
1
r
NEUROBLASTOMA:
Objective: To understand the clinical presentations , diagnosis , epidemiology ,
pathology , prognosis and treatment.
Neuroblastomas are embryonal cancers of the peripheral sympathetic nervous
system with heterogeneous clinical presentation and course, ranging from tumors
that undergo spontaneous regression to very aggressive tumors unresponsive to
very intensive multimodal therapy.
The causes of most cases remain unknown, and the outcomes for aggressive forms
of neuroblastoma remain poor.
EPIDEMIOLOGY:
Neuroblastoma is the most common extracranial solid tumor in children and the
most commonly diagnosed malignancy in infants.
Neuroblastoma accounts for >15% of the mortality from cancer in children.
The median age of children at diagnosis of neuroblastoma is 22 mo, and 90% of
cases are diagnosed by 5 yr of age.
The incidence is slightly higher in boys and in whites.
PATHOLOGY:
Neuroblastoma tumors, which are derived from neural crest cells, form a
spectrum with variable degrees of neural differentiation,ranging from tumors with
primarily undifferentiated small round cells (neuroblastoma) to tumors consisting
of mature and maturing Schwannian stroma with ganglion cells
(ganglioneuroblastoma or ganglioneuroma).
The tumors may resemble other small round blue cell tumors, such as
rhabdomyosarcoma, Ewing sarcoma, and non- Hodgkin lymphoma.
The prognosis of children with neuroblastoma varies with the histologic features of
the tumor, and prognostic factors include the presence and amount of Schwannian
stroma, the degree of tumor cell differentiation, and the mitosis-karyorrhexis index.
PATHOGENESIS:
The etiology of neuroblastoma in most cases remains unknown.
Familial neuroblastoma accounts for 1-2% of all cases, is associated with a
younger age at diagnosis, and is linked to mutations in the PHOX2Band ALK
genes. The BARD1 gene has also been identified as a major genetic contributor to
neuroblastoma risk.
Neuroblastoma is associated with other neural crest disorders, including
Hirschsprung disease, central hypoventilation syndrome, and neurofibromatosis
type I, and potentially congenital cardiovascular malformations.
Children with Beckwith-Wiedemann syndrome and hemihypertrophy also have a
higher incidence of neuroblastoma.
Increased incidence of neuroblastoma is associated with some maternal and
paternal occupational chemical exposures, farming, and work related to

2
electronics,although no single environmental exposure has been shown to directly
cause neuroblastoma. Amplification of MYCN is strongly associated with
advanced tumor stage and poor outcomes.
Hyperdiploidy confers better prognosis if the child is younger than 1 yr of age at
diagnosis.
CLINICAL MANIFESTATIONS:
The signs and symptoms of neuroblastoma reflect the tumor site and extent of
disease, and the symptoms of neuroblastoma can mimic many other disorders, a
fact that can result in a delayed diagnosis. Neuroblastoma may develop at any site
of sympathetic nervous system tissue. Approximately half of neuroblastoma
tumors arise in the adrenal glands, and most of the remainder originate in the
paraspinal sympathetic ganglia. Metastatic spread, which is more common in
children older than 1 yr of age at diagnosis, the most common sites of metastasis
are the regional or distant lymph nodes, long bones and skull, bone marrow, liver,
and skin. Lung and brain metastases are rare, occurring in >3% of cases.
Metastatic disease can cause a variety of signs and symptoms, including
fever,irritability, failure to thrive, bone pain, cytopenias, bluish subcutaneous
nodules, orbital proptosis, and periorbital ecchymoses.
