
1
THI QAR U. MEDICAL COLLEGE LECTURES 2017
DEPARTMENT OF INTERNAL MEDICINE Dr. FAEZ KHALAF, SUBSPACIALITY GIT
Primary biliary cirrhosis
Primary biliary cirrhosis (PBC) is a
chronic, progressive cholestatic liver
disease that predominantly
affects middle-aged women.
It is strongly associated with the presence of antimitochondrial antibodies
(AMA),
which are
diagnostic,
And is characterised by a granulomatous inflammation of the portal tracts, leading to progressive
damage and eventually loss of the small and middle-sized bile ducts.
This in turn leads to fibrosis and cirrhosis of the liver.
The condition typically presents with an insidious onset of itching and/or tiredness;
It may also be found incidentally as the result of routine blood tests.
Epidemiology
The prevalence of PBC varies across the world.
It is
relatively common in northern Europe
and North America but is rare in Africa and Asia.
There is a
strong female to- male predominance of 9 :1
it is also
more common amongst cigarette smokers
.
Clustering of cases has been reported, suggesting an environmental trigger in susceptible individuals.
Pathophysiology
Immune mechanisms are clearly involved. The condition is closely associated with other autoimmune no
hepatic diseases, such as
thyroid disease
, and there is a genetic association with
HLA-DR8
AMA are directed at pyruvate dehydrogenase complex
, a mitochondrial enzyme complex that plays a key role
in cellular energy generation.
PBC-specific ANA
(such as those directed at the nuclear pore antigen gp210) have a characteristic staining
pattern in immunofluorescence assays (selectively binding to the nuclear rim or nuclear dots), which means
that they should not be mistaken for the homogenously staining ANA seen in AIH.
Elevations in serum immunoglobulin levels are frequent but, unlike in AIH,
elevation is typically of IgM
.
AMA , IgM, ANA
Pathologically
chronic granulomatous inflammation
destroys the interlobular bile ducts
progressive lymphocyte-mediated inflammatory damage
Causes fibrosis, which spreads from the portal tracts to the liver parenchyma and
eventually leads to
cirrhosis
.
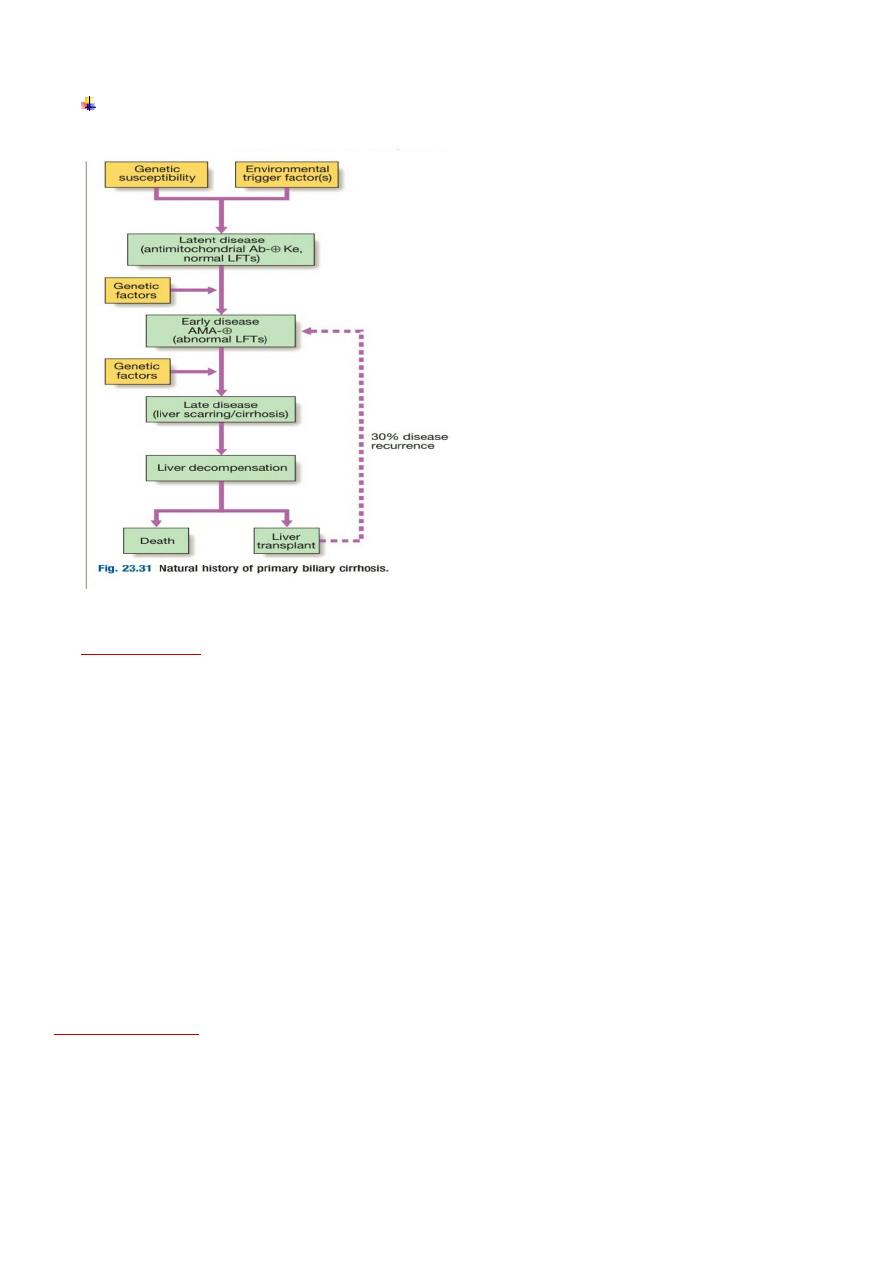
2
Natural history of primary biliary cirrhosis.
Clinical features
1. Systemic symptoms such as fatigue are common and may precede diagnosis for years.
2. Pruritus, which can be a feature of any cholestatic disease, is a common presenting complaint and
may precede jaundice by months or years.
3. Jaundice is rarely a presenting feature.
4. The itching is usually worse on the limbs.
