
ﺑ
ﺳ
م
ﷲ
ا
ﻟ
ر
ﺣ
ﻣ
ن
ﻟا
ر
ﺣ
ﯾ
م

}
Are group of inherited or acquired disorders,
presenting either with pure ataxia or in
association with other neurological and non-
neurological features.
}
DDx is wide

}
Causes of ataxia
1)
Structural
2)
Infection
3)
Degenerative
4)
Inflammatory (multiple sclerosis, Coeliac
disease)
5)
Metabolic and endocrine
6)
Vascular
7)
Inherited ataxias

}
Inherited ataxias:
are heterogeneous group
o
Friedreich ataxia:
childhood or adolescence
onset
v
Ataxia, nystagmus, dysarthria, spasticity,
areflexia, DM, optic atrophy and cardiac
abnormalities.
v
Autosomal recessive
o
Ataxia telangiectasia:
childhood onset
v
Progressive ataxia, telangiectasia on
conjunctivae, tendency to malignancies
v
Autosomal recessive



}
It is one of the
neurodegenerative
diseases
characterized by degeneration of motor neurons in
the
spinal cord
and
cranial nerve nuclei
, and of
pyramidal neurons in the
motor cortex.
}
M.N.D is a progressive disorder of unknown cause.

Clinical features
:
}
Usually after the age of 50 years
}
Very uncommon before the age of 30 years
}
Affects males more commonly than females
}
Limb muscle weakness, wasting,cramps,
occasionally fasciculation
}
Disturbance of speech/swallowing
(dysarthria/dysphagia)
}
Progressive over months

Signs include:
}
Wasting
and
fasciculation
of muscles
}
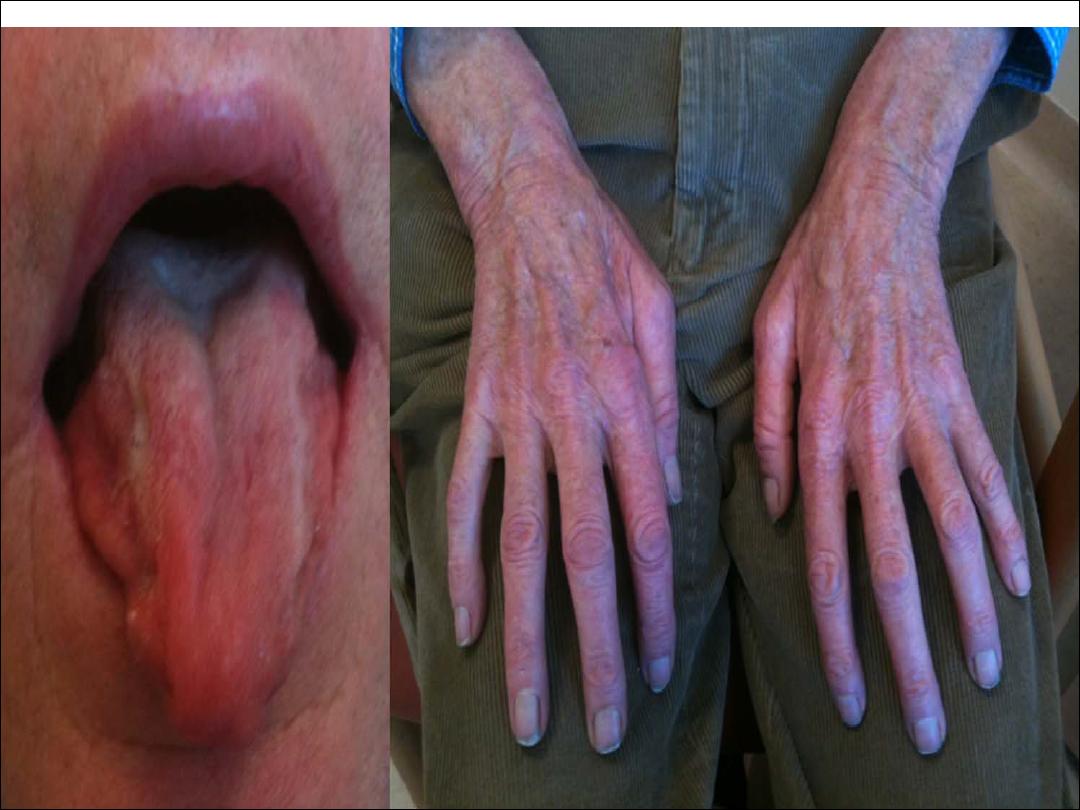
Weakness of muscles of limbs, tongue, face and palate
}
Pyramidal tract involvement causes spasticity,
exaggerated tendon reflexes, extensor plantar
responses
}
External ocular muscles and sphincters usually remain
intact
The presence of brisk reflexes in wasted
fasciculating limb muscles is typical.

