B-cell ALL (B-ALL) and Epstein-Barr viral infections.
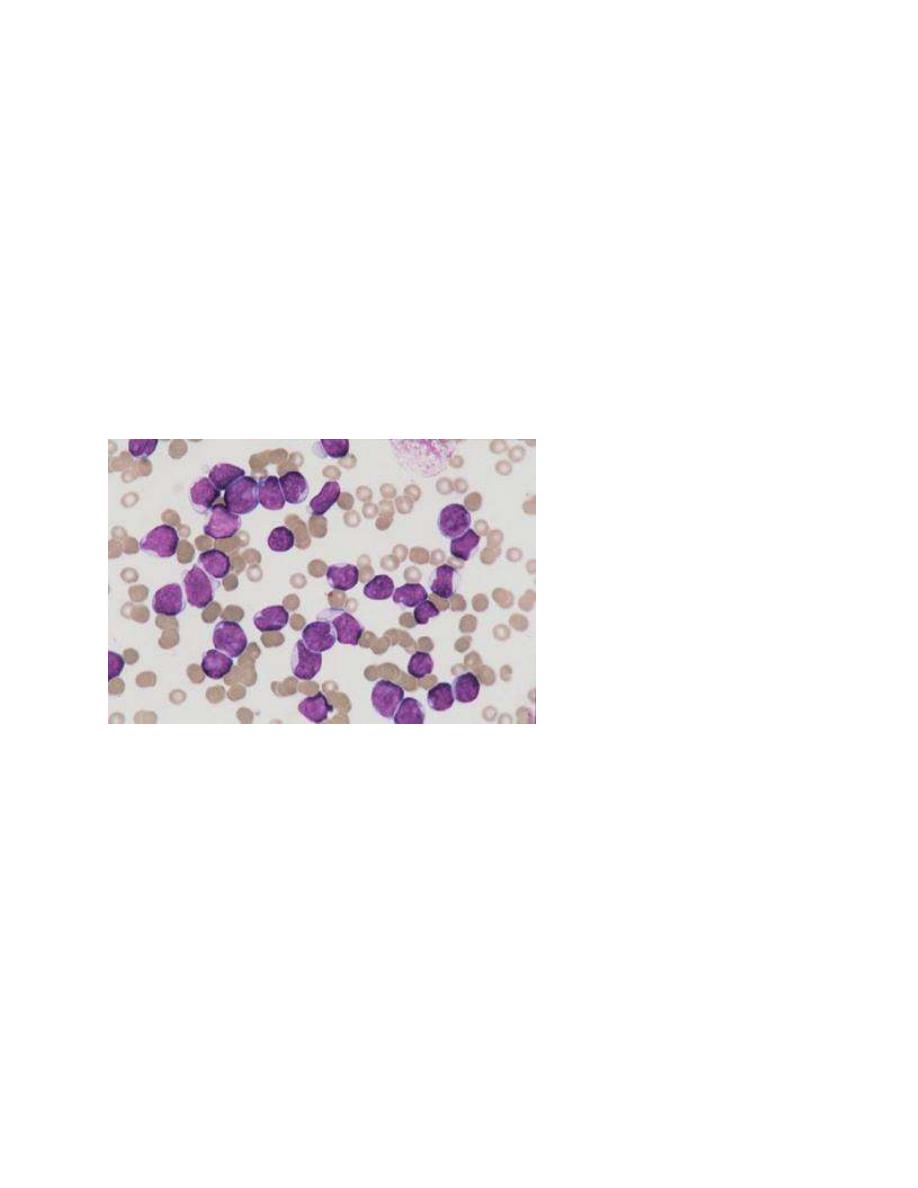
CELLULAR CLASSIFICATION:
The classification of ALL depends on examining the malignant cells in the bone marrow to
determine the morphology, phenotype as measured by cell membrane markers, and cytogenetic
and molecular genetic features.