5. Although there may be right upper abdominal discomfort, fever and rigors do not occur.
6. Bone pain or fractures can rarely result from Osteomalacia (fat-soluble vitamin malabsorption) or,
more commonly, from osteoporosis (hepatic osteodystrophy).
7. Initially, patients are well nourished but weight loss can occur as the disease progresses.
8. Scratch marks may be found in patients with severe pruritus.
9. Jaundice is only prominent late in the disease and can become intense.
10. Xanthomatous deposits occur in a minority, especially around the eyes.
11. Mild hepatomegaly is common and splenomegaly becomes increasingly common as portal
hypertension develops.
12. Liver failure may supervene
Associated diseases
1. Sicca syndrome
2. systemic sclerosis
3. coeliac disease
4. thyroid diseases.
Hypothyroidism should always be considered in patients with fatigue.

3
Diagnosis and investigations
The LFTs show a pattern of cholestasis
(Hypercholesterolemia is common and worsens as disease progresses; however, it is of no diagnostic value.
AMA is present in over 95% of patients
, and when it is absent the diagnosis should not be made without
obtaining histological evidence and considering cholangiography
(MRCP or ERCP) to exclude other biliary
disease
.
ANA and anti-smooth muscle antibodies are present in around 15% of patients
Ultrasound examination shows no sign of biliary obstruction.
Liver biopsy is only necessary if there is diagnostic uncertainty. The histological features of
PBC correlate poorly with the clinical features;
portal hypertension can develop before the histological onset of cirrhosis.
Management
The hydrophilic bile acid, ursodeoxycholic acid (UDCA), at a dose of 13–15 mg/kg/day improves bile flow,
replaces toxic hydrophobic bile acids in the bile acid pool, and reduces apoptosis of the biliary epithelium.
Clinically, UDCA improves LFTs, may slow down histological progression and has few side -effects it is
therefore widely used in the treatment of PBC and should be regarded as the optimal first-line treatment.
A significant minority of patients either fail to normalize their LFTs with UDCA or show an inadequate
response, and such patients have an increased risk of developing end-stage liver disease compared to
patients showing a full response.
There is currently no consensus as to how to treat such patients. Immunosuppressants, such as
corticosteroids, azathioprine, penicillamine and ciclosporin, have all been trialled in PBC.
Liver transplantation should be considered once liver failure has developed and may be indicated in patients
with intractable pruritus.
