
Bleeding
By dr Alaa sadiq
1

Bleeding
• Bleeding can be due to congenital or acquired abnormalities
in the clotting system.
• History and examination help to clarify the severity and
underlying cause of the bleeding.
• Normal bleeding is seen following surgery and trauma.
• Pathological bleeding occurs when structurally abnormal
vessels rupture or when a vessel is breached in the presence
of a defect in haemostasis.
• This may be due to a deficiency or dysfunction of platelets,
to the coagulation factors, or occasionally to excessive
fibrinolysis,which is most commonly observed following
therapeutic thrombolysis .
2

Clinical assessment
• ‘Screening’ blood tests do not reliably detect all causes of pathological
bleeding (e.g.von Willebrand disease, scurvy, certain anticoagulant
drugs
• A careful clinical evaluation is the key to diagnosis of bleeding
disorders
• It is important to consider the following:
Site of bleeding. Bleeding into muscle and joints, along with
retroperitoneal and intracranial haemorrhage, indicates a likely defect in
coagulation factors.
Purpura, prolonged bleeding from superficial cuts, epistaxis,
gastrointestinal haemorrhage or menorrhagia is more likely to be due to
thrombocytopenia, a platelet function disorder or von Willebrand
disease.
Recurrent bleeds at a single site suggest a local structural abnormality.
Duration of history. It may be possible to assess whether the disorder is
congenital or acquired.
3

Precipitating causes. Bleeding arising spontaneously
indicates a more severe defect than bleeding that occurs only
after trauma.
Surgery. Ask about operations. Dental
extractions,tonsillectomy and circumcision are stressful tests
of the haemostatic system. Immediate post-surgical bleeding
suggests defective platelet plug formation and primary
haemostasis; delayed haemorrhage is more suggestive of a
coagulation defect.
• In postsurgical patients, persistent bleeding from a single
site is more likely to indicate surgical bleeding than a
bleeding disorder.
Family history. While a positive family history may be present
in patients with inherited disorders, the absence of affected
relatives does not exclude a hereditary bleeding diathesis;
about one-third of cases of haemophilia arise in individuals
without afamily history, and deficiencies of factor VII, X
andXIII are recessively inherited.
4

• Recessive disorders are more common in cultures where
there is consanguineous marriage.
Drugs. Use of antithrombotic anticoagulant and fibrinolytic
drugs must belicited.
• Drug interactions with warfarin and drug-induced
thrombocytopenia should be considered.
• Some ‘herbal’ remedies may result in a bleeding diathesis.
• Clinical examination may reveal different patterns of skin
bleeding.
• Petechial purpura is minor bleeding into the dermis that is
flat and non-blanching .
• Petechiae are typically found in patients with
thrombocytopenia or platelet dysfunction.
• Palpable purpura occurs in vasculitis Ecchymosis, or bruising,
is more extensive bleeding into deeper layers of the skin.
5

• The lesions are initially dark red or purple but become
yellow as haemoglobin is degraded.
• Retroperitoneal bleeding presents with a flank haematoma.
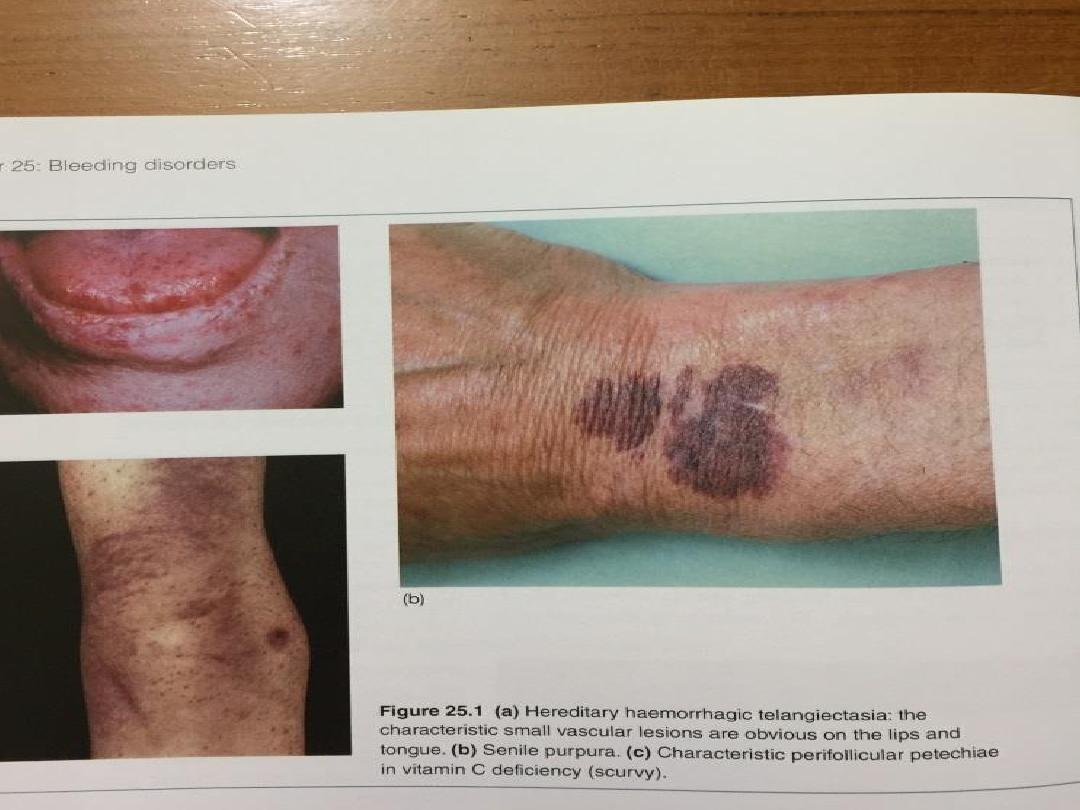
• Telangiectasia of lips and tongue points to hereditary
haemorrhagic telangiectasia .
• Joints should be examined for evidence of haemarthroses.
• A full examination is important, as it may give clues to an
underlying associated systemic illness such as a
haematological or other malignancy, liver disease, renal
failure, connective tissue disease and possible causes of
splenomegaly.
• If the patient has a history that is strongly suggestive of a
bleeding disorder and all the preliminary screening tests give
normal results, further investigations, such as measurement
of von Willebrand factor and assessment of platelet
function, should be performed
6

