Chloramphenicol It is poorly soluble in water. Chloramphenicol succinate, which is used for parenteral administration, is highly water-soluble. Mechanism of Action & Antimicrobial ActivityChloramphenicol is a potent inhibitor of microbial protein synthesis. It binds reversibly to the 50S subunit of the bacterial ribosome and inhibits peptide bond formation.Chloramphenicol is a bacteriostatic broad-spectrum antibiotic that is active against both aerobic and anaerobic gram-positive and gram negativeorganisms. It is active also against Rickettsiae but not Chlamydiae . H influenzae, Neisseria meningitidis , and some strains of bacteroides are highly susceptible, and for these organisms, chloramphenicol may be bactericidal.Low-level resistance to chloramphenicol may emerge from large populations of chloramphenicol-susceptible cells by selection of mutants that are less permeable to the drug. Clinically significant resistance is due to production of chloramphenicol acetyltransferase, a plasmid-encoded enzyme that inactivates the drug.
PharmacokineticsAfter oral administration, crystalline chloramphenicol is rapidly and completely absorbed. Chloramphenicol palmitate is a pro drug that is hydrolyzed in the intestine to yield free chloramphenicol.The parenteral formulation is a pro drug, chloramphenicol succinate, which hydrolyzes to yield free chloramphenicol, giving blood levels somewhat lower than those achieved with orally administered drug. Chloramphenicol is widely distributed to virtually all tissues and body fluids, including the central nervous system and cerebrospinal fluid, such that the concentration of chloramphenicol in brain tissue may be equal to that in serum. The drug penetrates cell membranes readily.Most of the drug is inactivated either by conjugation with glucuronic acid (principally in the liver) or by reduction to inactive aryl amines. Active chloramphenicol (about 10% of the total dose administered) and its inactive degradation products (about 90% of the total) are eliminated in the urine. A small amount of active drug is excreted into bile and feces. The systemic dosage of chloramphenicol need not be altered in renal insufficiency, but it must be reduced markedly in hepatic failure. Newborns less than a week old and premature infants also clear chloramphenicol less well, and the dosage should be reduced to 25 mg/kg/d.
Clinical UsesBecause of potential toxicity, bacterial resistance, and the availability of many other effective alternatives, chloramphenicol is rarely used now. It may be considered for treatment of serious rickettsial infections such as typhus and Rocky Mountain spotted fever. It is an alternative to a β-lactam antibiotic for treatment of bacterial meningitis occurring in patients who have major hypersensitivity reactions to penicillin. The dosage is 50–100 mg/kg/d in four divided doses.Chloramphenicol is used topically in the treatment of eye infections because of its broad spectrum and its penetration of ocular tissues and the aqueous humor. It is ineffective for chlamydial infections.
Adverse ReactionsAdults occasionally develop gastrointestinal disturbances, including nausea, vomiting, and diarrhea. This is rare in children. Oral or vaginal candidiasis may occur as a result of alteration of normal microbial flora.Chloramphenicol causes immune suppression and aplastic anemia, a rare consequence (1 in 24,000 to 40,000 courses of therapy).Chloramphenicol administration by any route, rarly caused idiosyncratic reaction unrelated to dose, although it occurs more frequently with prolonged use. It tends to be irreversible and can be fatal.Newborn infants lack an effective glucuronic acid conjugation mechanism for the degradation and detoxification of chloramphenicol. Consequently, when infants are given dosages above 50 mg/kg/d, the drug may accumulate, resulting in the gray baby syndrome , with vomiting, flaccidity, hypothermia, gray color, shock,and vascular collapse. To avoid this toxic effect, chloramphenicol should be used with caution in infants and the dosage limited to 50 mg/kg/d (or less during the first week of life) in full-term infants more than 1 week old and 25 mg/kg/d in premature infants.