Morphology is usually adequate alone to establish a diagnosis, but the other studies are
essential for disease classification, which can have a major influence on the prognosis and the
choice of appropriate therapy.

2
Phenotypically, surface markers show that approximately 85% of cases of ALL are classified as
B lymphoblastic leukemia , approximately 15% are T-lymphoblastic leukemia, and
approximately 1% are derived from mature B cells. The rare leukemia of mature B cells is
termed Burkitt leukemia and is one of the most rapidly growing cancers in humans, requiring a
different therapeutic approach than other subtypes of ALL.
A small percentage of children with leukemia have a disease characterized by surface markers of
both lymphoid and myeloid derivation.
Chromosomal abnormalities are used to subclassify ALL into prognostic groups.
The polymerase chain reaction( PCR) and fluorescence in situ hybridization techniques can be
used to detect small numbers of malignant cells at diagnosis as well as during follow-up
(minimal residual disease [MRD] .
The development of DNA microanalysis makes it possible to analyze the expression of
thousands of genes in the leukemic cell. This technique promises to further enhance the
understanding of the biology and to provide clues to the therapeutic approach of ALL.
CLINICAL MANIFESTATIONS:
The initial presentation of ALL usually is nonspecific and relatively brief.
Anorexia, fatigue, malaise, and irritability often are present, as is an intermittent, low-grade
fever. Bone or joint pain, particularly in the lower extremities, may be present.
Less commonly, symptoms may be of several months’ duration, may be localized predominantly
to the bones or joints, and can include joint swelling. Bone pain is severe and can wake the
patient at night.
As the disease progresses, signs and symptoms of bone marrow failure become more obvious
with the occurrence of pallor, fatigue, exercise intolerance, bruising, or epistaxis, as well as
fever, which may be caused by infection or the disease.
Organ infiltration can cause lymphadenopathy, hepatosplenomegaly, testicular enlargement, or
central nervous system (CNS) involvement (cranial neuropathies, headache, seizures).
Respiratory distress may be due to severe anemia or mediastinal node compression of the
airways.
On physical examination, findings of pallor, listlessness, purpuric and petechial skin lesions, or
mucous membrane hemorrhage can reflect bone marrow failure . The proliferative nature of the
disease may be manifested as lymphadenopathy, splenomegaly,or, less commonly,hepatomegaly.
In patients with bone or joint pain, there may be clear tenderness over the bone or objective
evidence of joint swelling and effusion and with marrow involvement, deep bone pain may be
present but tenderness will not be elicited.
Rarely, patients show signs of increased intracranial pressure that indicate leukemic involvement
of the CNS which include papilledema , retinal hemorrhages, headache ,seizers and cranial
nerve palsies.
Respiratory distress usually is related to anemia but can occur in patients with an obstructive
airway problem (wheezing) as the result of a large anterior mediastinal mass, this problem is
most typically seen in adolescent boys with T-cell ALL (T-ALL).
T-ALL also usually has a higher leukocyte count.
B-lymphoblastic leukemia is the most common immunophenotype, with onset at 1-10 yr of age.
Hepatosplenomegaly is seen in 30-40% of patients.
In all types of leukemia, CNS symptoms are seen at presentation in 5% of patients (5-10% have
blasts in the cerebrospinal fluid [CSF].
3
Testicular involvement is rarely evident at diagnosis, but prior studies indicate occult
involvement in 25% of boys. There is no indication for testicular biopsy.
DIAGNOSIS:
The diagnosis of ALL depend on the history , examination , investigations and is strongly
suggested by complete blood count and blood film that indicate bone marrow failure. Anemia
and thrombocytopenia are seen in most patients. Leukemic cells might not be reported in the
peripheral blood in routine laboratory examinations. The median leukocyte count at presentation
is 33,000/μL, although 75% of patients have counts <20,000/μL; many patients with ALL
present with total leukocyte counts of <10,000/μL ,thrombocytopenia is seen in 75% of patients
When the results of an analysis of peripheral blood suggest the possibility of leukemia, the bone
marrow should be examined to establish the diagnosis. It is important that all studies necessary
to confirm a diagnosis and adequately classify the type of leukemia be performed, including
bone marrow aspiration and biopsy, flow cytometry, cytogenetics, and molecular studies.
