
Brain tumor
&
Raised intracranial pressure
By
Dr.Ali Abbas Almusawi
Consultant Neurosurgeon
FRCS Glasg,FRCSED,FACS,FIBMS

Brain tumors are responsible for
approximately 2% of all cancer
deaths. Central nervous system
tumours comprise the most common
group of solid tumours in young
patients, accounting for 20% of all
paediatric neoplasms. The overall
incidence of brain tumours is 8–10
per 100 000 population per year.
Brain tumors

Glioma
Neuroectodermal tumors arise from cells
derived from neuroectodermal origin. Gliomas
comprise the majority of cerebral tumours and
arise from the neuroglia cells. There are four
distinct types of glial cells: astrocytes, oligo-
dendroglia, ependymal cells and neuroglial
precursors. Each of these gives rise to tumours
with different biological and anatomical charac-
teristics. The neuroepithelial origin of microglia
is in question.

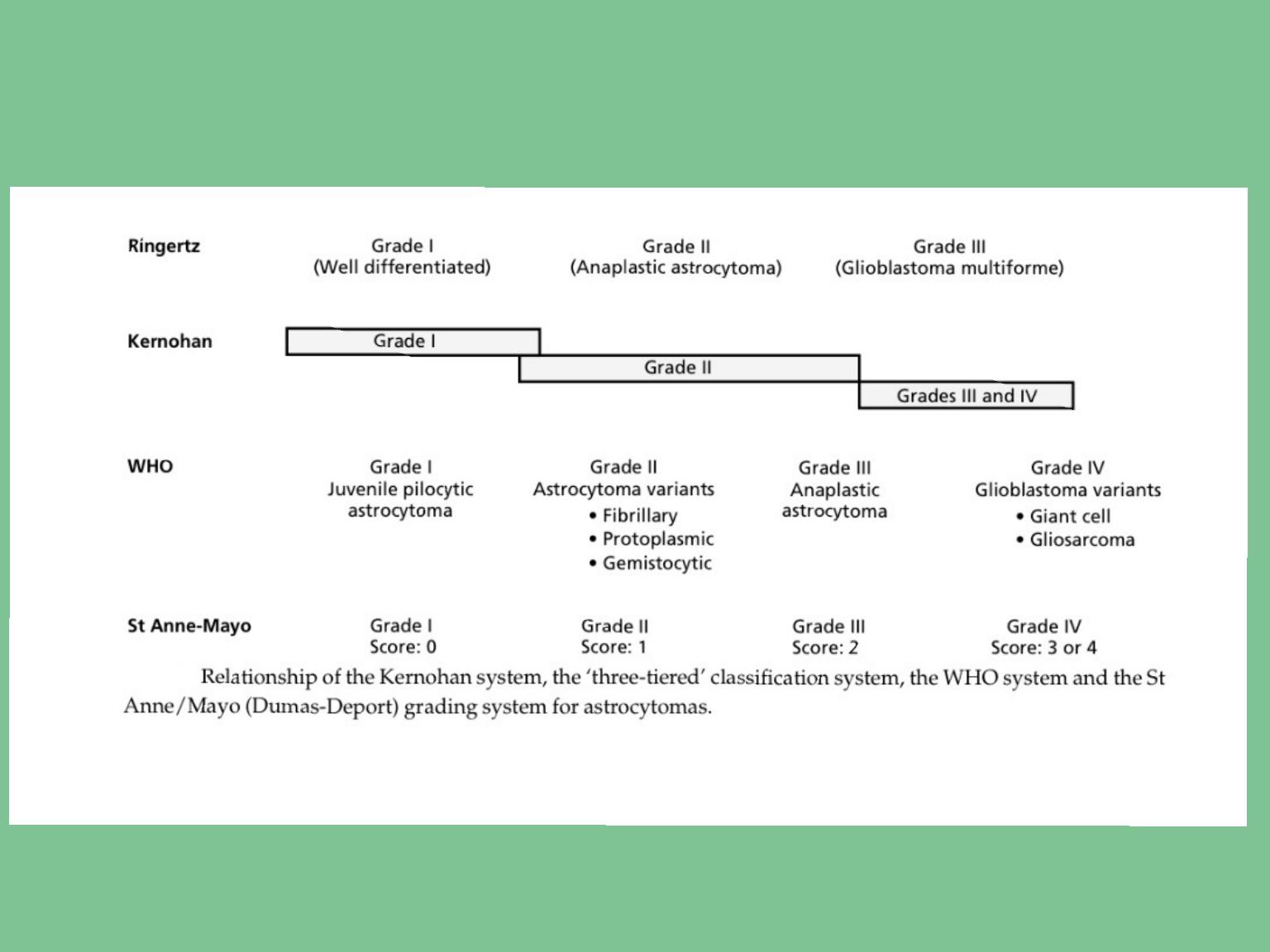
Classification of brain Tumour
1.NEUROEPITHELIAL TUMOUR
Gliomas:
Astrocytoma
Oligodendrocytoma
Ependymoma
Choroid plexus Tumour
Pineal Tumors
Neuronal Tumour :ganglioglioma,gangliocytoma,neuroblastoma
Medulloblastoma
2.NERVE SHEATH TUMOUR:acoustic neuroma
3.MENINGEAL TUMOUR:meningioma
4.PITUITARY TUMOUR
5.GERM CELL TUMOURS:germinoma,teratoma
6.LYMPHOMAS
7.TUMOUR LIKE MALFORMATION:
craniopharyngioma,epidermoid,dermoid,colloid cyst
8.METASTATIC TUMORS
9.LOCAL EXTENSIONS FROM REGIONAL TUMOUR: glomus
jugular,carcinomas of ethmoid