Tetracyclines
All tetracyclines have the basic structure: tetracyclic nucleus to which a variety of functional groups are attached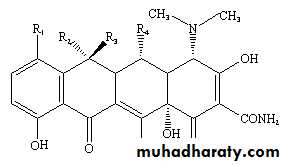
Free tetracyclines are crystalline amphoteric substances of low solubility. They are available as hydrochlorides, which are more soluble. Such solutions are acid and, with the exception of chlortetracycline, fairly stable. Tetracyclines chelate divalent metal ions, which can interfere with their absorption and activity. A newly approved tetracycline analog, tigecycline, is a glycylcycline and a semisynthetic derivative of minocycline.
Mechanism of Action & Antimicrobial Activity:
Tetracyclines are broad-spectrum bacteriostatic antibiotics that inhibit protein synthesis. Tetracyclines are active against many gram-positive and gram negative bacteria, including certain anaerobes, rickettsiae, chlamydiae, and mycoplasmas. The antibacterial activities of most tetracyclines are similar except that tetracycline-resistant strains may be susceptible to doxycycline and minocycline.
Differences in clinical efficacy for susceptible organisms are minor and attributable largely to features of absorption, distribution, and excretion of individual drugs.
Resistance
Three mechanisms of resistance to tetracyclines included:
(1) impaired influx or increased efflux by an active transport protein pump
(2) ribosome protection due to production of proteins that inibit tetracycline binding to the ribosome
(3) enzymatic inactivation.
Pharmacokinetics
Tetracyclines differ in their absorption after oral administration and in their elimination.Absorption after oral administration is approximately 30% for chlortetracycline; 60–70% for tetracycline, oxytetracycline, demeclocycline, and methacycline; and 95–100% for doxycycline and minocycline.
Tigecycline is poorly absorbed orally and must be administered intravenously.
A portion of an orally administered dose of tetracycline remains in the gut lumen, alters intestinal flora, and is excreted in the feces. Absorption occurs mainly in the upper small intestine and is impaired by food (except doxycycline and minocycline); by divalent cations (Ca 2+ , Mg 2+ , Fe 2+ ) or Al 3+ ; by dairy products and antacids, which contain multivalent cations; and by alkaline pH. Specially buffered tetracycline solutions are formulated for intravenous administration.
Tetracyclines are 40–80% bound by serum proteins.
Oral dosages of 500 mg every 6 hours of tetracycline hydrochloride or oxytetracycline produce peak blood levels of 4–6 mcg/mL.
Intravenously injected tetracyclines give somewhat higher levels, but only temporarily. Peak levels of 2–4 mcg/mL are achieved with a 200-mg dose of doxycycline or minocycline.
Tetracyclines are distributed widely to tissues and body fluids except for cerebrospinal fluid, where concentrations are 10–25% of those in serum. Minocycline reaches very high concentrations in tears and saliva, which makes it useful for eradication of the meningococcal carrier state.
Tetracyclines cross the placenta to reach the fetus and are also excreted in milk.
As a result of chelation with calcium, tetracyclines bound to-and damage-growing bones and teeth.
Tetracyclines are excreted mainly in bile and urine. Concentrations in bile exceed those in serum tenfold. Some of the drug excreted in bile is reabsorbed from the intestine (enterohepatic circulation) and may contribute to maintenance of serum levels. 10-50% of various tetracyclines is excreted into the urine, mainly by glomerular filtration. Ten to forty percent of the drug is excreted in feces.
Doxycycline and tigecycline, in contrast to other tetracyclines, are eliminated by nonrenal mechanisms, do not accumulate significantly, and require no dosage adjustment in renal failure.
Tetracyclines are classified as short-acting (chlortetracycline, tetracycline, oxytetracycline)
T 1/2= 6-8h, intermediate-acting (demeclocycline and methacycline) t1/2= 12h, or long-acting (doxycycline and minocycline) t1/2= 16-18h.
The almost complete absorption and slow excretion of doxycycline and minocycline
allow for once-daily dosing for certain indications, but by convention these two drugs are usually dosed twice daily.
