

Hereditary Clotting Factor
Deficiency (Bleeding Disorders )
Dr. Yahya Altufaily
Professor of Pediatrics
Babylon Medical College

Hemophilia A (factor VIII deficincy ) and hemophilia B (factor IX
definciency) are the most common and serious congenital coagulation
factor defiencies .
The clinical findings in hemophilia A and hemophilia B are identical.
Hemophilia C is the bleeding disorder associated with reduced levels
of factor 11 .
Reduced levels of the contact factors(factor 12 ,high molecular-weight
kininogen and prekallikrein) are associated with significant prolongation
of PTT , but are not associated with hemorrhage .

Factor VIII or Factor IX
Deficiency (Hemophilia A or B)
Deficiencies of factors VIII and IX are the most common sever
inherited bleeding disorders.
Pathophysiology:
Factor VIII and IX with phospholipid and calcium , they form the
tenase or factor X activating complex.
Factor X being activated by either the complex of factor VIII and
IX or the complex of tissue factor and factor VII .
Prothrombin time (PT) measures the activation of factor X by
factor VII and is therefore normal in patients with factor VIII and
IX deficiency.

In hemophilia A and B ,due to inadequate thrombin generation
leads to failure to form a tightly cross- linked fibrin clot support the
platelet plug .
When untreated bleeding occurs in closed space, such as joint
cessation of bleeding may be the result of tamponade.
With open wounds ,in which tamponade cannot occur, bleeding
may result in significant blood loss.
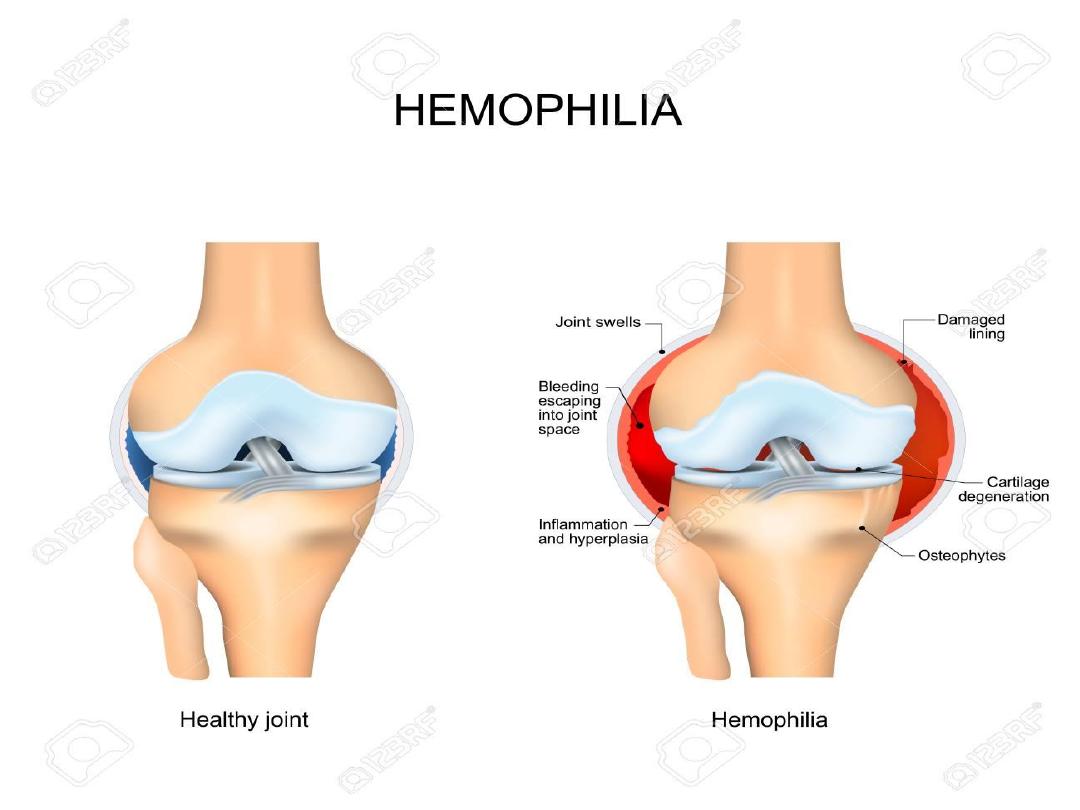

Clinical Manifestations:
Neither factor VIII nor factor IX crosses the placenta , bleeding
symptoms may be present from birth or may occur in the fetus.
Only 2% of neonates with hemophilia sustain intracranial hemorrhages,
and 30% of male infants with hemophilia bleed with circumcision.
In the absence of a positive family history (30% of hemophilia A occurs
by spontaneous mutation), hemophilia may go undiagnosed in the
newborn.
Obvious symptoms, such as easy bruising, intramuscular hematomas,
and hemarthroses, begin when the child starts to cruise.
Bleeding from minor traumatic lacerations of the mouth (torn frenulum)
may persist for hours or days and may cause the parents to seek medical
evaluation.