Pathology
Macroscopic changes
An astrocytoma may arise in any part of the brain,
although it usually occurs in the cerebrum in adults and
the cerebellum in children.
A low-grade tumour in the cerebral hemi- spheres
invades diffusely into the brain. The tu- mour does not
have a capsule and there is no distinct tumour margin.
The low-grade gliomas are usually relatively avascular
with a firm fibrous or rubbery consistency
the macroscopic appearance of a high- grade tumour, the
glioblastoma multiforme, is characterized by a highly
vascular tumour margin with necrosis in the centre of the
tumour

Microscopical
The low-grade astrocytoma is characterized by an
increased cellularity, composed entirely of
astrocytes (Fig. 6.2). Intermediate-grade tumours
show nuclear pleomorphism, mitotic figures are
frequent, and there is increased vascularity,
The major histological features of glioblastoma
multiforme are endothelial proliferation and
necrosis. The anaplastic astrocytoma is char-
acterized by nuclear pleomorphism and mitoses,
which are absent in the astrocytoma.

Clinical presentation
The presenting features can be classified under: • raised
intracranial pressure
• focal neurological signs
• epilepsy.
The duration of the symptoms and the progression and
evolution of the clinical presentation will depend on the grade
of the tumour —that is, its rate of growth.
patient presenting with a low-grade astrocytoma (Grade I or II)
may have a history of seizures extending over many years
Patients with the higher-grade tumours present with a shorter
history and glioblastoma multiforme is characterized by a short
illness of weeks or a few months.

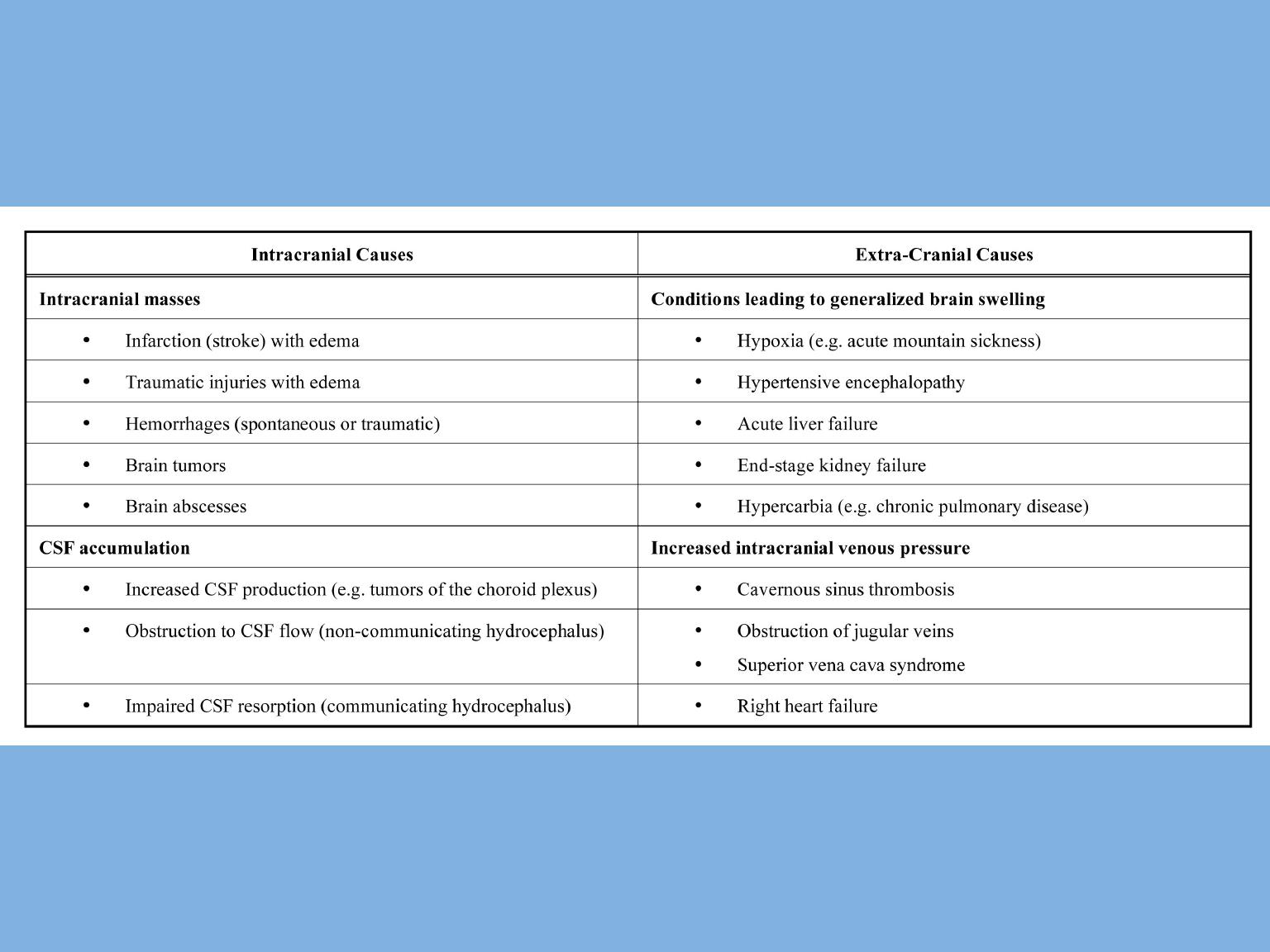
Raised intracranial pressure
Raised intracranial pressure is due to the tumour mass,
surrounding cerebral edema and hydrocephalus due to
blockage of the CSF pathways
The major symp- toms are headache, nausea and vomiting, and
drowsiness.
Headache is the most common symptom in patients with
cerebral astrocytoma and occurs in nearly three-quarters of
patients; vomiting occurs in about one-third. The headaches are
usually gradually progressive and although frequently worse on
the side of the tumors, they may be bitemporal and diffuse.
Characteristically, the headache is worse on waking and im-
proves during the day. Nausea and vomiting occur as the
intracranial pressure increases, and the patient frequently
indicates that vomiting may temporarily relieve the severe
headache. Drowsiness, that is, a deterioration of conscious
state, is the most important symptom and sign of raised
intracranial pressure. The extent of impairment of conscious
state will be related to the severity of raised intracranial
pressure. An alert patient with severely raised intracranial
pressure may rapidly deteriorate and become deeply un-
conscious when there is only a very small further rise in the
pressure within the cranial cavity.

