
1
Organophosphorus insecticides and nerve agents
Organophosphorus compounds are widely used as pesticides,
especially in developing countries. Case fatality following deliberate
ingestion is high (5–20%). Nerve agents, developed for chemical
warfare, are derived from OP insecticides and are much more toxic.
They are commonly classified as G (originally synthesised in Germany)
or V (‘venomous’) agents. The ‘G’ agents, such as tabun, sarin and
soman, are volatile, absorbed by inhalation or via the skin, and
dissipate rapidly after use. ‘V’ agents, such as VX, are contact poisons
unless aerosolised, and contaminate ground for weeks or months. The
toxicology and management of nerve agent and pesticide poisoning are
similar.
Mechanism of toxicity
OP compounds inactivate acetylcholinesterase (AChE), resulting in the
accumulation of acetylcholine (ACh) in cholinergic synapses. Initially,
spontaneous hydrolysis of the OP–enzyme complex allows reactivation
of the enzyme but, subsequently, loss of a chemical group from the OP–
enzyme complex prevents further enzyme reactivation. After this
process (termed ‘ageing’) has taken place, new enzyme needs to be
synthesised before function can be restored. The rate of ‘ageing’ is an
important determinant of toxicity and especially rapid after exposure to
nerve agents (soman in particular), which cause ‘ageing’ within
minutes.
Clinical features and management
OP poisoning causes an acute cholinergic phase, which may
occasionally be followed by the intermediate syndrome or
2
organophosphate-induced delayed polyneuropathy (OPIDN). The onset,
severity and duration of poisoning depend on the route of exposure
and agent involved.
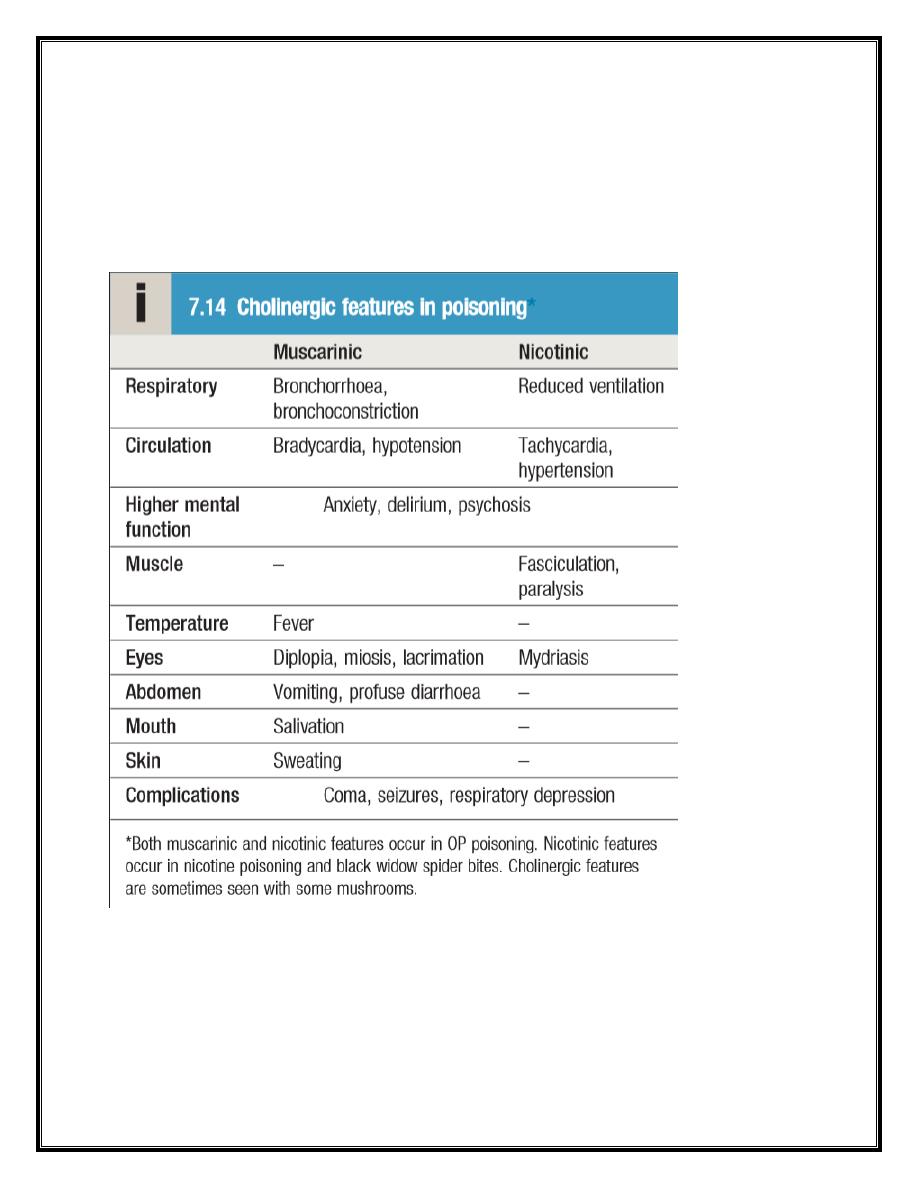
Acute cholinergic syndrome
This usually starts within a few minutes of exposure and nicotinic or
muscarinic features may be present. Vomiting and profuse diarrhoea
are typical following ingestion. Bronchoconstriction, bronchorrhoea and
salivation may cause severe respiratory compromise. Excess sweating

3
and miosis are characteristic and the presence of muscular
fasciculations strongly suggests the diagnosis, although this feature is
often absent, even in serious poisoning. Subsequently, generalised
flaccid paralysis may develop and affect respiratory and ocular muscles,
resulting in respiratory failure. Ataxia, coma, convulsions, cardiac