Thrombocytopenia (low platelet count)
• A reduced platelet count may arise by one of two
mechanisms:
• • decreased or abnormal production (bone marrow failure
and hereditary thrombocytopathies
• increased consumption following release into the circulation
(immune-mediated, DIC or sequestration).
• Spontaneous bleeding does not usually occur until the
platelet count falls below 20 × 109/L, unless their function is
also compromised.
• Purpura and spontaneous bruising are characteristic but
there may also be oral, nasal, gastrointestinal or
genitourinary bleeding.
• Severe thrombocytopenia (< 10 × 109/L) may result in
retinal haemorrhage and potentially fatal intracranial
bleeding, but this is rare.
7

•A blood film is the single most useful initial
investigation. Examination of the bone marrow may
reveal increased megakaryocytes in consumptive
causes of thrombocytopenia, or the underlying cause
of bone marrow failure in leukaemia, hypoplastic
anaemia or myelodysplasia.
•Treatment (if required) depends on the underlying
cause.
•Platelet transfusion is rarely required and is usually
confined to patients with bone marrow failure and
platelet counts below 10 × 109/L, or to clinical
situations with actual or predicted serious
haemorrhage.
8

Disorders of primary haemostasis
• The initial formation of the platelet plug also known as
‘primary haemostasis’) may fail in thrombocytopenia , von
Willebrand disease and also in platelet function disorders
and diseases affecting the vessel wall.
Vessel wall abnormalities
• Vessel wall abnormalities may be:
1. congenital, such as hereditary haemorrhagic
telangiectasia
2. acquired, as in a vasculitis or scurvy.
9

Hereditary haemorrhagic
telangiectasia
• Hereditary haemorrhagic telangiectasia (HHT) is a
dominantly inherited condition
• Telangiectasia and small aneurysms are found on the
fingertips, face and tongue, and in the nasal passages, lung
and gastrointestinal tract.
• A significant proportion of these patients develop larger
pulmonary arteriovenous malformations (PAVMs) that cause
arterial hypoxaemia due to a right-to-left shunt.
• These predispose to paradoxical embolism, resulting in
stroke or cerebral abscess. All patients with HHT should be
screened for PAVMs; if these are found, ablation by
percutaneous embolisation should be considered. Patients
present either with recurrent bleeds, particularly epistaxis,
or with iron deficiency due to occult gastrointestinal
bleeding.
10

• Treatment can be difficult because of the multiple bleeding
points but regular iron therapy often allows the marrow to
compensate for blood loss.
• Local cautery or laser therapy may prevent single lesions
from bleeding.
11
12

Ehlers–Danlos disease
• Vascular Ehlers–Danlos syndrome is a rare autosomal
dominant disorder (1/100 000) which results in fragile blood
vessels and organ membranes, leading to bleeding and
organ rupture.
• The diagnosis should be considered when there is a history
of bleeding but normal laboratory tests.
Scurvy
• Vitamin C deficiency affects the normal synthesis of collagen
and results in a bleeding disorder characterised by petechial
haemorrhage, bruising and subperiosteal bleeding. The key
to diagnosis is the dietary history .
13