Focal neurological deficit
Patients presenting with tumours involving the frontal lobes frequently
have pseudopsychiatric problems, personality change and mood
disturbance. These changes are particularly char- activistic of the
‘butterfly glioma’, so called because it involves both frontal lobes by
spreading across the corpus callosum, giving it a characteristic
macroscopic
Limb paresis
Field defects associated with Tumors of the temporal, occipital and
parietal lobes are common, but may be evident only on careful testing
Dysphasia
Epileptic seizures
Seizures are the most frequent initial symptom in patients with cerebral
astrocytoma and occur in 50–75% of all patients. Tumours adjacent to
the cortex are more likely to be associated with epilepsy than those
deep to the cortex and tumours involving the occipital lobe are less
likely to cause epilepsy than those which are more anteriorly placed.

Investigations
Computerized tomography
CT scan or MRI of the brain are the essential radiological
investigations an accurate diagnosis can be made in nearly all
tumours. Low-grade gliomas show decreased density on the CT
scan; this does not enhance with contrast and there is little or no
surrounding oedema. Calcification may be present. High-grade
gliomas are usually large and enhance vividly following
intravenous injection of contrast material . The enhancement is
often patchy and non- uniform and frequently occurs in a broad,
irregular rim around a central area of lower density. Although
tumour cysts may occur in the high- grade tumours, the central
area of low density surrounded by the contrast enhancement is
usually due to tumour necrosis. High-grade tumours are
surrounded by marked cerebral oedema and there is frequently
considerable distortion of the lateral ventricles. Compression of
the lateral ven- tricle in one hemisphere, with pressure extending
across the midline, may result in an obstructive hydrocephalus
involving the opposite lateral ventricle.

MRI
When used with gadolinium contrast
enhancement, MRI improves the visualization and
anatomical localization of the tumours (Figs 6.8
and 6.9). MRI has the advantage of being more
sensitive than CT scan, enabling the detection of
small tumours and particularly low-grade gliomas
that might be missed by CT scan. MRI provides
better anatomical detail and is more useful in
visualizing skull base, posterior fossa and
brainstem tumours.

Cerebral angiography
This was the standard study in most patients with
astrocytomas prior to the introduction of CT. It
provides helpful information on the vascular supply
of the tumours but is now only rarely indicated.
Plain X-rays
Plain X-rays of the skull do not need to be
performed as a routine. The most common
abnormality is erosion of the sella turcica due to long-
standing raised intracranial pressure. Radiologically
visible calcification is present in about 8% of patients
with astrocyte-derived gliomas.

Management
Following the presumptive diagnosis of a glioma the
management involves:
• surgery
• radiotherapy
• other adjuvant treatments.
Surgery
Surgery is performed with three principal aims. • To make a
definite diagnosis.
• Tumour reduction to alleviate the symptoms of raised
intracranial pressure.
• Reduction of tumour mass as a precursor to adjuvant
treatments.
The patient is started on glucocorticoid steroid therapy (e.g.
dexamethasone) when presenting with clinical features of
raised intracranial pressure with the aim of decreasing the
cerebral oedema prior to surgery.