Serum bilirubin remains the most reliable marker of declining liver function. Transplantation is associated
with an excellent 5-year survival of over 80%,
although the disease will recur in over one third of patients at 10 years.
Pruritus
This is the main symptom requiring treatment. The cause is unknown but up-regulation of opioid receptors
and increased levels of endogenous opioids may play a role. First-line treatment is with the anion-binding
resin
cholestyramine
,
which probably acts by binding potential pruritogens in the intestine and increasing
their excretion in the stool. A dose of 4–16 g/day orally is used. The powder is mixed in orange
juice and the main dose (8 g) taken before and after breakfast, when maximal duodenal bile acid
concentrations occur.
Cholestyramine may bind other drugs in the gut (most obviously, UDCA), and adequate spacing should be
used between drugs. Cholestyramine is sometimes ineffective, especially in complete biliary obstruction, and
can be difficult for some patients to tolerate.
Alternative treatments include
rifampicin
(150 mg/day, titrated up to a maximum of 600 mg/day as required
and contingent on there being no deterioration in LFTs),
naltrexone
(an opioid antagonist; 25 mg/day initially, increasing up to 300 mg/day),
plasmapheresis and a liver support device (e.g. a molecular adsorb recirculating system
(MARS)).
Fatigue
Fatigue affects about
one-third
of patients with PBC.
The cause is unknown but it may reflect intracerebral changes due to cholestasis.
Unfortunately, once
depression, hypothyroidism and coeliac disease have been
excluded, there is currently no specific treatment.
The impact on patients’ lives can be substantial.
4
Malabsorption
Prolonged cholestasis is associated with steatorrhea and malabsorption of fat-soluble
vitamins, which should be replaced as necessary. Coeliac disease should be excluded since
its incidence is increased in PBC.
Bone disease
Osteopenia and osteoporosis are common, and normal post-menopausal bone loss is
accelerated.
Baseline bone density should be measured and treatment
started with replacement
calcium and vitamin D3.
Bisphosphonates should be used if there is evidence of osteoporosis.
Osteomalacia is rare.
Overlap syndromes
AMA-negative PBC (‘autoimmune cholangitis’)
A few patients demonstrate the clinical, biochemical and histological features of PBC but do not have
detectable AMA in the serum.
Serum transaminases, serum Ig levels and titres of ANA tend to be higher than in AMA positive PBC.
However, the clinical course mirrors classical PBC and these patients should be considered to have a variant
of PBC
PBC/autoimmune hepatitis overlap
A few patients with AMA and cholestatic LFTs have elevated transaminases, high serum immunoglobulins
and interface hepatitis on liver histology; in such individuals a trial of corticosteroid therapy may be
beneficial.
Primary sclerosing cholangitis
(PSC) is a
cholestatic liver disease caused by diffuse inflammation and fibrosis;
it can involve the entire biliary tree and leads to the gradual obliteration of intrahepatic and extrahepatic bile ducts,
and ultimately biliary cirrhosis, portal hypertension and hepatic failure.
Although considered as an autoimmune disease, evidence for an autoimmune pathophysiology is weaker than is the
case for PBC and autoimmune hepatitis. The incidence is about
6.3/100 000
in Caucasians.
Cholangiocarcinoma develops in about 10–30% of patients during the course of the disease.
PSC is twice as common in young men.
Most patients present at age
25–40
years, although the condition
may be diagnosed at any age and is an
important cause of chronic liver disease in children
.
The generally accepted diagnostic criteria are:
1.
generalized beading and stenosis of the biliary system on cholangiography (Fig. 23.32)
2.
absence of choledocholithiasis (or history of bile duct surgery)
3.
exclusion of bile duct cancer, by prolonged follow-up.
The term ‘secondary sclerosing cholangitis’ is used to describe the typical bile duct changes described above
when a clear predisposing factor for duct fibrosis can be identified.