ALL is diagnosed by a bone marrow evaluation that demonstrates >25% of the bone marrow
cells as a homogeneous population of lymphoblasts.
Initial evaluation also includes CSF examination. If lymphoblasts are found and the CSF
leukocyte count is elevated, overt CNS or meningeal leukemia is present. This finding reflects a
worse stage and indicates the need for additional CNS and systemic therapies. The staging
lumbar puncture may be performed in conjunction with the first dose of intrathecal
chemotherapy, if the diagnosis of leukemia was previously established from bone marrow
evaluation.
Additional investigations include : CXR to detect mediastinal mas , serum electrolytes ,calcium ,
phosphate , renal and liver function .
kemia of Childhood
DIFFERENTIAL DIAGNOSIS:
Elevation of the lactate dehydrogenase is often a clue to the diagnosis of ALL.
When only pancytopenia is present, aplastic anemia (congenital or acquired) and myelofibrosis
should be considered.
Failure of a single cell line, as seen in transient erythroblastopenia of childhood, immune
thrombocytopenia,and congenital or acquired neutropenia, is rarely the presenting feature of
ALL.

4
A high index of suspicion is required to differentiate ALL from infectious mononucleosis in
patients with acute onset of fever and lymphadenopathy and from juvenile idiopathic arthritis in
patients with fever, bone pain but often no tenderness, and joint swelling. These presentations
also can require bone marrow examination.
ALL must be differentiated from AML and other malignant diseases that invade the bone
marrow and can have clinical and laboratory findings similar to ALL, including neuroblastoma,
rhabdomyosarcoma, Ewing sarcoma, and retinoblastoma.
TREATMENT:
The single most important prognostic factor in ALL is the treatment: without effective therapy,
the disease is fatal. Survival is also related to age and subtype .
It vary with the clinical risk feature .
The standard risk and more favorable characteristics include:
1-Age between 1 and 10 yr old. 2- A leukocyte count of <50,000/μL.3-A rapid response to
therapy. 4-Hyperdiploidy. 5-Trisomy of specific chromosomes (4, 10, and 17).
6- Rearrangements of the ETV6-RUNX1genes.
High risk and additional characteristics that adversely affect outcome include:
1- Children who are younger than 1 or older than 10 yr of age. 2- Who have an initial leukocyte
count of >50,000/μL. 3- T-cell immunophenotype . 4- A slow response to initial therapy.
5-Chromosomal abnormalities including t(4:11) ,t(9:22) Philadelphia chromosom posative ,
11q23 MLL gen rearrangements and Hypodiploidy , pointed a poorer outcome.
The outcome for patients at higher risk can be improved by administration of more intensive
therapy despite the greater toxicity of such therapy.
However, the poor outcome of Philadelphia chromosome positive ALL with t(9;22) has
dramatically changed by the addition of imatinib to an intensive chemotherapy backbone.
Standard treatment involves chemotherapy for 2-3 yr and most achieve remission at the end of
the induction phase.
MRD can be quantitative and can provide an estimate of the burden of leukemic cells present in
the marrow. Higher levels of MRD present at the end of induction suggest a poorer prognosis
and higher risk of subsequent relapse.
Therapy for ALL intensifies treatment in patients with evidence of MRD at the end of induction
.Initial therapy, termed remission induction, is designed to eradicate the leukemic cells from the
bone marrow.
During this phase, therapy is given for 4 wk and consists of vincristine weekly, a corticosteroid
such as dexamethasone or prednisone, and usually a single dose of a long-acting, pegylated
asparaginase preparation. Patients at higher risk also receive daunomycin at weekly intervals.