Other adjuvant therapies
Chemotherapy
Radiotherapy
Hyperthermia
Immunotherapy
Photodynamic therapy
Gene therapy

Oligodendroglioma
Oligodendrogliomas are responsible for
approximately 5% of all gliomas and occur
throughout the adult age group with a
maximal incidence in the 5th decade. The
tumour is rare in children.

Pathology
Oligodendrogliomas have the same spectrum
of histological appearance as astrocytomas,
rang- ing from very slow growing, benign
tumours to a more rapidly growing, malignant
v a r i e t y w i t h a b u n d a n t m i t o t i c f i g u r e s ,
endothelial prolifera- tion and foci of necrosis.
Calcium deposits are found by histological
e x a m i n a t i o n
i n
u p
t o
9 0 %
o f
oligodendrogliomas

Clinical presentation
The presenting features are essentially the same as
for the astrocyte group but, as these tumours are
more likely to be slow growing, epilepsy is common,
occurring in 80% of patients and seen as an initial
symptom in 50%. The features of raised intracranial
pressure and focal neurologi- cal deficits are each
present in approximately one-third of patients.
Radiological investigation
CT scanning and MRI are the fundamental inves-
tigations. They will confirm the diagnosis of an
intracranial tumour and in many cases the diag- nosis
of oligodendroglioma will be highly probable.
Calcification will be present in 90% of cases and over
half show contrast enhancement

Treatment and results
Treatment involves:
• surgicalresection
• radiotherapy
• otheradjuvanttreatments.
The standard treatment for oligodendroglioma has been an
aggressive resection of the tumour followed by radiation
therapy, although radio- therapy would now not be given to
low-grade tu- mours, and utilized only for the intermediate-
or high-grade oligodendroglial tumours. Oligoden-
drogliomas have been shown to be more sensi- tive to
chemotherapy than the astrocytoma tumours,

Ependymoma
Ependymomas are glial neoplasms arising from the
ependyma and constitute approximately 5% of all gliomas.
Approximately two-thirds of ependymomas occur in the
infratentorial com- partment and most of these present in
children, adolescents and young adults. The supraten-
torial ependymomas occur mostly in adults.
Pathology
The tumour arises from the ependyma of the ven-
tricle and, although predominantly intraventric- ular, the
tumour often invades into the adjacent cerebellum,
brainstem or cerebral hemisphere

Clinical presentation
Posterior fossa ependymomas
Patients present with features of raised intracranial
pressure due to hydrocephalus as a result of
obstruction of the 4th ventricle, ataxia due to
cerebellar involve- ment, and occasionally features of
brainstem pressure or infiltration.
Supratentorial tumours
Virtually all patients with supratentorial ependy-
momas present with features of raised intracranial
pressure, often due to hydrocephalus as a result of
obstruction of the CSF pathways. Ataxia is common
and focal neurological deficits may occur due to
involvement of the underlying cere- bral hemisphere.

Radiological investigation
The CT scan and MRI will show a tumour that arises in the ventricle
and enhances after admin- istration of intravenous contrast.
Calcification is common in tumours arising from the lateral
ventricles. .. There is frequently associated hydrocephalus
Treatment
The treatment of ependymomas is initially surgical, with an attempt
to perform a radical macroscopic resection of the tumour
Postoperative radiation therapy is advisable and, as these tumours
may spread through the CSF.pathways, sometimes whole neuraxis
radiation is recommended.
The prognosis is related to the degree of anaplasia of the tumour and
for intratentorial tu- mours varies from 20% to 50% 5-year survival.
The prognosis for the supratentorial tumours is better, particularly in
adults.

Pineal Tumours
•germinoma
• teratoma
• pineocytoma • pineoblastoma • miscellaneous:•
glioma •cyst
Germinoma is the most common pineal region tumour and is
similar in histological appearance to germinoma of the
gonads and mediastinum; it occurs predominantly in males.
Clinical presentation
Patients with pineal tumours present with:
• raised intracranial pressure
• neurological signs dueto focal compression •endocrine
disturbance.

Neurological signs
ataxia and distortion of the quadrigeminal plate,
produces limitation of upgaze, convergence paresis
with impairment of reaction of pupils to light and
accommodation (Parinaud’s syndrome), and may
result in convergence-retraction nystagmus on
upgaze (Koerber–Salius–Elschnig syndrome).
Endocrine disturbance.
are uncommon but
include precocious puberty in 10% of patients, almost
invariably male, and diabetes insipidus in 10%. The
endocrine effects can either be due to direct tumour
involvement of the hypothalamus or result from the
secondary effects of hydrocephalus.

Radiological investigations
CT scan and MRI will show a pineal region tumour and will
often suggest the correct pathol- ogical diagnosis
Management
This consists of surgery and radiotherapy.
A ventriculoperitoneal shunt or drainage of CSF by a 3rd
ventriculostomy may be required if
the hydrocephalus is severe.

Metastatic tumours
Metastatic tumours are responsible for approxi- mately
15% of brain tumours in clinical series but up to 30% of
brain tumours reported by patholo- gists
carcinoma of the lung
carcinoma of the breast
metastatic melanoma
carcinoma of the kidney gastrointestinal carcinoma
The presenting features are similar to those described for
other intracranial tumours:
• raisedintracranialpressure
• focalneurologicalsigns
• epilepticseizures.