Although bleeding may occur in any area of the body,
the hallmark of hemophilic bleeding is hemarthrosis.
Bleeding into the joints may be induced by minor trauma; many
hemarthroses are spontaneous.
The earliest joint hemorrhages appear most often in the ankle.
In the older child and adolescent, hemarthroses of the knees and
elbows are also common.

The child's early joint hemorrhages are recognized only after major
swelling and fluid accumulation in the joint space,
but older children complain of a warm, tingling sensation in the
joint as the first sign of an early joint hemorrhage.
Repeated bleeding episodes into the same joint in a patient with
severe hemophilia may result in a “target” joint.
Recurrent bleeding may then become spontaneous because of the
underlying pathologic changes in the joint

Although most muscular hemorrhages are clinically evident
because of localized pain or swelling, bleeding into the iliopsoas
muscle requires specific mention .
A patient may lose large volumes of blood into the iliopsoas
muscle, leading to hypovolemic shock, with only a vague area of
referred pain in the groin.
The hip is held in a flexed, internally rotated position due to
irritation of the iliopsoas.
The diagnosis is made clinically from the inability to extend the hip
but must be confirmed with ultrasonography or CT scan.

Life- threatening bleeding in the patient with hemophilia is caused
by bleeding into vital structures (central nervous system, upper
airway) or by exsanguination (external trauma, gastrointestinal or
iliopsoas hemorrhage).
Argent treatment with clotting factor concentrate for these life-
threatening hemorrhages is essential.
If head trauma is of sufficient concern to suggest radiologic
evaluation, factor replacement should precede radiologic
evaluation.

Life-threatening hemorrhages require replacement therapy to
achieve a level equal to that of normal plasma (100 IU/dL , or
100%).
Patients with mild hemophilia who have factor VIII or factor IX
levels >5 IU/dL
usually do not have spontaneous hemorrhages and
may experience prolonged bleeding after dental work, surgery, or
injuries from moderate trauma and
may not be diagnosed until they are older.

Laboratory Findings and Diagnosis :
A reduced level of factor VIII or factor IX will result in a laboratory
finding of a prolonged PTT.
In severe hemophilia, the PTT value is usually 2-3 times the upper limit
of normal.
Other screening tests of the hemostatic mechanism (platelet count,
bleeding time, PT, thrombin time) are normal.
Unless the patient has an inhibitor to factor VIII or IX, the mixing of
normal plasma with patient plasma results in correction of PTT value.

The specific assay for factors VIII and IX will confirm the
diagnosis of hemophilia.
If correction does not occur on mixing, an inhibitor may be present.
In 25–35% of patients with hemophilia who receive infusions of
factor VIII or factor IX, a factor-specific antibody may develop
(inhibitors).
In such patients, the quantitative Bethesda assay for inhibitors
should be performed to measure the antibody titer.

Differential Diagnosis :
Severe thrombocytopenia;
severe platelet function disorders, such as Bernard-Soulier
syndrome and Glanzmann thrombasthenia; type 3 (severe) von
Willebrand disease; and
vitamin K deficiency.

Genetics and Classification :
Hemophilia occurs in approximately 1 : 5,000 males, with 85%
having factor VIII deficiency and
10–15% having factor IX deficiency.
Hemophilia shows no apparent racial predilection, appearing in all
ethnic groups.

Severe hemophilia is characterized as having <1% activity of the
specific clotting factor, and bleeding is often spontaneous.
Moderate hemophilia have factor levels of 1–5%
and usually require mild trauma to induce bleeding.
Mild hemophilia have levels >5%, may go many years before the
condition is diagnosed, and frequently require significant trauma to cause
bleeding.
Hemostatic level for factor VIII is >30–40%, and for factor IX, it is
>25–30%.
Lower limit of levels for factors VIII and IX in normal individuals is
approximately 50%.

The genes for factors VIII and IX are carried near the terminus of
the long arm of the X chromosome and are therefore X-linked
traits.
Approximately 45–50% of patients with severe
hemophilia A have the same mutation, which can be detected in the
blood of patients or carriers and in the amniotic fluid by molecular
techniques.
In the newborn, factor VIII values may be artificially elevated
because of the acute-phase response elicited by the birth process.
This artificial elevation may cause a mildly affected patient to have
normal or near-normal levels of factor VIII.