repolarisation abnormalities and torsades de pointes may occur.
Management
The airway should be cleared of excessive secretions, breathing and
circulation assessed, high-flow oxygen administered and intravenous
access obtained. Appropriate external decontamination is needed.
Gastric lavage or activated charcoal may be considered if the patient
presents sufficiently early. Seizures should be treated. The ECG, oxygen
saturation, blood gases, temperature, urea and electrolytes, amylase
and glucose should be monitored closely. Early use of sufficient doses
of atropine is potentially life-saving in patients with severe toxicity.
Atropine reverses ACh-induced bronchospasm, bronchorrhoea,
bradycardia and hypotension. When the diagnosis is uncertain, a
marked increase in heart rate associated with skin flushing after a 1 mg
intravenous dose makes OP poisoning unlikely. In OP poisoning,
atropine (2 mg IV) should be administered and this dose should be
doubled every 5–10 minutes until clinical improvement occurs. Further
bolus doses should be given until secretions are controlled, the skin is
dry, blood pressure is adequate and heart rate is > 80 bpm. Large doses
may be needed, but excessive doses may cause anticholinergic effects.
In severe poisoning requiring atropine, an oxime such as pralidoxime
chloride or obidoxime is generally recommended, if available, although
efficacy is debated. This may reverse or prevent muscle weakness,

4
convulsions or coma, especially if given rapidly after exposure. Oximes
reactivate AChE that has not undergone ‘ageing’ and are therefore less
effective with dimethyl compounds and nerve agents, especially soman.
Oximes may provoke hypotension, especially if administered rapidly.
Intravenous magnesium sulphate has been reported to increase
survival in animals and in small human studies of OP poisoning;
however, further clinical trial evidence is needed before this can be
recommended routinely. Ventilatory support should be instituted
before the patient develops respiratory failure. Benzodiazepines may
be used to treat agitation, fasciculations and seizures and for sedation
during mechanical ventilation. Exposure is confirmed by measurement
of plasma or red blood cell cholinesterase activity but antidote use
should not be delayed pending results. Plasma cholinesterase is
reduced more rapidly but is less specific than red cell cholinesterase.