Radiological investigations
CT scan or MRI will diagnose the metastatic tumour and
will show whether the deposits are solitary or multiple
Treatment
Steroid
medication (e.g. dexamethasone) will control
cerebral oedema and should be commenced immediately if
there is raised intracra- nial pressure.
Surgery
to remove the metastasis is indicated if: • there is a
solitary metastasis in a surgically accessible position
• there is no systemic spread.
Radiotherapy
, together with steroid medication to control
cerebral oedema, is used to treat patients with multiple
cerebral metastases and may be advisable following the
excision of a single metastasis

Leptomeningeal metastases
Meningeal carcinomatosis is widespread, multi- focal seeding of
the leptomeninges by systemic cancer.
Chordomas
Chordomas are rare tumours arising from noto- chord cell nests.
They may arise throughout the craniospinal axis but occur
predominantly at the ends of the axial skeleton in:
• thebasioccipitalregion
• thesacrococcygealregion.
Clinical presentation
The majority of intracranial chordomas arise be- tween 20 and 60
years of age. The clinical features result from the widespread
tumour extension and include:
• raised intracranial pressure, causing headaches and vomiting
• multiple cranial nerve palsies, often unilateral • nasopharyngeal
obstruction.

Posterior fossa tumours
Sixty per cent of paediatric brain tumours occur in the posterior
fossa. The relative incidence of the tumours is:
1 cerebellarastrocytoma30%
2 medulloblastoma (infratentorial neuroectodermal tumour) 30%
3 ependymoma 20%
4 brainstem glioma 10%
5 miscellaneous 10%:
(a) choroid plexus papilloma (b)haemangioblastoma
(c) epidermoid, dermoid
(d) chordoma.

Meningioma
The tumour arises from the arachnoid layer of the
meninges, principally the arachnoid villi and
granulations
Meningiomas are the most common of the benign
brain tumours and constitute about 15% of all in-
tracranial tumours, being about one-third of the
number of gliomas. Although they may occur at any
age, they reach their peak incidence in mid- dle age,
are very uncommon in children and occur more
frequently in women than men.

The major histological types are
:
Syncytial or meningotheliomatous
The transitional type
The fibroblastic type
Angiomatous meningiomas
Malignant meningiomas
Clinical presentation
Meningiomas present with features of: •
raisedintracranialpressure
• focalneurologicalsigns
• epilepsy.

Radiological investigations
The CT scan appearance shows a tumour of slightly
increased density prior to contrast; it enhances
vividly and uniformly following intravenous contrast.
Hyperostosis of the cranial vault may be a focal
process at the site of
the tumour attachment or, as seen with en plaque
meningioma, a more diffuse sclerosis. These bone
changes may also be seen on plain skull X-ray.
Magnetic resonance imaging will demonstrate
meningiomas following the intravenous injection of
gadolinium contrast

Preoperative management
Meningiomas are frequently surrounded by severe
cerebral oedema and patients should be treated with
high-dose steroids (dexamethasone) prior to surgery
if possible. Preoperative embolization of the tumour
vasculature may be considered advisable in some
anterior basal and sphenoidal wing tumours where
the major vascular supply is not readily accessible in
the early stages of the operation.

Treatment
The treatment of meningiomas is total surgical excision,
including obliteration of the dural at- tachment. Although
this objective is usually pos- sible there are some
situations where complete excision is not possible
because of the position of the tumour. Tumours arising
from the clivus, in front of the brainstem or those
situated within the cavernous sinus, are notoriously
difficult to excise without causing serious morbidity.
Radiation
therapy may be used to treat residual tumours
following subtotal resection, in order to reduce the risk of
recurrent growth.
Stereotactic radiotherapy
has been used to treat small
meningiomas (less than 3 cm in diam- eter), particularly if
the tumours are located in portions not easily amenable
to surgery, or in the elderly or medically infirm patient.

Acoustic neuroma
Acoustic schwannomas arise from the 8th cranial
nerve and account for 8% of intracranial tumours
CPA in decreasing frequency, are:
• meningioma
• metastatic tumour
• exophytic brainstem glioma • epidermoid tumour.

Clinical manifestations of pituitary tumours.
‘Mass’ effects
Headaches (especially acromegaly) Superior extension
Chiasmal syndrome (impaired visual acuity and fields)
Hypothalamic syndrome (disturbance in thirst, appetite, satiety, sleep and temperature regulation;
diabetes insipidus —uncommon; inappropriate ADH syndrome —uncommon)
Obstructive hydrocephalus Lateral extension
Cranial 3rd, 4th, 6th, diplopia Cranial 5th, facial pain Temporal lobe dysfunction
Inferior extension Nasopharyngeal mass CSF rhinorrhoea
‘Endocrine’ effects
Hyperpituitarism
GH —gigantism/acromegaly
PRL —hyperprolactinaemic syndrome ACTH —Cushing’s disease
TSH —thyrotoxicosis
Hypopituitarism
GH —child: shortness of stature,
hypoglycaemia
PRL —adult female: failure of postpartum
lactation
ACTH —hypocortisolism (Addison’s) TSH —hypothyroidism
LH/FSH —hypogonadism
Acute deterioration
Pituitary apoplexy

Treatment of pituitary Tumour
1 Operative procedures:
(a) trans-sphenoidal excision (b)
transcranial excision.
2 Radiotherapy.
3 Medical treatment with antisecretory
drugs.