}
No ocular involvement
}
No sphenctor dysfunction
}
No objective sensory deficit
}
No intellectual impairment in most cases
}
No CSF abnormality
}
No cerebellar involvement

}
There are four clinical pattern of M.N.D:
1-
Progressive muscular atrophy
2- Progressive bulbar palsy
3- Pseudobulbar palsy
4- Amyotrophic lateral sclerosis
Diagnosis:
}
Alternative diagnoses need to be carefully excluded in
particular, potentially treatable disorders.
}
Investigations may include EMG&NCS, thyroid function
test, brain and cervical MRI.

}
Differential diagnosis:
1-cervical spondylosis
2-multifocal motor neuropathy with conduction block
3- Thyrotoxicosis
4-diabetic amyotrophy
5-chronic lead poisoning
6-spinal cord tumor

Treatment :
}
The glutamate antagonist,
riluzole 100mg/d
, has
recently been shown to have a small effect in
prolonging life expectancy by about two months.
}
Psychological and physical support.
}
splints, walking aids, wheelchairs.
}
Feeding by percutaneous gastrostomy.
}
non-invasive ventilatory support may help distress
from weak respiratory muscles.


MYOPATHIES
}
Skeletal muscle diseases, or myopathies, are
disorders with structural changes or functional
impairment of muscle.
}
The most common clinical findings of a myopathy are
proximal, symmetric limb weakness (arms or legs or
both) with preserved reflexes and sensation
. Other
symptoms and signs of muscle disease include
myotonia
(delayed muscle relaxation) and muscle pain
(myalgia)
.

}
Classification of myopathies:
1-Muscular dystrophies
2- Inflammatory myopathies
3-Inherited metabolic myopathies
4-Mitochondrial myopathies
5-Periodic paralyses (muscle channelopathies)
6-Endocrine myopathies
7-Drugs and toxin

}
Classification of myopathies:
1-Muscular dystrophies
2- Inflammatory myopathies
3-Inherited metabolic myopathies
4-Mitochondrial myopathies
5-Periodic paralyses (muscle channelopathies)
6-Endocrine myopathies
7-Drugs and toxin

Duchenne Muscular Dystrophy
}
This is X-linked recessive disorder.
}
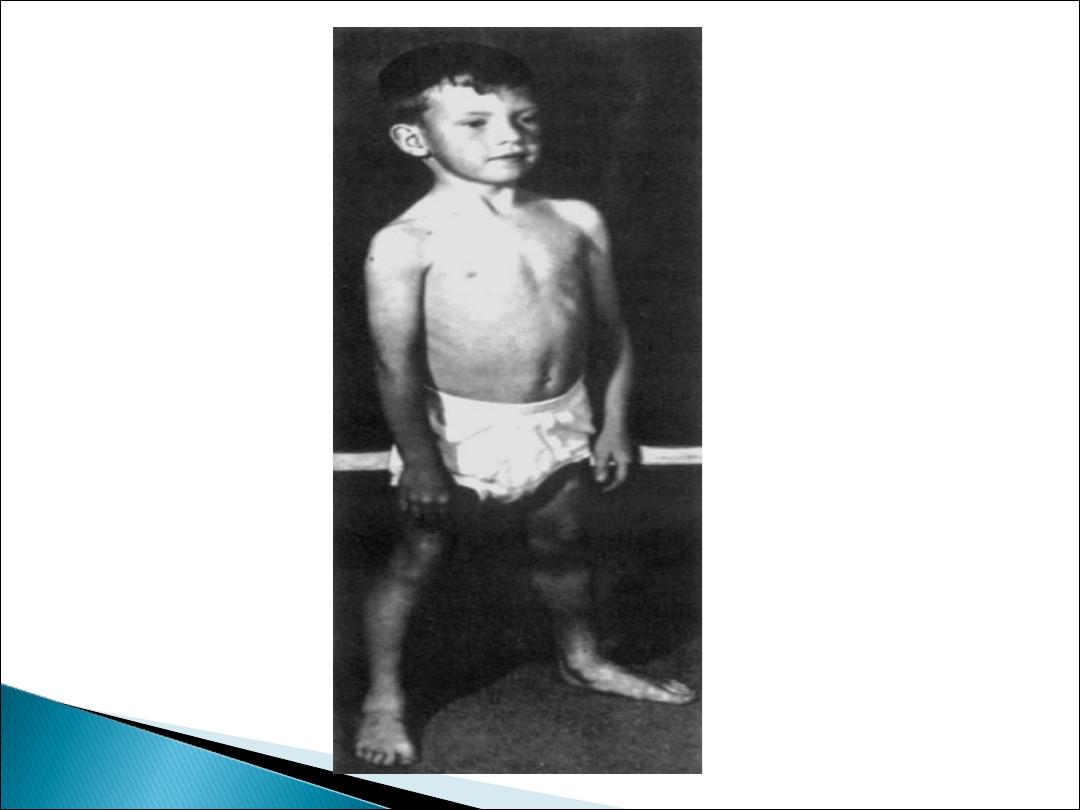
The disorder usually becomes apparent between ages 3
and 5 years. The boys fall frequently and have difficulty
keeping up with friends when playing. Running, jumping,
and hopping are invariably abnormal.
}
On getting up from the floor, the patient uses his hands
to climb up himself [
Gower‘s sign
].
}
He may start toe walking.