Values correlate poorly with the severity of clinical features but are
usually < 10% in severe poisoning, 20–50% in moderate poisoning and >
50% in subclinical poisoning. The acute cholinergic phase usually lasts
48–72 hours, with most patients requiring intensive cardiorespiratory
support and monitoring. Cholinergic features may be prolonged over
several weeks with some lipid-soluble agents.
Intermediate syndrome
About 20% of patients with OP poisoning develop weakness that
spreads rapidly from the ocular muscles to those of the head and neck,
proximal limbs and the muscles of respiration, resulting in ventilatory
failure. This ‘intermediate syndrome’ generally develops 1–4 days after
exposure, often after resolution of the acute cholinergic syndrome, and
may last 2–3 weeks. There is no specific treatment and supportive care
is needed, including maintenance of airway and ventilation.

5
Organophosphate-induced delayed polyneuropathy
Organophosphate-induced delayed polyneuropathy (OPIDN) is a rare
complication that usually occurs 2–3 weeks after acute exposure. It is a
mixed sensory/motor polyneuropathy, affecting long myelinated
neurons especially, and appears to result from inhibition of enzymes
other than AChE. Early clinical features are muscle cramps followed by
numbness and paraesthesiae, proceeding to flaccid paralysis of the
lower and subsequently the upper limbs, with foot and wrist drop and a
high-stepping gait, progressing to paraplegia. Sensory loss may also be
present but is variable. Initially, tendon reflexes are reduced or lost but
mild spasticity may develop later. There is no specific therapy for
OPIDN. Regular physiotherapy may limit deformity caused by muscle-
wasting. Recovery is often incomplete and may be limited to the hands
and feet, although substantial functional recovery after 1–2 years may
occur, especially in younger patients.
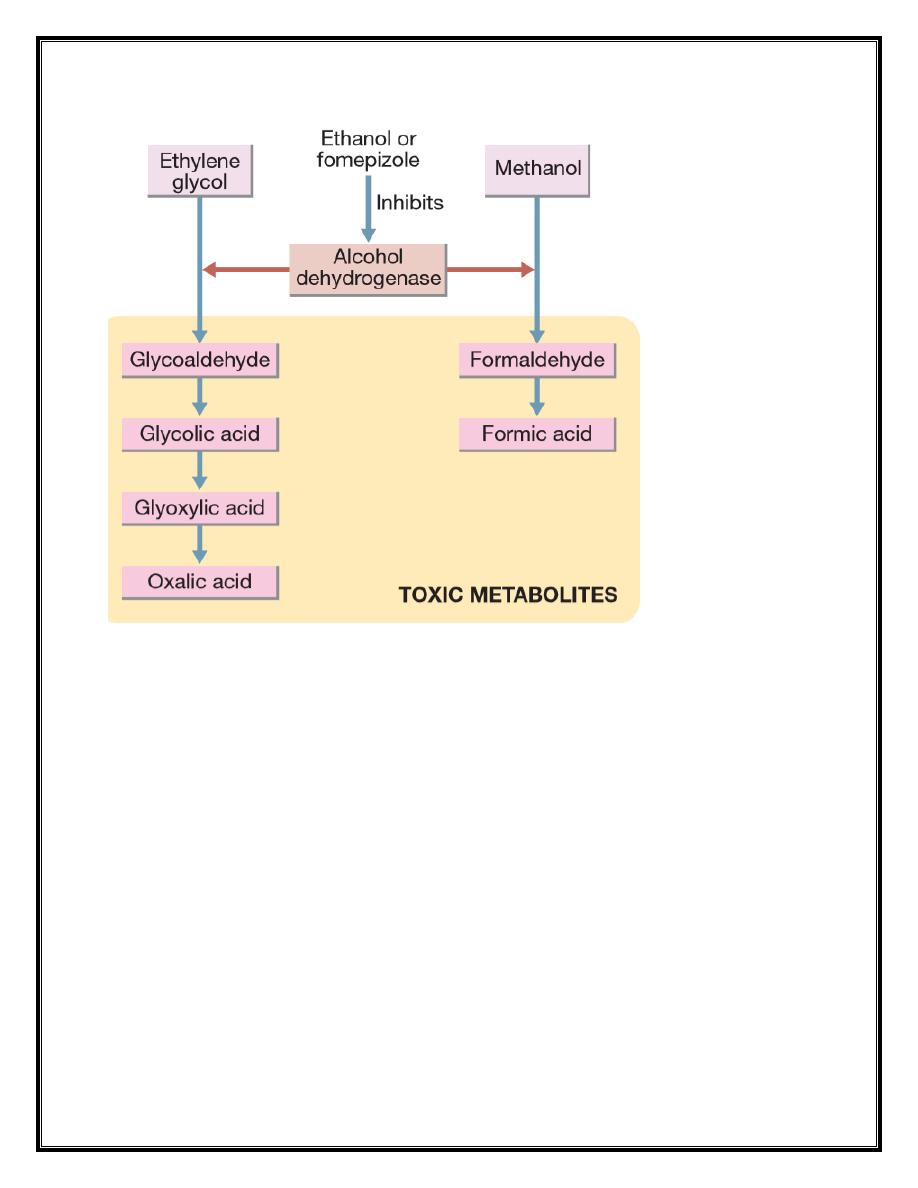
Methanol and ethylene glycol
Ethylene glycol is found in antifreeze, brake fluids and, in lower
concentrations, windscreen washes. Methanol is present in some
antifreeze products and commercially available industrial solvents, and
in low concentrations in some screen washes and methylated spirits. It
may also be an adulterant of illicitly produced alcohol. Both are rapidly
absorbed after ingestion. Methanol and ethylene glycol are not of high
intrinsic toxicity but are converted via alcohol dehydrogenase to toxic
metabolites that are largely responsible for their clinical effects.
6
Clinical features
Early features of poisoning with either methanol or ethylene glycol