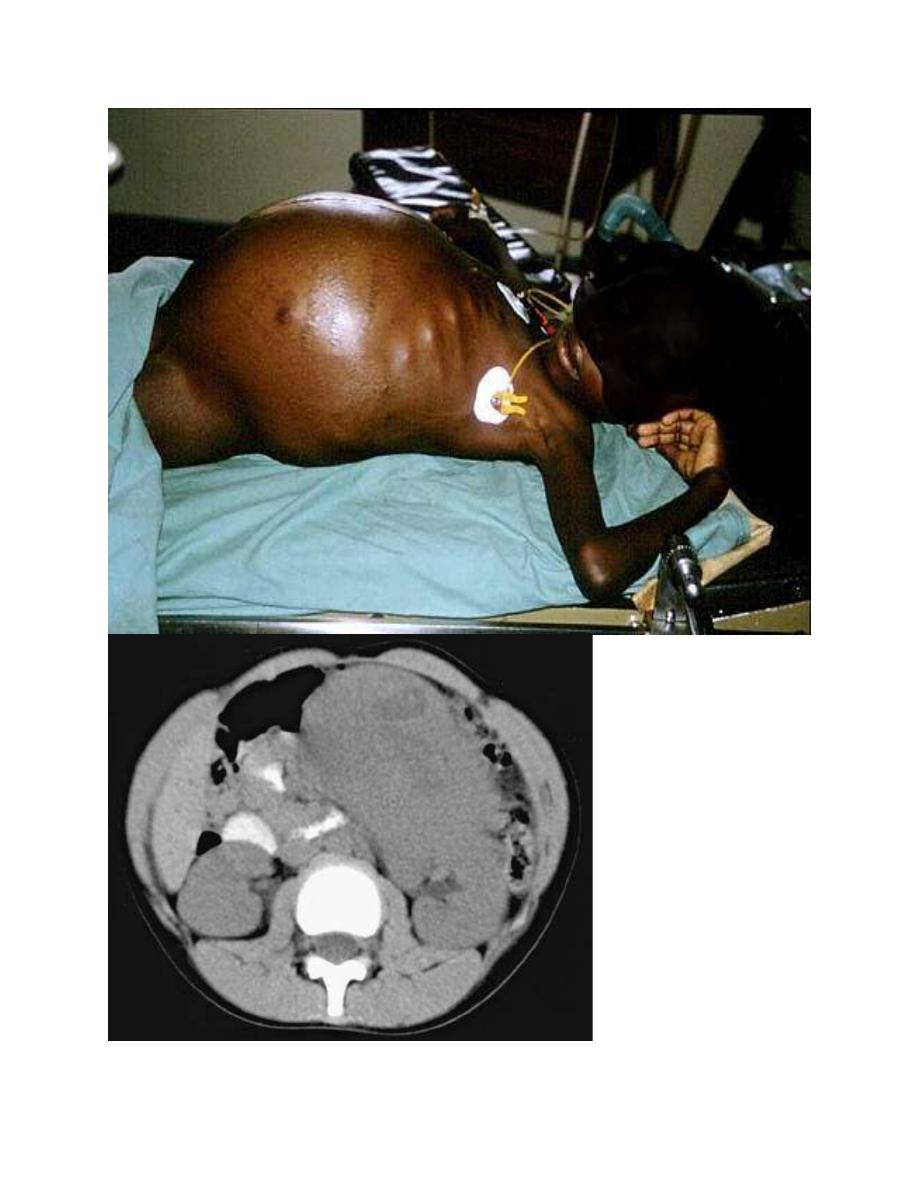
Localized disease can manifest as an asymptomatic mass or can cause Paraspinal
neuroblastoma tumors can invade the neural foramina, causing spinal cord and
nerve root compression.
Neuroblastoma can also be associated with a paraneoplastic syndrome of
autoimmune origin, termed opsoclonus–myoclonus–ataxia syndrome, in which
patients experience rapid, uncontrollable jerking eye and body movements, poor
coordination,and cognitive dysfunction. Some tumors produce catecholamines that
can cause increased sweating and hypertension, and some release vasoactive
intestinal peptide, causing a profound secretory diarrhea. Children with extensive
tumors can also experience tumor lysis syndrome and disseminated intravascular
coagulation.
Infants younger than 1 yr of age also can present in unique fashion, termed stage
4S, with widespread subcutaneous tumor nodules, massive liver involvement,
limited bone marrow disease, and a small primary tumor without bone involvement
or other metastases.