}
By age 12, most patients are wheelchair dependent
}
Serum CK (creatine kinase) levels are invariably
elevated to between 20 and 100 times normal
.
}
Patients with Duchenne dystrophy used to die within 10
years of diagnosis, but with improved general care they
are now living into the third decade.
}
Cardiac involvement may include conduction defect,
cardiomyopathy.
}
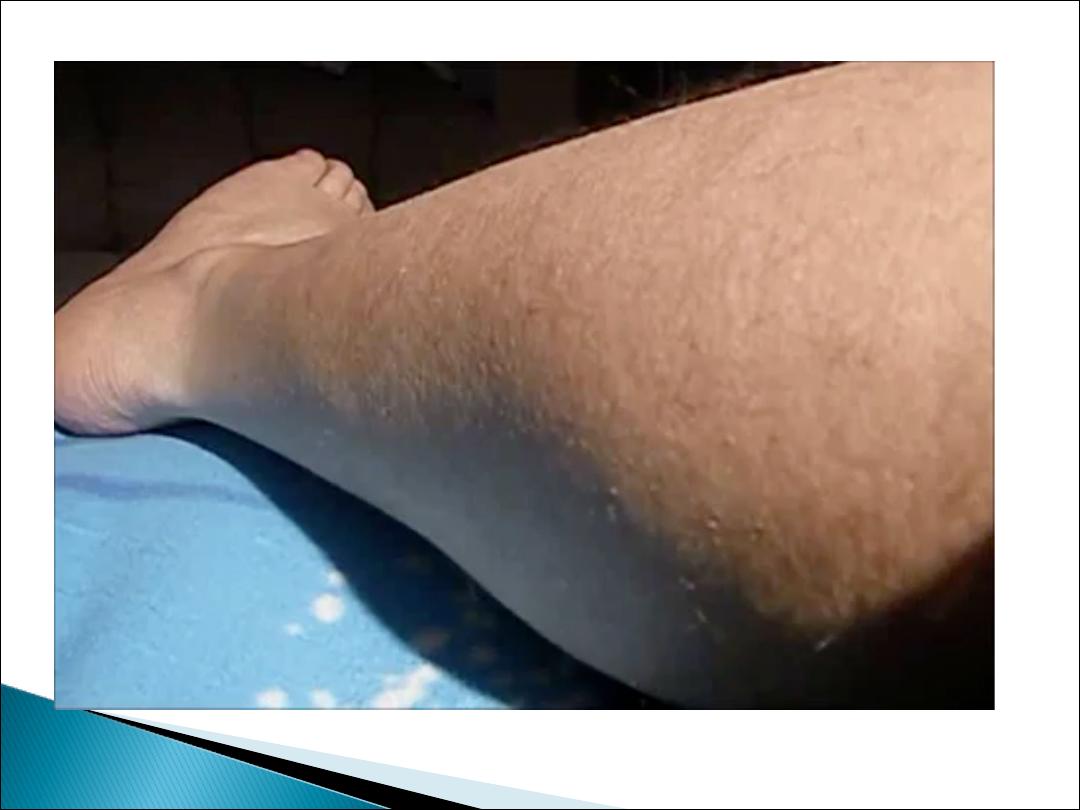
Have bilateral calf muscle enlargement (
pseudo
hypertrophy)

Myotonic dystrophy
}
Autosomal dominant inheritance.
}
Onset at any age, usually around 20 years
}
Affected patients have a typical "hatchet-faced"
appearance due to temporalis, masseter, and facial
muscle atrophy and weakness.
}
Frontal baldness is also characteristic of the
disease.
}
Bilateral ptosis.

}
Distal limb weakness.
}
Myotonia is demonstrable by percussion of the
thenar eminence, the tongue, and wrist extensor
muscles. Myotonia causes a slow relaxation of
hand grip after a forced voluntary closure.
}
Lens opacities.
}
cardiac conduction abnormalities
}
Hypogonadism.