Craniopharyngioma
This tumour may occur at any age, although nearly half
occur in the first 20 years of life. They are thought to
arise from the epithelial remnants of Rathke’s pouch.
The tumours occur in the region of the pitu- itary fossa
and extend through the suprasellar cisterns to the
hypothalamus. The majority are cystic, and the fluid is
often yellow and sparkling with cholesterol crystals.
The cyst may be larger than the solid component,
which is often pale and crumbly, consisting of epithelial
debris.
adamantinous type resembles adamantino- ma of the
jaw and is encountered in virtually all children. The
papillary type, so-called adult craniopharyngioma,
occurs in about one-third of adults and is rare in
children.

Raised intracranial pressure

Cerebral blood flow
Between physiological ranges in blood pressure, the
brain is able to maintain a constant cerebral blood
flow. This is achieved by a process called
autoregulation whereby the brain adjusts the in-
tracranial vascular resistance by altering vessel
diameter and tone
CPP=MAP-ICP
Thus in order to maintain cerebral perfusion in the
presence of raised ICP, the systemic blood pressure
needs to be elevated.

Clinical symptoms and signs of raised intracranial
pressure
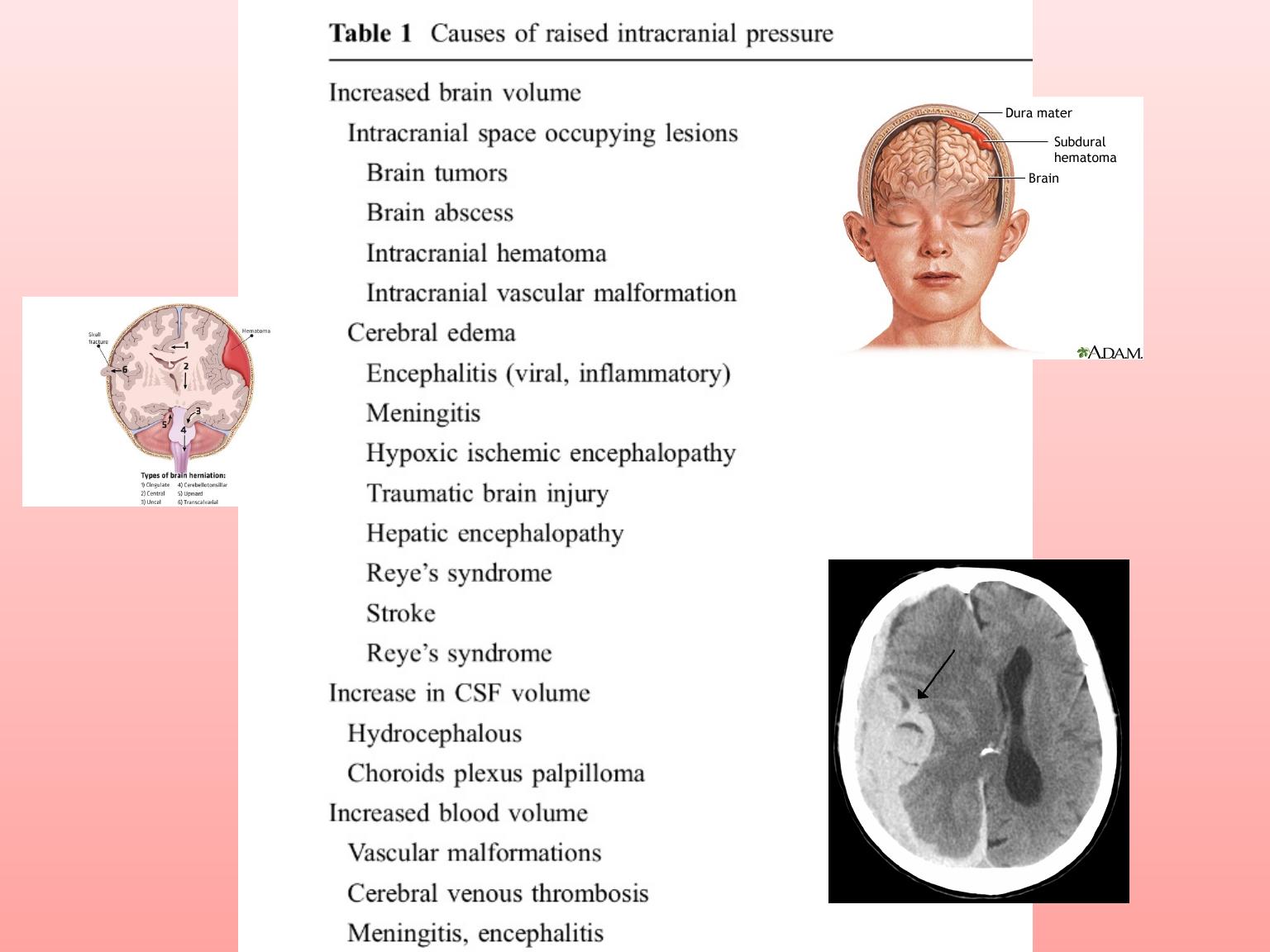
The common causes of raised intracranial pressure are:
• space-occupying lesion—cerebral tumour (and edema),
abscess, intracranial hematoma
• hydrocephalus
• benign intracranial hypertension.
The clinical features will be determined in large part by
the underlying cause of the raised pressure. However,
some of the clinical symptoms and signs will be the same,
no matter what the cause of the raised pressure. The
major features are:
• headache
• nausea and vomiting
• drowsiness
• papilloedema.
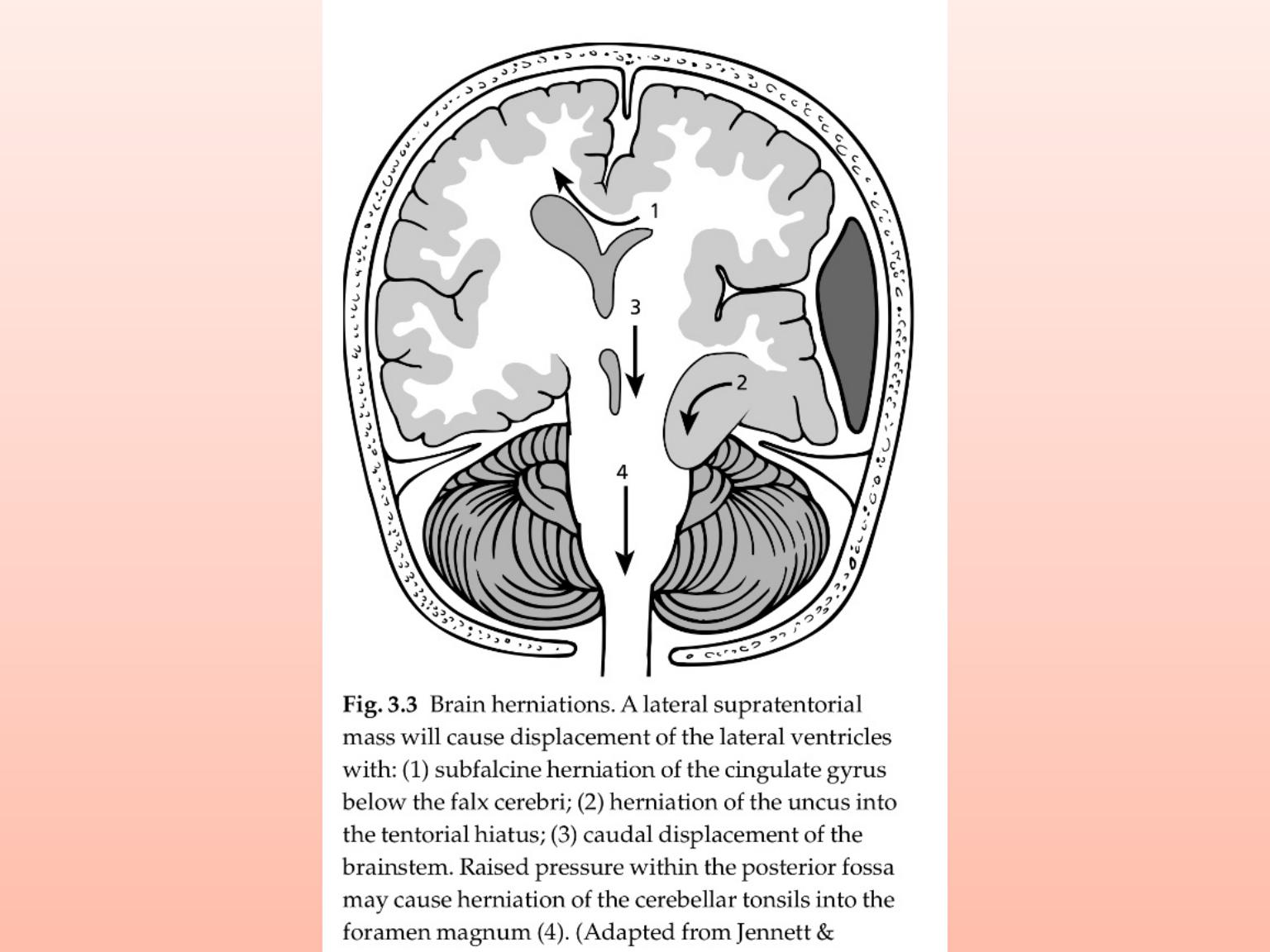

Headache
. The headache associated with in- creased intracranial
pressure is usually worse on waking in the morning and is relieved by
vomiting. Intracranial pressure increases during sleep, probably from
vascular dilatation due to carbon dioxide retention. The cause of the
headache in raised intracranial pressure is probably traction on the
pain-sensitive blood vessels and compres- sion of the pain-sensitive
dura at the base of the cranium.
Nausea and vomiting.
The nausea and vomiting is usually worse
in the morning.
Papilloedema
. The definitive sign of raised intracranial pressure,
papilloedema is due to transmission of the raised pressure along the
subarachnoid sheath of the optic nerve