DIAGNOSIS:
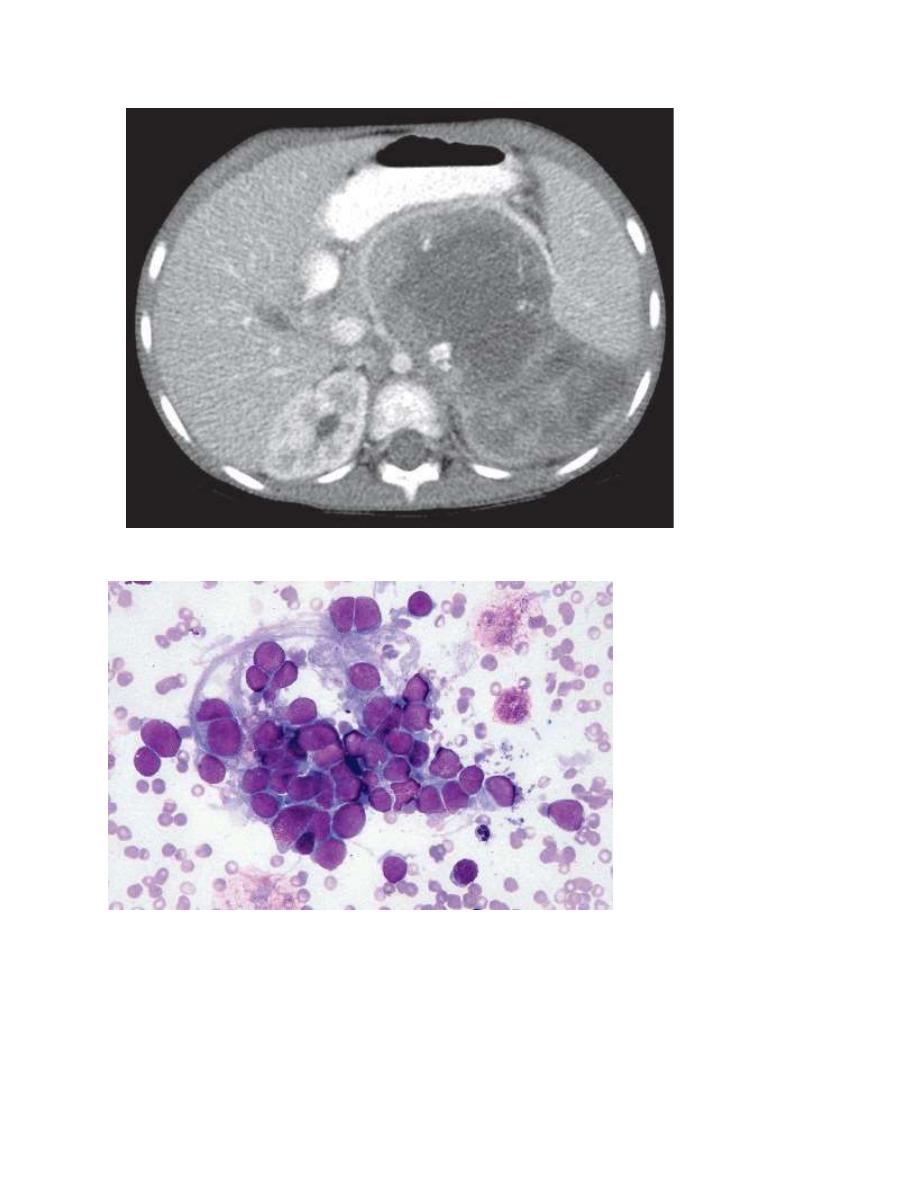
Neuroblastoma is usually discovered as a mass or multiple masses on plain
radiography, CT, or MRI . The mass often contains calcification and hemorrhage
that can be appreciated on plain radiography or CT.
Prenatal diagnosis of neuroblastoma on maternal ultrasound scans is sometimes
possible.

3
Tumor markers, including catecholamine metabolites homovanillic acid and
vanillylmandelic acid, are elevated in the urine of approximately 95% of cases and
help to confirm the diagnosis.
A pathologic diagnosis is established from tumor tissue obtained by biopsy.
Neuroblastoma can be diagnosed without a primary tumor biopsy if small round
blue tumor cells are observed in bone marrow samples and the levels of
vanillylmandelic acid or homovanillic acid are elevated in the urine.
Evaluations for metastatic disease should include CT or MRI of the chest and
abdomen, bone scans to detect cortical bone involvement,and at least 2
independent bone marrow aspirations and biopsies to evaluate for marrow disease.
MRI of the spine should be performed in cases with suspected spinal cord
compression, but imaging of the brain with either CT or MRI is not routinely
performed unless indictated by the clinical presentation.
ed with Neuroblastoma
.
.
Periorbital metastases of neuroblastoma with ecchymoses and proptosis.