include vomiting, ataxia, drowsiness, dysarthria and nystagmus. As toxic
metabolites are formed, metabolic acidosis, tachypnoea, coma and
seizures may develop. Toxic effects of ethylene glycol include
ophthalmoplegia, cranial nerve palsies, hyporeflexia and myoclonus.
Renal pain and acute tubular necrosis occur because of renal calcium
oxalate precipitation. Hypocalcaemia, hypomagnesaemia and
hyperkalaemia are common. Methanol poisoning causes headache,
delirium and vertigo. Visual impairment and photophobia develop,
associated with optic disc and retinal oedema and impaired pupil
reflexes. Blindness may be permanent, although some recovery may

7
occur over several months. Pancreatitis and abnormal liver function
have also been reported.
Management
Urea and electrolytes, chloride, bicarbonate, glucose, calcium,
magnesium, albumin, plasma osmolarity and arterial blood gases
should be measured in all patients with suspected methanol or
ethylene glycol toxicity. The osmolar and anion gaps should be
calculated. Initially, poisoning is associated with an increased osmolar
gap, but as toxic metabolites are produced, an increased anion gap
develops, associated with metabolic acidosis. The diagnosis can be
confirmed by measurement of ethylene glycol or methanol
concentrations but assays are not widely available. An antidote, ideally
fomepizole but otherwise ethanol, should be administered to all
patients with suspected significant exposure while awaiting the results
of laboratory investigations. These block alcohol dehydrogenase and
delay the formation of toxic metabolites until the parent drug is
eliminated in the urine or by dialysis. The antidote should be continued
until ethylene glycol or methanol concentrations are undetectable.
Metabolic acidosis should be corrected with sodium bicarbonate (e.g.
250 mL of 1.26% solution, repeated as necessary). Convulsions should
be treated with an intravenous benzodiazepine. In ethylene glycol
poisoning, hypocalcaemia should be corrected only if there are severe
ECG features or if seizures occur, as this may increase calcium oxalate
crystal formation. In methanol poisoning, folinic acid should be
administered to enhance the metabolism of the toxic metabolite,
formic acid. Haemodialysis or haemodiafiltration should be used in
severe poisoning, especially if renal failure is present or there is visual
loss in the context of methanol poisoning. It should be continued until

8
acute toxic features are no longer present and ethylene
glycol/methanol concentrations are undetectable.