Factor IX levels are physiologically low in the newborn.
Through lyonization of the X chromosome, some female carriers of
hemophilia A or B have sufficient reduction of factor VIII or factor IX
to produce mild bleeding disorders.
Levels of these factors should be determined in all known or potential
carriers to assess the need for treatment in the event of surgery or
clinical bleeding.
Because factor VIII is carried in plasma by von Willebrand factor, the
ratio of factor VIII to VWF is sometimes used to diagnose carriers of
hemophilia but may give false-positive or false-negative results.
.

Treatment :
Patients with hemophilia are best managed through comprehensive
hemophilia care centers.
Early, appropriate therapy is the hallmark of excellent hemophilia care
When mild to moderate bleeding occurs, values of factor VIII or factor
IX must be raised to hemostatic levels, in the 35–50% range.
For life-threatening or major hemorrhages, the dose should aim to
achieve levels of 100% activity.
Calculation of the dose of recombinant factor VIII (FVIII) or
recombinant factor IX is as fallows :
Dose of rF VIII(IU) = % Desired (rise in F VIII) × Body weight (Kg) ×
0.5

Prophylaxis is the standard of care for most children with severe
hemophilia, to prevent spontaneous bleeding and early joint
deformities.
If target joints develop, “secondary” prophylaxis is often initiated.
With mild factor VIII hemophilia, the patient's endogenously
produced factor VIII can be released by the administration of
desmopressin acetate .
In patients with moderate or severe factor VIII deficiency, the
stored levels of factor VIII in the body are inadequate, and
desmopressin treatment is ineffective

A concentrated intranasal form of desmopressin acetate, , can also
be used to treat patients with mild hemophilia A.
The dose is 150 µg (1 spray) for children weighing <50 kg and 300
µg (2 sprays) for children and young adults weighing >50 kg.
Desmopressin is not effective in the treatment of factor IX–
deficient hemophilia.

Preliminary trials of factor IX gene therapy are underway with
some encouraging initial results.
Mucosal bleeding may require adjunct use of an antifibrinolytic
such as aminocaproic acid or tranexemic acid.
Once-weekly prophylactic subcutaneous injections of
emicizumab (humanized monoclonal antibody) may be able to
reduce the rate of bleeding in patients with or without factor VIII
inhibitors.

Prophylaxis:
Many patients are now given lifelong prophylaxis to prevent
spontaneous joint bleeding and such programs are initiated with the
1st or 2nd joint hemorrhage.
Treatment is usually provided every 2-3 days to maintain a
measurable plasma level of clotting factor (1–2%) when assayed
just before the next infusion (trough level).
Newer long-acting formulations of factor IX are available that
extend dosing to every week or every other week.
In the older child who is not given primary prophylaxis, secondary
prophylaxis is frequently initiated if a target joint develops.

Supportive Care :
Although it is easy to tell parents that their child should avoid
trauma, this advice is not practical in active children and
adolescents.
Effective measures include anticipatory guidance, including the use
of car seats, seatbelts, and bike helmets and the avoidance of high-
risk behaviors.
Older boys should be counseled to avoid violent contact sports .
Boys with severe hemophilia often sustain hemorrhages in the
absence of known trauma.
Early psychosocial intervention helps the family achieve a
balance between overprotection and permissiveness.

Patients with hemophilia should avoid aspirin and other
nonsteroidal antiinflammatory drugs (NSAIDs) that affect platelet
function.
The child with a bleeding disorder should receive the appropriate
vaccinations against hepatitis B, even though recombinant
products may avoid exposure to transfusion-transmitted diseases.
Patients exposed to plasma-derived products should be screened
periodically for hepatitis B and C, HIV, and abnormalities in
liver function.

Chronic Complications :
Chronic arthropathy .
The development of an inhibitor to either factor VIII or factor IX .
The risk of transfusion-transmitted infectious diseases.

Inhibitor Formation :
Failure of a bleeding episode to respond to appropriate replacement
therapy is usually the first sign of an inhibitor.
Inhibitors develop in approximately 25–35% of patients with
hemophilia A but only 2–3% of patients with hemophilia B, many
of
Many patients who have an inhibitor will lose it with continued
regular infusions.

Others have a higher titer of antibody with subsequent infusions
and may need to go through desensitization (immune tolerance
induction) programs, in which high doses of factor VIII for
hemophilia A or factor IX for hemophilia B are infused .
Factor IX immune tolerance programs have resulted in nephrotic
syndrome in some patients.