4
T
able 498-3
International
Neuroblastoma
Staging System

5
The International Neuroblastoma Staging System (INSS) is currently used to
stage patients with neuroblastoma after initial surgical resection .
Stage 1 tumors are confined to the organ or structure of origin and are completely
resected.
Stage 2 tumors extend beyond the structure of origin but not across the midline,
either with (stage 2B) or without (stage 2A) ipsilateral lymph node involvement.
Stage 3 tumors extend beyond the midline, with or without bilateral lymph node
involvement.
Stage 4 tumors are disseminated, with metastases to bones, bone marrow, liver,
distant lymph nodes, and other organs.
Stage 4S refers to neuroblastoma in children younger than 1 yr of age with
dissemination to liver, skin, and/or bone marrow without bone involvement and
with a primary tumor that would otherwise be staged as stage 1 or 2.
TREATMENT:
The patient’s age and tumor stage are combined with cytogenetic and molecular
features of the tumor to determine the treatment risk group and estimated prognosis
for each patient .
The usual treatment for children with low-risk neuroblastoma is surgery for stages
1and 2 and observation for stage 4S with cure rates generally >90% without further
therapy.
Children with spinal cord compression at diagnosis also may require urgent
treatment with chemotherapy, surgery, or radiation to avoid neurologic damage.
Stage 4S neuroblastomas have a very favorable prognosis, and many regress
spontaneously without therapy. Chemotherapy or resection of the primary tumor
does not improve survival rates, but for infants with massive liver involvement and
respiratory compromise, small doses of cyclophosphamide or low-dose hepatic
irradiation may alleviate symptoms. For children with stage 4S neuroblastoma who
require treatment for symptoms, the survival rate is 81%.
Treatment of intermediate-risk neuroblastoma includes surgery ,chemotherapy,
and, in some cases, radiation therapy. The chemotherapy usually includes
moderate doses of cisplatin or carboplatin, cyclophosphamide,etoposide, and
doxorubicin given for several months. Radiation therapy is used for tumors with
incomplete response to chemotherapy. Children with intermediate-risk
neuroblastoma,including children with stage 3 disease and infants with stage 4
disease and favorable characteristics, have an excellent prognosis and >90%
survival with this moderate treatment.
Children with high-risk neuroblastoma have long-term survival rates between 25%
and 35% with current treatment that consists of intensive chemotherapy, high-dose
chemotherapy with autologous stem cell rescue, surgery, radiation, and 13-cis-
retinoic acid . Induction chemotherapy for children with high-risk neuroblastoma

6
includes combinations of cyclophosphamide, topotecan, doxorubicin, vincristine,
cisplatin, and etoposide.
After completion of induction chemotherapy, resection of the residual primary
tumor is followed by high-dose chemotherapy with autologous stem cell rescue
and focal radiation therapy to tumor sites.
Cases of high-risk neuroblastoma are associated with frequent relapses, and
children with recurrent neuroblastoma have a <50% response rate to alternative
chemotherapy regimens.
Wilms Tumor:
Wilms tumor (WT), also known as nephroblastoma, is the most common primary
malignant renal tumor of childhood.
It is the second most common malignant abdominal tumor in childhood.
The most common sites of metastases are the lungs, regional lymph nodes, and
liver.
Histologically, the classic WT is made up of varying proportions of blastemal,
stromal, and epithelial cells, recapitulating stages of normal renal development.
The treatment includes surgery and chemotherapy with or without radiotherapy.
The use of the current, multimodality treatment has dramatically improved the cure
rate of WT from <30% to approximately 90% .
EPIDEMIOLOGY:
WT accounts for 6% of pediatric malignancies and more than 95% of kidney
tumors in children. Approximately 75% of the cases occur in children younger
than 5 yr with a peak incidence at 2-3 yr of age. It can arise in 1 or both kidneys;
the incidence of bilateral WTs is 7%. Most cases are sporadic, but approximately
2% of patients have a family history. In 8-10% of patients, WT is observed in the
context of hemihypertrophy, aniridia, genitourinary anomalies, and a variety of
rare syndromes, including Beckwith- Wiedemann syndrome and Denys-Drash
syndrome .An earlier age of diagnosis and an increased incidence of bilateral
disease are generally observed in syndromic and familial cases.
ETIOLOGY: GENETICS AND MOLECULAR BIOLOGY:
WT is thought to be derived from incompletely differentiated renal mesenchyme,
and tumors are typically composed of the undifferentiated and partially
differentiated cells that normally arise from renal mesenchyme.