Long-standing papilloedema
from prolonged raised
intracranial pressure will subsequently develop into secondary
optic atrophy.
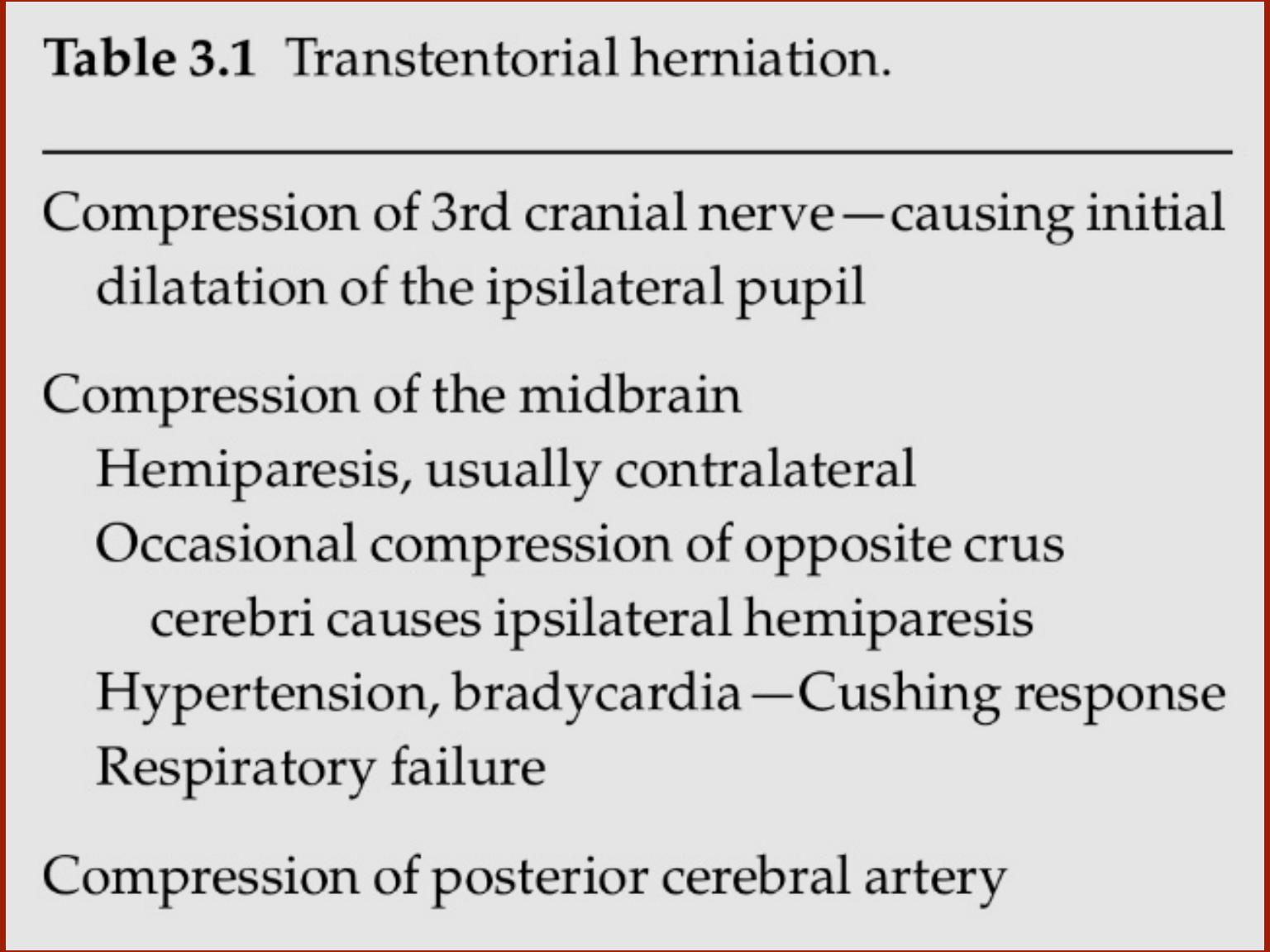
Cushing reflex:
Hypertension/bradycardia/irregular respiration
Sixth nerve palsy,
causing diplopia, may occur in raised
intracranial pressure due to stretching of the 6th nerve by
caudal displacement of the brainstem.
In an infant, raised intracranial pressure will cause a tense,
bulging fontanelle.

Measurement of intracranial pressure
The most common indications are:
• Head injury
• Following major intracranial surgery, when measurement of
the intracranial pressure may help in the management of
patients
•In the assessment of dementia and benign intracranial
hypertension
The intracranial pressure may be recorded from the ventricle,
brain substance, subdural or extradural space.
The
intracranial catheters are attached by a transducer to a
continuous recorder. There are now numerous monitoring
devices with various degrees of technical sophistication.

Management of raised intracranial pressure