Platelet function disorders
• Bleeding may result from thrombocytopenia or from congenital or
acquire abnormalities of platelet function. The most common acquired
disorders are iatrogenic, resulting from the use of aspirin, clopidogrel,
dipyridamole and the IIb/IIIa inhibitors to prevent arterial thrombosis .
• Inherited platelet function abnormalities are relatively rare.
• Congenital abnormalities may be due to deficiency of the membrane
glycoproteins, e.g. Glanzmann’s thrombasthenia (IIb/IIIa) or Bernard–
Soulier disease (Ib), or due to the presence of defective platelet
granules, e.g. a deficiency of dense (delta) granules giving rise to
storage pool disorders.
• Apart from Glanzmann’s thrombasthenia, these conditions are mild
disorders, with bleeding typically occurring after trauma or surgery but
rarely spontaneously.
• Glanzmann’s is an autosomal recessive condition associated with a
variable but often severe bleeding disorder.
14

•These conditions are usually managed by local
mechanical measures, but antifibrinolytics, such as
tranexamic acid, may be useful and, in severe
bleeding, platelet transfusion may be required.
•Recombinant VIIa is licensed for the treatment of
resistant bleeding in Glanzmann’s thrombasthenia.
15

Idiopathic thrombocytopenic purpura
• Idiopathic thrombocytopenic purpura (ITP) is mediated by
autoantibodies, most often directed against the platelet
membrane glycoprotein IIb/IIIa, which sensitise the platelet,
resulting in premature removal from the circulation by cells
of the reticulo-endothelial system.
• It is not a single disorder; some cases occur in isolation while
others are associated with underlying immune dysregulation
in conditions such as connective tissue diseases, HIV
infection, B cell malignancies, pregnancy and certain drug
therapies. However, the clinical presentation and
pathogenesis are similar, whatever the cause of ITP
16

Clinical features and investigations
• The presentation depends on the degree of thrombocytopenia.
• Spontaneous bleeding typically occurs only when the platelet count is
below 20 ×109/L. At higher counts, the patient may complain of easy
bruising or sometimes epistaxis or menorrhagia.
• Many cases with counts of more than 50 × 109/L are discovered by
chance.
• In adults, ITP more commonly affects females and may have an
insidious onset.
• Unlike ITP in children, it is unusual for there to be a history of a
preceding viral infection.
• Symptoms or signs of a connective tissue disease may be apparent at
presentation or emerge several years later.
• Patients aged over 65 years should have a bone marrow examination
to look for an accompanying B cell malignancy and appropriate
autoantibody testing performed if a diagnosis of connective tissue
disease is likely.
HIV testing should be considered.
17

• The peripheral blood film is normal, apart from a greatly
reduced platelet number, whilst the bone marrow reveals an
obvious increase in megakaryocytes
18

Management
• Many patients with stable compensated ITP and a platelet
count of more than 30 × 109/L do not require treatment to
raise the platelet count, except at times of increased
bleeding risk, such as surgery and biopsy.
• First-line therapy for patients with spontaneous bleeding is
with prednisolone
1 mg/kg daily to suppress antibody production and inhibit
phagocytosis of sensitised platelets by reticuloendothelial
cells.
• Administration of intravenous immunoglobulin (IVIg) can
raise the platelet count by blocking antibody receptors on
reticuloendothelial cells, and is combined with corticosteroid
therapy if there is severe haemostatic failure or a slow
response to steroids alone.
• Persistent or potentially lifethreatening bleeding should be
treated with platelet transfusion in addition to the other
therapies.
19

• The condition may become chronic, with remissions and
relapses.
• Relapses should be treated by reintroducing corticosteroids.
• If a patient has two relapses, or primary refractory disease,
splenectomy is considered,
• Splenectomy produces complete remission in about 70% of
patients and improvement in a further 20–25%, so that,
following splenectomy, only 5–10% of patients require
further medical therapy.
• If severe thrombocytopenia with or without significant
bleeding persists despite splenectomy, second-line therapy
with the thrombopoietin analogue romiplostim or the
thrombopoietin receptor agonist eltrombopag should be
considered
• Low-dose corticosteroid therapy, immunosuppressants such
as rituximab, ciclosporin and tacrolimus should be
considered in cases where the approaches above are
ineffective.
20

Coagulation disorders
• Coagulation factor deficiency may be congenital or acquired,
and may affect one or several of the coagulation factors
Inherited disorders are almost uniformly related to
decreased synthesis, as a result of mutation in the gene in
coagulation.
• Von Willebrand disease is the most common inherited
bleeding disorder.
• Haemophilia A and B are the most common single
coagulation factor deficiencies, but inherited deficiencies of
all the other coagulation factors are seen.
• Acquired disorders may be due to under-production (e.g. in
liver failure), increased consumption(e.g. in disseminated
intravascular coagulation) or inhibition of function (such as
heparin therapy or immune inhibitors of coagulation, e.g.
acquired haemophilia A).
21

22

23