Clinical Uses
A tetracycline is used in the treatment of infections caused by rickettsiae, Mycoplasma pneumonia , chlamydiae, and some spirochetes.They are used in combination regimens to treat gastric and duodenal ulcer disease caused by Helicobacter pylori .
They may be used in various G+ve and G-ve bacterial infections: vibrio infections.
Tetracycline in combination with other antibiotics indicated for plague, tularemia, and brucellosis.
Tetracyclines are sometimes used in the treatment or prophylaxis of protozoal infections.
Other uses include treatment of acne, exacerbations of bronchitis, community-acquired pneumonia, Lyme disease, relapsing fever, leptospirosis, and some nontuberculous mycobacterial infections (eg, Mycobacterium marinum ). Tetracyclines formerly were used for a variety of common infections, including bacterial gastroenteritis and urinary tract infections.
However, many strains of bacteria causing these infections are now resistant, and other agents have largely supplanted tetracyclines.
Adverse Reactions
A. Gastrointestinal Adverse Effects: Nausea, vomiting, and diarrhea are the most common reasons for discontinuing tetracycline medication.B. Bony Structures and Teeth: Tetracyclines are readily bound to calcium deposited in newly formed bone or teeth in young children. When a tetracycline is given during pregnancy, it can be deposited in the fetal teeth, leading to fluorescence, discoloration, and enamel dysplasia; it can also be deposited in bone, where it may cause deformity or growth inhibition. Because of these effects, tetracyclines are generally avoided in pregnancy. If the drug is given for long periods to children younger than 8 years, similar changes can result.
C. Other Toxicities
-Tetracyclines can impair hepatic function, especially during pregnancy, in patients with preexisting hepatic insufficiency and when high doses are given intravenously.
-Hepatic necrosis has been reported with daily doses of 4 g or more intravenously.
-Renal tubular acidosis and other renal injury. Tetracyclines given along with diuretics may produce nitrogen retention. Tetracyclines other than doxycycline may accumulate to toxic levels in patients with impaired kidney function.
Intravenous injection can lead to venous thrombosis.
-Intramuscular injection produces painful local irritation and should be avoided.
-Systemically administered tetracycline, especially demeclocycline, can induce sensitivity to sunlight or ultraviolet light, particularly in fair-skinned persons.
-Dizziness, vertigo, nausea, and vomiting have been noted particularly with doxycycline at doses above 100 mg.
MacrolidesThe macrolides are a group of closely related compounds characterized by a macrocyclic lactone ring (usually containing 14 or 16 atoms) to which deoxy sugars are attached. The prototype drug, erythromycin was obtained in 1952 from Streptomyces erythreus Clarithromycin and azithromycin are semisynthetic derivatives of erythromycin.
ErythromycinMechanism of Action & Antimicrobial ActivityErythromycin and other macrolides may be bacteriostatic or bactericidal, particularly at higher concentrations, for susceptible organisms. Inhibition of protein synthesis occurs via reversibly binding to 50s subunit of the ribosome à inhibits translocation during protein synthesis, as a result, peptidyl-tRNA is dissociated from the ribosome. Erythromycin also inhibits the formation of the 50S ribosomal subunit. -Erythromycin is active against susceptible strains of gram-positive organisms, especially pneumococci, streptococci, staphylococci, and corynebacteria. Mycoplasma pneumoniae , L pneumophila , Chlamydia trachomatis , Chlamydia psittaci, Chlamydia pneumoniae , H pylori , Listeria monocytogenes, and certain mycobacteria (Mycobacterium kansasii, Mycobacterium scrofulaceum) are also susceptible. -Gram negative organisms such as Neisseria sp, Bordetella pertussis, Bartonellahenselae, and Bartonella quintana as well as some Rickettsia species,Treponema pallidum , and Campylobacter species are susceptible. -Haemophilus influenzae is somewhat less susceptible. Resistance to erythromycin is usually plasmid-encoded. Three mechanisms were identified: (1) reduced permeability of the cell membrane or active efflux; (2) production (by Enterobacteriaceae) of esterases that hydrolyze macrolides; (3) modification of the ribosomal binding site (so-called ribosomal protection) by chromosomal mutation.