Foci of benign, undifferentiated mesenchyme (nephrogenic rests) that persist
abnormally in the kidney into postnatal life are observed in approximately 1% of
children in the general population, but are present in up to 90% of children who
have a family history of WT, develop bilateral tumors, or display features of WT-
related syndromes.

7
Nephrogenic rests usually regress or differentiate ,but those that persist can
become malignant.
Genetic mutations have been detected in a third of WTs ,located on chromosome
11 .
Germline mutations are usually associated with WT in the context of genitourinary
anomalies or the WAGR (Wilms, aniridia, genitourinary anomalies, mental
retardation) syndrome.
WT is occasionally observed in families with a predisposition to
pleuropulmonary blastoma, with mutations of the gene, located at 14q31.are
observed in these families. A family history of WT is noted in approximately 2%
of WT patients, and predisposition is inherited as an autosomal dominant trait with
incomplete penetrance.
CLINICAL PRESENTATION:
The most common initial clinical presentation for WT is the incidental discovery
of an asymptomatic abdominal mass by parents while bathing or clothing an
affected child or by a physician during a routine physical examination.
Functional defects in paired organs like the kidney, with good functional reserve,
are also unlikely to be detected early. Hypertension is present in approximately
25% of tumors at presentation and has been attributed to increased renin activity.
Abdominal pain, gross painless hematuria, and fever are other frequent findings
at diagnosis. Occasionally, rapid abdominal enlargement and anemia occur as a
result of bleeding into the renal parenchyma or pelvis. WT thrombus extends into
the inferior vena cava in 4-10% of patients, and rarely into the right atrium.
Patients might also have microcytic anemia from iron deficiency or anemia of
chronic disease, polycythemia, elevated platelet count, and acquired deficiency of
von Willebrand factor or factor VII deficiency.
4 Staging of mor
Stage I Tumor confined to the kidney and completely resected. Renal capsule or
sinus vessels not involved. Tumor not ruptured or biopsied. Regional lymph nodes
examined and negative.
Stage II Tumor extends beyond the kidney but is completely resected with
negative margins and lymph nodes. At least 1 of the following has occurred: (a)
penetration of renal capsule, (b) invasion of renal sinus vessels.
Stage III Residual tumor present following surgery confined to the abdomen,
including gross or microscopic tumor; spillage of tumor preoperatively or
intraoperatively; biopsy prior to nephrectomy, regional lymph node metastases;
tumor implants on the peritoneal surface; extension of tumor thrombus into the
inferior vena cava including thoracic vena cava and heart.
Stage IV Hematogenous metastases (lung, liver, bone, brain, etc.) or lymph node
metastases outside the abdominopelvic region.
Stage V Bilateral renal involvement by tumor.
8

9
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS:
An abdominal mass in a child should be considered malignant until diagnostic
imaging, laboratory findings, and pathology can define its true nature.
Imaging studies include plain abdominal radiography, abdominal ultrasonography,
and CT of the abdomen to define the intrarenal origin of the mass and differentiate
it from adrenal masses (e.g., neuroblastoma) and other masses in the abdomen.
Abdominal ultrasonography helps differentiate solid from cystic masses. WT
might show focal areas of necrosis or hemorrhage and hydronephrosis caused by
obstruction of the renal pelvis by the tumor. Ultrasonography with Doppler
imaging of renal veins and the inferior vena cava is a useful first study that not
only can look for WT but also can evaluate the collecting system and demonstrate
tumor thrombi in the renal veins and inferior vena cava.
CT is useful to define the extent of the disease, integrity of the contralateral kidney,
and metastasis.
MRI requires sedation in young children and is not routinely used; it may be
helpful in defining an extensive tumor thrombus that extends up to the level of the
hepatic veins or even into the right atrium, and to distinguish WT from
nephrogenic rests.
Chest CT is more sensitive than chest radiography to screen for pulmonary
metastasis, and is preferably performed before surgery because effusions and
atelectasis can confound the interpretation of postoperative imaging studies.
A bone scan is performed if the histologic diagnosis confirms clear cell sarcoma
of the kidney to look for bone metastasis.
Regional spread and metastatic lesions can be visualized on positron emission
tomography( PET )/CT scanning.