Rituximab, corticosteroids, and other immunosuppressives have all
been used as alternate therapy for patients with high inhibitor titers
in whom immune tolerance programs have failed.
If desensitization fails, bleeding episodes are treated with either
recombinant factor VIIa or activated prothrombin complex
concentrates (factor VIII inhibitor bypassing activity).
The use of these products bypasses the inhibitor in many cases but
may increase the risk of thrombosis.
Emicizumab may be another approach for patients with inhibitors

Von Willebrand Disease :
It is the most common inherited bleeding disorder,
Prevalence cited at 1 : 100 to 1 : 10,000 depending on the criteria
used for diagnosis.
Patients with VWD typically present with mucosal bleeding.
A family history of either VWD or bleeding symptoms and
confirmatory laboratory testing are also required for the diagnosis
of VWD.
Epistaxis, easy bruising, and menorrhagia in women are common
complaints.


Symptoms are variable and do not necessarily correlate well with VWF
levels.
Surgical bleeding, particularly with dental extractions or
adenotonsillectomy, is another common presentation.
Severe type 3 VWD may present as mild hemophilia with joint bleeds
and intracranial hemorrhage.
Most patients will have a family history of bleeding. Women are more
likely to be diagnosed with VWD because of the potential for symptoms
with menorrhagia, but men and women are equally likely to have VWD.
However, diagnosis based on symptoms may be difficult, since minor
bruising and epistaxis are not uncommon in childhood .

Classification :
VWD may be caused by quantitative or qualitative defects in VWF.
Mild to moderate quantitative defects are classified as type 1
VWD,
Severe quantitative defects, in which there is no detectable VWF
protein, are classified as type 3 VWD.
The qualitative defects are grouped together as type 2 VWD.

Type 1 VWD is the most common type, accounting for 60–80% of
all VWD patients.
Typical symptoms include mucosal bleeding, such as epistaxis and
menorrhagia, as well as easy bruising and potentially surgical
bleeding.
Patients with type 1 VWD may have low VWF as a result of
increased clearance of their VWF, or type 1C VWD .
Diagnosis of this subtype is important because treatment of these
patients with desmopressin is likely to be ineffective, necessitating
administration of VWF-containing products

.
Type 3 VWD is the most severe form and presents with symptoms
similar to those seen in mild hemophilia.
In type 3 VWD the VWF protein is completely absent.
In addition to mucosal bleeding, patients may experience joint
bleeds or central nervous system hemorrhage.
Some physicians elect to treat patients with prophylaxis, or
modified prophylaxis following injury, given that these
patients typically have very low FVIII (<10 IU/dL).

Laboratory Diagnosis
There are no reliable screening tests for VWD.
Patients with significant bleeding may present with anemia, and
some patients with type 2B VWD and platelet-type pseudo-VWD
may have thrombocytopenia.
The partial thromboplastin time may be prolonged if FVIII is low
but especially in type 1 VWD it is often normal, precluding use of
the PTT as a screening test.

Platelet function analysis has been considered as a screening test
for VWD, but suboptimal sensitivity and
specificity render results difficult to interpret.
Bleeding times are similarly unreliable in diagnosis of VWD.
No single test can reliably diagnose VWD;these include VWF:Ag,
which measures the total amount of VWF protein present,
and VWF activity test, typically using the ristocetin cofactor
activity assay (VWF:RCo ).

VWF levels can be influenced by external factors.
Blood type has long been known to affect VWF, with lower VWF
levels seen in people with blood group O.
Stress, exercise, and pregnancy all increase VWF levels;
therefore a single normal VWF level does not necessarily rule out
the presence of VWD.
Certain diseases, such as hypothyroidism , and medications, such
as valproic acid, can lower VWF levels in affected patients.
Repeat testing may be required to rule out or confirm a diagnosis
of VWD.

Treatment
Treatment of VWD depends on the type of VWD present and the reason
for treatment.
In general, type 1 VWD patients may be treated with
desmopressin , which increases the amount of circulating VWF by
release from storage.
The exceptions are the rare type 1 patient who lacks a response to
desmopressin and patients with type 1C VWD who do respond with an
increase in VWF levels, but whose rapid clearance of circulating
endogenous VWF results in a rapid return to baseline levels.
Treatment of types 2 and 3 VWD requires VWF-containing concentrates
similar to the treatment of hemophilia.

For all types of VWD, adjunct therapy should be considered when
possible, such as the use of antifibrinolytics for oral surgery or
hormonal treatment for menorrhagia.
Local treatment of epistaxis, such as nasal cautery or packing, may
be helpful in some circumstances.
Iron therapy for patients with iron-deficiency anemia may also be
required.

THANK YOU