PharmacokineticsErythromycin base is destroyed by stomach acid and must be administered with enteric coating. Food interferes with absorption.Stearates are fairly acid-resistant and somewhat better absorbed. The erythromycin estolate is the best-absorbed oral preparation.Absorbed drug is distributed widely except to the brain and cerebrospinal fluid. It traverses the placenta and reaches the fetus.The serum half-life is approximately 1.5 hours normally and 5 hours in patients with anuria. Adjustment for renal failure is not necessary. Erythromycin is not removed by dialysis.Large amounts of an administered dose are excreted in the bile and lost in feces, and only 5% is excreted in the urine.
Clinical Uses-Erythromycin is a drug of choice in corynebacterial infections (diphtheria, corynebacterial sepsis, erythrasma); -Respiratory, neonatal, ocular, or genital chlamydial infections; and in treatment of community-acquired pneumonia because its spectrum of activity includes pneumococcus, M pneumoniae , and L pneumophila. -Erythromycin is also useful as a penicillin substitute in penicillin allergicindividuals with infections caused by staphylococci , streptococci, or pneumococci. -Emergence of erythromycin resistance in strains of group A streptococci and pneumococci (penicillin-non-susceptible pneumococci in particular) has made macrolides less attractive as first line agents for treatment of pharyngitis, skin and soft tissue infections, and pneumonia. -Erythromycin has been recommended as prophylaxis against endocarditis during dental procedures in individuals with valvular heart disease, although clindamycin,which is better tolerated, has largely replaced it. The oral dosage of erythromycin base, stearate, or estolate is 0.25–0.5 g every 6 hours (for children, 40 mg/kg/d). The dosage of erythromycin ethylsuccinate is 0.4–0.6 g every 6 hours. Oral erythromycin base (1 g) is sometimes combined with oral neomycin or kanamycin for preoperative preparation of the colon.The intravenous dosage of erythromycin gluceptate or lactobionate is 0.5–1.0 g every 6 hours for adults and 20-40 mg/kg/d for children. The higher dosage is recommended when treating pneumonia caused by L pneumophila .Adverse ReactionsAnorexia, nausea, vomiting, and diarrhea are common. Gastrointestinalintolerance, which is due to a direct stimulation of gut motility, is themost common reason for discontinuing erythromycin and substitutinganother antibiotic.Erythromycins, particularly the estolate, can produce acute cholestatic hepatitis (fever, jaundice, impaired liver function), probably as a hypersensitivity reaction. Other allergic reactions include fever, eosinophilia, and rashes.
ClarithromycinClarithromycin is derived from erythromycin by addition of a methyl group and has improved acid stability and oral absorption compared with erythromycin. Clarithromycin and erythromycin are similar with respect to antibacterial activity except that clarithromycin is more active against Mycobacterium avium. Clarithromycin also has activity against H pylori, Mycobacterium leprae , Toxoplasma gondii, and H influenzae . Erythromycin-resistant streptococci and staphylococci are also resistant to clarithromycin.The longer half-life of clarithromycin (6 hours) compared with erythromycin permits twice-daily dosing.The recommended dosage is 250–500 mg twice daily or 1000 mg of the extended-release formulation once daily. Clarithromycin penetrates most tissues well, with concentrations equal to orexceeding serum concentrations.Clarithromycin is metabolized in the liver. The major metabolite is 14-hydroxyclarithromycin, which also has antibacterial activity.Portions of active drug and this major metabolite are eliminated inthe urine, and dosage reduction is recommended for patients with creatinine clearances less than 30 mL/min..The advantages of clarithromycin compared with erythromycin are lower incidence of gastrointestinal intolerance and less frequentdosing.