The causes of secondary sclerosing cholangitis
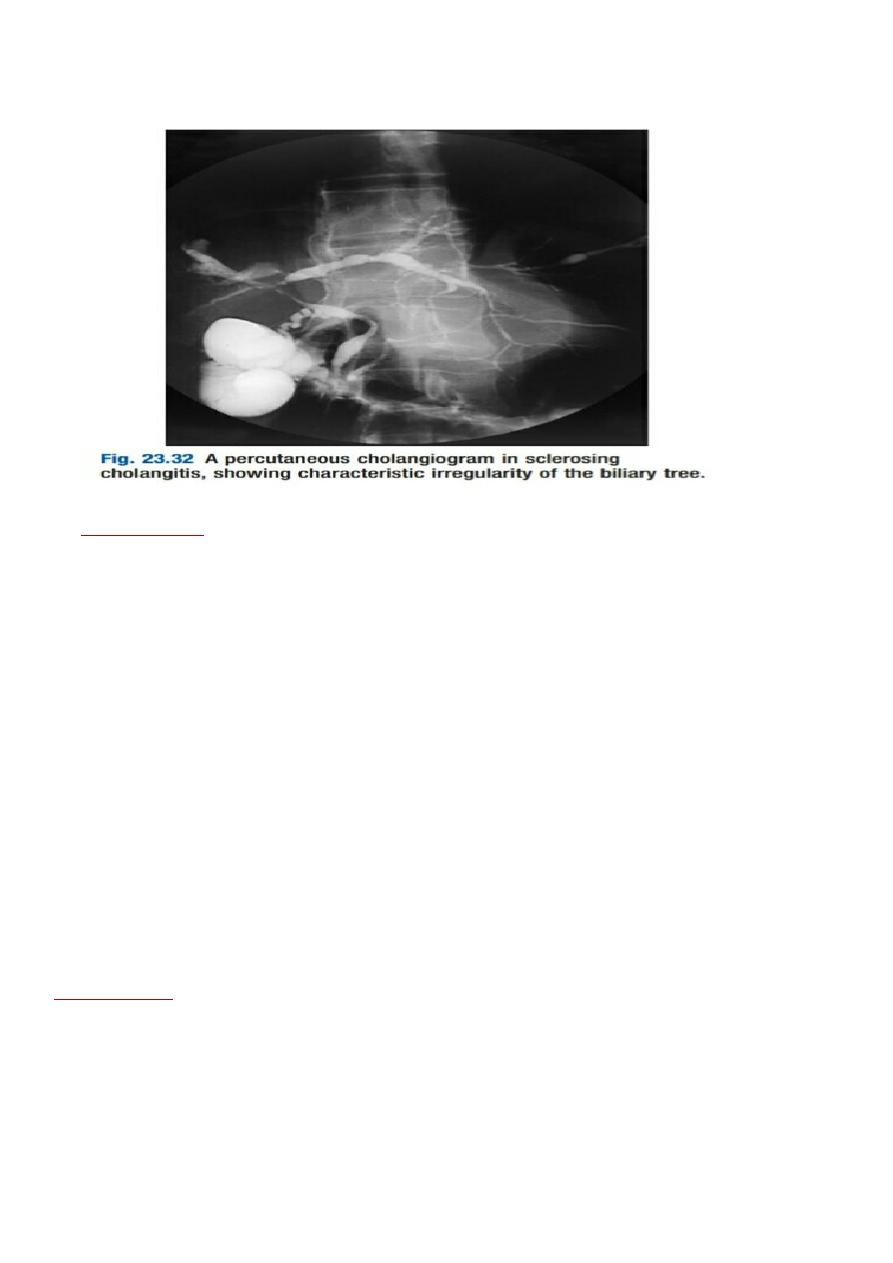
5
Pathophysiology
The cause of PSC is unknown but there is a close association with
IBD particularly ulcerative colitis
. About
two-thirds
of patients have coexisting ulcerative colitis, and
PSC is the most common form of chronic liver
disease in ulcerative colitis
.
Between
3% and 10% of patients
with ulcerative colitis develop PSC, particularly those with extensive colitis
or pancolitis.
The prevalence of primary sclerosing cholangitis is lower in patients with Crohn’s colitis (about 1%).
Patients with
PSC and ulcerative colitis are at greater risk of colorectal neoplasia than those with ulcerative
colitis alone
, and those who develop colorectal neoplasia are at greater risk of Cholangiocarcinoma.
It is currently believed that PSC is an immunologically mediated disease, triggered in genetically susceptible
individuals by toxic or infectious agents, which may gain access to the biliary tract through a leaky, diseased
colon.