The diagnosis is usually made by imaging studies and confirmed by histology at
the time of nephrectomy.
Although biopsy is a reliable diagnostic tool, it is discouraged as it results in
disease upstaging.
A core needle biopsy obtained via a posterior approach should be performed in
cases of unusual presentation (older age, signs of infection, inflammation) or
unusual imaging findings (significant adenopathy, no renal parenchyma seen,
intratumoral calcification).

10
TREATMENT:
There are 2 major schools of thought in the management of WT, the first
recommends surgery prior to initiating treatment and the second recommends
preoperative chemotherapy. Each approach has advantages and limitations but they
have similar outcomes. Early surgery provides accurate diagnosis and staging, and
can facilitate risk-adapted therapy. Preoperative chemotherapy can make surgery
easier and reduces the risk of intraoperative tumor rupture and hemorrhage.
Surgery entails a radical nephrectomy with meticulous dissection to avoid rupture
of the tumor capsule and lymph node sampling despite the absence of abnormal
nodes on preoperative imaging studies or intraoperative assessment.
Partial nephrectomy is performed in patients with bilateral disease or with
unilateral WT and a predisposing syndrome such as Denys-Drash and WAGR, so
as to minimize the risk of future renal failure.
Prognostic factors for risk-adapted therapy include age, stage, tumor weight, and
loss of heterozygosity at chromosomes 1p and 16q. Histology plays a major role in
risk stratification of WT. Absence of anaplasia is considered a favorable histologic
finding but presence of anaplasia is further classified as focal or diffuse, both of
which are unfavorable histologic findings.
Patients with favorable histologic findings of WT have a good outcome and are
generally treated in the outpatient setting.
Nephrectomy alone may be sufficient for patients younger than 2 yr of age with
stage I disease and a tumor weighing <550 g.
Patients with stages I and II disease receive chemotherapy with 2 drugs,
vincristine and actinomycin D ( dactinomycin), every 1-3 wk for a total of 18 wk .
Patients with stage III or IV disease receive chemotherapy with 3 drugs
(vincristine, doxorubicin, and actinomycin D) every 1-3 wk for a total of 24 wk
and radiation therapy.
Patients with regional lymph node metastases, residual disease after surgery, or
tumor rupture receive radiation therapy to the flank or abdomen, and those with
lung metastases receive radiation therapy to the lungs.
Anaplastic histology (focal and diffuse) accounts for approximately 11% of WT
cases.
Patients with diffuse anaplasia, in particular, have a poor outcome & treated with
intensive chemotherapy regimens that include vincristine, cyclophosphamide,
doxorubicin, etoposide, carboplatin, and ifosfamide, in addition to radiation
therapy.

11
RECURRENT DISEASE:
Approximately 15% of WT patients with favorable histology and 50% of those
with anaplastic histology suffer relapse; most relapses occur early (within 2 yr of
diagnosis).
Factors associated with a favorable outcome after relapse include low stage (I/II)
at diagnosis, treatment with vincristine and actinomycin D only, no prior
radiotherapy, favorable histology, relapse to lung only, and interval from
nephrectomy to relapse 12 mo or longer.
Patients with recurrent WT who previously received only vincristine and
actinomycin D had a 4 yr survival of approximately 80%, whereas those who
previously received the 3 drug regimen of vincristine, actinomycin D, and
doxorubicin had a 4 yr survival of only 50%.
Other agents used to treat recurrent WT include doxorubicin, carboplatin,
cyclophosphamide, ifosfamide, etoposide, and topotecan.
Metachronous WT may not represent tumor relapse but instead may indicate
development of a new tumor in the opposite kidney.
PROGNOSIS:
Despite some adverse risk factors that decrease prognosis (metastases, unfavorable
histology, recurrent disease, and loss of heterozygosity of both 1p and 16q), most
children with WT have a very favorable prognosis.
Overall, the survival of children with WT approaches 90%, with some prognostic
factors (low stage, favorable histology, young age, low tumor weight) conferring
even better outcomes.
LATE EFFECTS:
Late complications are a consequence of treatment type and intensity; the use of
radiotherapy and anthracyclines increases the risk of these complications.
Clinically significant late sequelae include musculoskeletal effects, cardiac
toxicity, pulmonary disease, reproductive problems
,
renal dysfunction, and the
development of second malignant neoplasms.