With this approach, 98%of patients are in remission, as defined by <5% blasts in the marrow
and a return of neutrophil and platelet counts to near-normal levels after 4-5 wk of treatment.
Intrathecal chemotherapy is always given at the start of treatment and at least once more during
induction.
The second phase of treatment, consolidation, focuses on intensive CNS therapy in
combination with continued intensive systemic therapy in an effort to prevent later CNS relapses.
Intrathecal chemotherapy is given repeatedly by lumbar puncture.
The likelihood of later CNS relapse is thereby reduced to <5%.
A small percentage of patients with features that predict a high risk of CNS relapse may receive
irradiation to the brain in later phases of therapy. This includes patients who, at the time of

5
diagnosis,have lymphoblasts in the CSF and either an elevated CSF leukocyte count or physical
signs of CNS leukemia, such as cranial nerve palsy.
Subsequently, many regimens provide 14-28 wk of therapy, with the drugs and schedules used
varying depending on the risk group of the patient. This period of treatment is often termed
intensification and includes phases of aggressive treatment (delayed intensification) as well as
relatively nontoxic phases of treatment ( maintenance).
Multiagent chemotherapy, including such medications as cytarabine, methotrexate,
asparaginase, and vincristine, is used during these phases to eradicate residual disease.
Finally, patients enter the maintenance phase of therapy, which lasts for 2-3 yr, depending on
the protocol used.
Patients are given daily mercaptopurine and weekly oral methotrexate, usually with intermittent
doses of vincristine and a corticosteroid.
A small number of patients with particularly poor prognostic features, such as those with
induction failure or extreme hypodiploidy, may undergo bone marrow transplantation during the
first remesion.
Adolescents and young adults with ALL have a poor prognosis compared to children younger
than 15 yr old.
Treatment of Relapse:
Relapse occurs in the bone marrow in 15-20% of patients with ALL. Intensive chemotherapy
with agents not previously used in the patient followed by allogeneic stem cell transplantation
can result in long-term survival for some patients with bone marrow relapse .
The incidence of CNS relapse has decreased to <5% since introduction of preventive CNS
therapy.
CNS relapse may be discovered at the time of a routine lumbar puncture in the asymptomatic
patient.
Symptomatic patients with relapse in the CNS usually present with signs and symptoms of
increased intracranial pressure and can present with isolated cranial nerve palsies.
The diagnosis is confirmed by demonstrating the presence of leukemic cells in the CSF.
The treatment includes intrathecal medication and cranial or craniospinal irradiation.
Systemic chemotherapy also must be used, because these patients are at high risk for subsequent
bone marrow relapse.
Most patients with leukemic relapse confined to the CNS do well, especially those in whom the
CNS relapse occurs longer than 18 mo after initiation of chemotherapy.
Testicular relapse occurs in less than 2% of boys with ALL, usually after completion of therapy.
Such relapse occurs as painless swelling of 1 or both testes.
The diagnosis is confirmed by biopsy of the affected testis.
Treatment includes systemic chemotherapy and possibly local irradiation.
A high proportion of boys with a testicular relapse can be successfully retreated, and the
survival rate of these patients is good.
6
SUPPORTIVE CARE:
Patients with high white blood counts are especially prone to tumor lysis syndrome as therapy is
initiated.
The kidney failure associated with very high levels of serum uric acid can be prevented or treated
with allopurinol or urate oxidase.
Chemotherapy often produces severe myelosuppression, which can require erythrocyte and
platelet transfusion and which always requires a high index of suspicion and aggressive empiric
antimicrobial therapy for sepsis in febrile children with neutropenia.
Patients must receive prophylactic treatment for Pneumocystis jiroveci pneumonia during
chemotherapy and for several months after completing treatment.
PROGNOSIS:
Improvements in therapy and risk stratification have resulted in significant increases in survival
rates, with current data showing overall 5 yr survival around 90% .
However, survivors are more likely to experience significant chronic medical conditions
compared to siblings, including musculoskeletal, cardiac, and neurologic conditions.