A close link with HLA haplotype A1 B8
DR3 DRW52A has been identified.
Perinuclear antineutrophil cytoplasmic antibodies (ANCA)
have been detected in the
sera of
60–80% of patients with PSC with or without ulcerative colitis, and in 30–40% of patients with
ulcerative colitis alone.
The antibody is not specific for PSC and is found in other chronic liver diseases
(e.g. 50% of patients with
autoimmune hepatitis
Clinical features
The diagnosis is often made incidentally when persistently raised serum ALP is discovered in an individual
with ulcerative colitis. Common symptoms include
fatigue, intermittent jaundice, weight loss, right upper quadrant abdominal pain and pruritus.
Attacks of acute cholangitis are uncommon and usually follow biliary instrumentation.
Physical examination is abnormal in about 50% of symptomatic patients; the most
common findings are
jaundice and hepatomegaly/splenomegaly.
The condition may be associated with many other diseases

6
Investigations
Biochemical screening usually reveals
a cholestatic pattern of LFTs
but ALP and bilirubin levels may vary
widely in individual patients during the course of the disease.
For example, ALP and bilirubin values increase during acute cholangitis, decrease after therapy, and
sometimes fluctuate for no apparent reason.
Modest elevations in serum transaminases are usually seen, whereas hypoalbuminemia and clotting
abnormalities are found only at a late stage.
In addition to ANCA, low titres of serum ANA and anti-smooth muscle antibodies may be found in PSC but
have no diagnostic significance;
serum AMA is absent
.
The key investigation is now
MRCP,
which is usually diagnostic,
revealing multiple irregular stricturing and
dilatation .
ERCP should be reserved for patients in whom therapeutic intervention is likely to be necessary and should
follow MRCP.
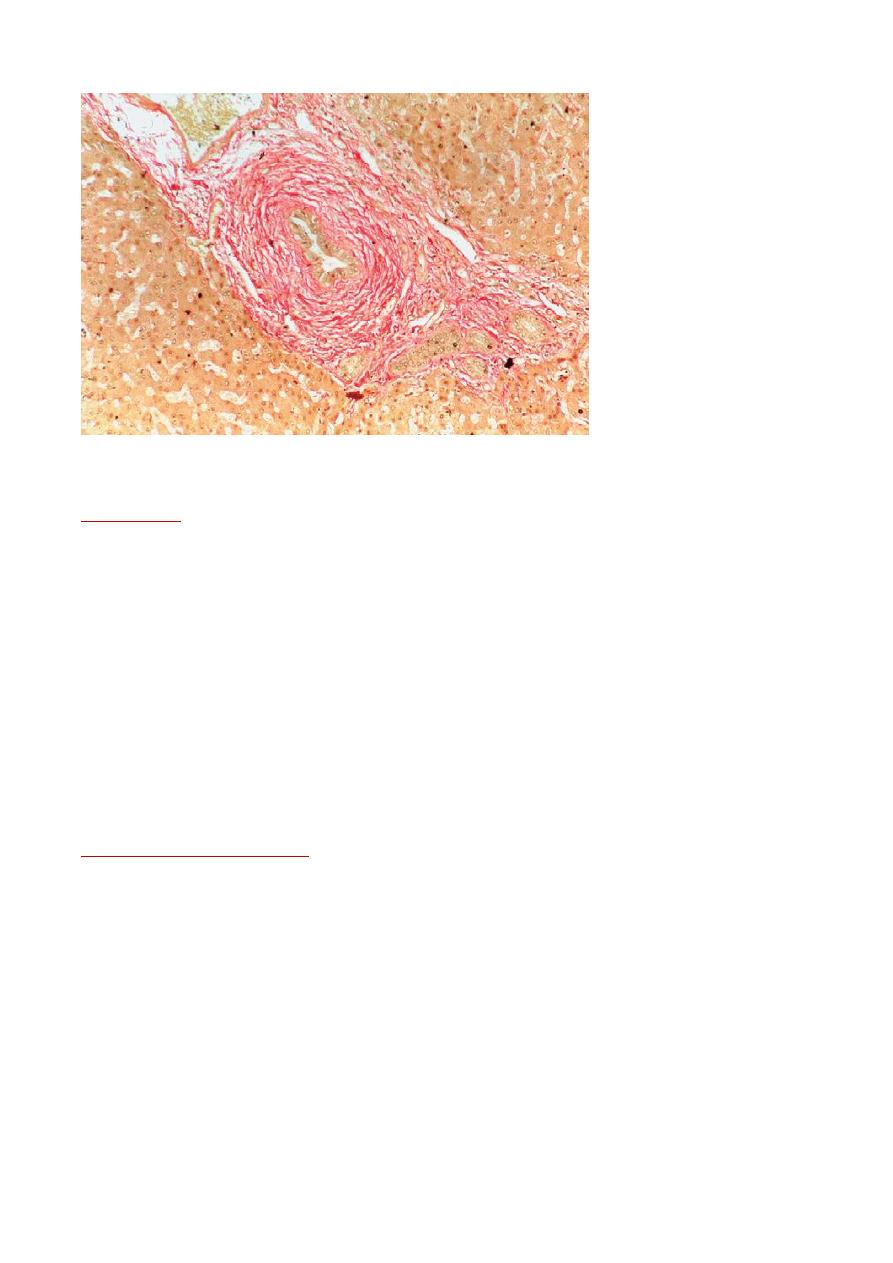
On liver biopsy the characteristic early features of PSC are periductal ‘
onion-skin’
fibrosis and inflammation,
with portal oedema and bile ductular proliferation resulting in expansion of the portal tracts .
Later, fibrosis spreads, progressing inevitably to biliary cirrhosis; obliterative cholangitis leads to the so -
called
‘vanishing bile duct syndrome’
.
7
Management
There is
no cure for PSC
, but management of cholestasis and its complications and specific treatment of the
disease process are indicated. UDCA is widely used, although the evidence to support this is limited.
UDCA
may have benefit in terms of reducing colon carcinoma risk.
The course of PSC is variable. In symptomatic patients, median survival from presentation to death or liver