AzithromycinAzithromycin, is derived from erythromycin by addition of a methylated nitrogeninto the lactone ring. Its spectrum of activity, mechanism of action, and clinical uses are similar to those of clarithromycin. Azithromycin is active against M avium complex and T gondii . Azithromycin is slightly less active than erythromycin and clarithromycin againststaphylococci and streptococci and slightly more active against H influenzae . Azithromycin is highly active against Chlamydia sp. Azithromycin differs from erythromycin and clarithromycin mainly in pharmacokinetic properties, azithromycin penetrates into most tissues(except cerebrospinal fluid) and phagocytic cells extremely well, withtissue concentrations exceeding serum concentrations by 10- to100-fold. The drug is slowly released from tissues (tissue half-life of 2–4 days) to produce an elimination half-life approaching 3 days. These unique properties permit once-daily dosing and shortening of the duration of treatment in many cases. For example, a single 1-g dose of azithromycin is as effective as a 7-day course of doxycyclinefor chlamydial cervicitis and urethritis. Community-acquired pneumoniaAzithromycin is rapidly absorbed and well tolerated orally. It should be administered 1 hour before or 2 hours after meals.
Clindamycin Clindamycin is a chlorine-substituted derivative of lincomycin, an antibiotic that is elaborated by Streptomyces lincolnensis .Mechanism of Action & Antibacterial ActivityClindamycin: inhibits protein synthesis like erythromycin. - Streptococci, staphylococci, and pneumococci are inhibited by clindamycin.-Gram-negative aerobic species are intrinsically resistant because of poor permeability of the outer membrane.-Enterococci and gram negative aerobic organisms are resistant.- Bacteroides sp and other anaerobes are susceptible. Resistance to clindamycin, which onfers cross resistance to macrolides , is due to:(1) mutation of the ribosomal receptor site; (2) modification of the receptor by a constitutively expressed methylase (3) enzymatic inactivation of clindamycin.
PharmacokineticsAbsorbed orally, oral dosages of clindamycin, 0.15–0.3 g every 8 hours (10–20 mg/kg/d for children), yield serum levels of 2–3 mcg/mL. When administered intravenously, 600 mg of clindamycin every 8 hours gives levels of 5–15 mcg/mL. The drug is about 90% protein bound.Clindamycin penetrates well into most tissues, with brain and cerebrospinal fluid being important exceptions. It penetrates well into abscesses and is actively taken up and concentrated by phagocytic cells. Clindamycin is metabolized by the liver, and both active drug and active metabolites are excreted in bile and urine. The half-life is about 2.5 hours in normal individuals, increasing to 6 hours in patients with anuria. No dosage adjustment is required for renal failure.
Clinical UsesClindamycin is indicated for the treatment of Skin and soft-tissue infections caused by streptococci and staphylococci. It is often active against community-acquired strains of methicillin-resistantS aureus , an increasingly common cause of skin and soft tissue infections. Clindamycin is also indicated for treatment of anaerobic infections caused by Bacteroides sp and other anaerobes that often participate in mixed infections. Clindamycin, sometimes in combination with an aminoglycoside or cephalosporin, is used to treat penetrating wounds of the abdomen and the gut; infections originatingin the female genital tract, eg, septic abortion, pelvic abscesses, or pelvic inflammatory disease; and lung abscesses.Clindamycin is now recommended rather than erythromycin for prophylaxis of endocarditis in patients with valvular heart disease who are undergoing certain dental procedures. Clindamycin plus primaquine is an effective alternative to trimethoprim-sulfamethoxazole for moderate to moderately severe Pneumocystis jiroveci pneumonia in AIDS patients. It is also used in combination with pyrimethamine forAIDS-related toxoplasmosis of the brain.Adverse EffectsCommon adverse effects are diarrhea, nausea, and skin rashes. Impaired liver function (with or without jaundice) and neutropenia sometimes occur. Administration of clindamycin is a risk factor for diarrhea and colitis due to C difficile .