transplantation is about 12 years.
About
75% of asymptomatic patients survive 15 years or more
.
Most patients die from liver failure
, about
30% die from bile duct carcinoma
, and the remainder die from
colonic cancer or complications of colitis.
Immunosuppressive agents, including prednisolone, azathioprine, methotrexate and ciclosporin, have been
tried; generally, results have been disappointing.
Symptomatic patients often have pruritus. Management is as for PBC. Fatigue appears to be less prominent
than in PBC, although it is still present in some patients.
Management of complications
Broad-spectrum antibiotics (e.g.
ciprofloxacin
) should be given for
acute attacks of cholangitis
but have no
proven value in preventing attacks.
If cholangiography shows a well-defined obstruction to the extrahepatic bile ducts
(‘dominant stricture’), mechanical relief can be obtained by placement of a plastic stent or by balloon
dilatation performed at ERCP.
It is important, in this situation, actively to consider the possibility
Cholangiocarcinoma (the differential
diagnosis for dominant extrahepatic stricture).
Fat-soluble vitamin replacement is necessary in jaundiced patients.
Metabolic bone disease (
usually osteoporosis
) is a common complication that requires treatment
Primary sclerosing cholangitis. Note onion-skin scarring arrows) surrounding a bile duct.

8
Surgical treatment
Surgical resection of the extrahepatic bile duct and biliary reconstruction have a
limited role in the
management of non-cirrhotic patients with dominant extrahepatic disease.
Orthotropic transplantation is the only surgical option in patients with advanced liver disease;
5-year
survival is 80–90% in most centres.
Unfortunately, the condition may recur in the graft and there are no identified therapies able to
prevent this.
Cholangiocarcinoma is a contraindication to transplantation.
Colon carcinoma risk can be increased in patients following transplant
because of the
effects of immune suppression .
IgG4-associated cholangitis
This recently reported disease is closely related to autoimmune pancreatitis
(which is present in more than 90% of the patients. IgG4-associated cholangitis
(IAC)
often presents with
obstructive jaundice (due to either hilar structuring/
intrahepatic sclerosing cholangitis
or a low bile duct
stricture), and Cholangiographic appearances suggest PSC with or without hila Cholangiocarcinoma.
The
serum IgG4 is often raised and liver biopsy shows a lymphoplasmacytic infiltrate, with IgG4-positive
plasma cells
. An important observation is that, compared to PSC,
IAC appears to respond well to steroid therapy