
Bleeding disorders
TUCOM
Dep. of Medicine
5th year
Dr. Hasan I. Sultan
22- 1- 2019

Bleeding disorders
1. Define haemostasis
2. Explain the stages haemostasis
3. Enumerate the causes bleeding disorders
4. Clarify the clinical assessment of patient with bleeding
disorders
5. Outline the investigations of bleeding disorders
6. Discuss the following conditions: Hereditary
haemorrhagic telangiectasia, Inherited coagulation
disorders and Von Willebrand disease.
7. Review the acquired bleeding disorders
8. Outline the management of bleeding disorders

Haemostasis
Haemostasis is the physiologic balance of procoagulant and
anticoagulant forces that maintain both liquid blood flow
and the structural integrity of the vasculature, but must be
able to form a localized clot at the site of vascular injury in
order to prevent excessive bleeding.
This is achieved by complex interactions between the
vascular endothelium, platelets, coagulation factors, natural
anticoagulants and fibrinolytic enzymes. Dysfunction of any
of these components may result in haemorrhage or
thrombosis.
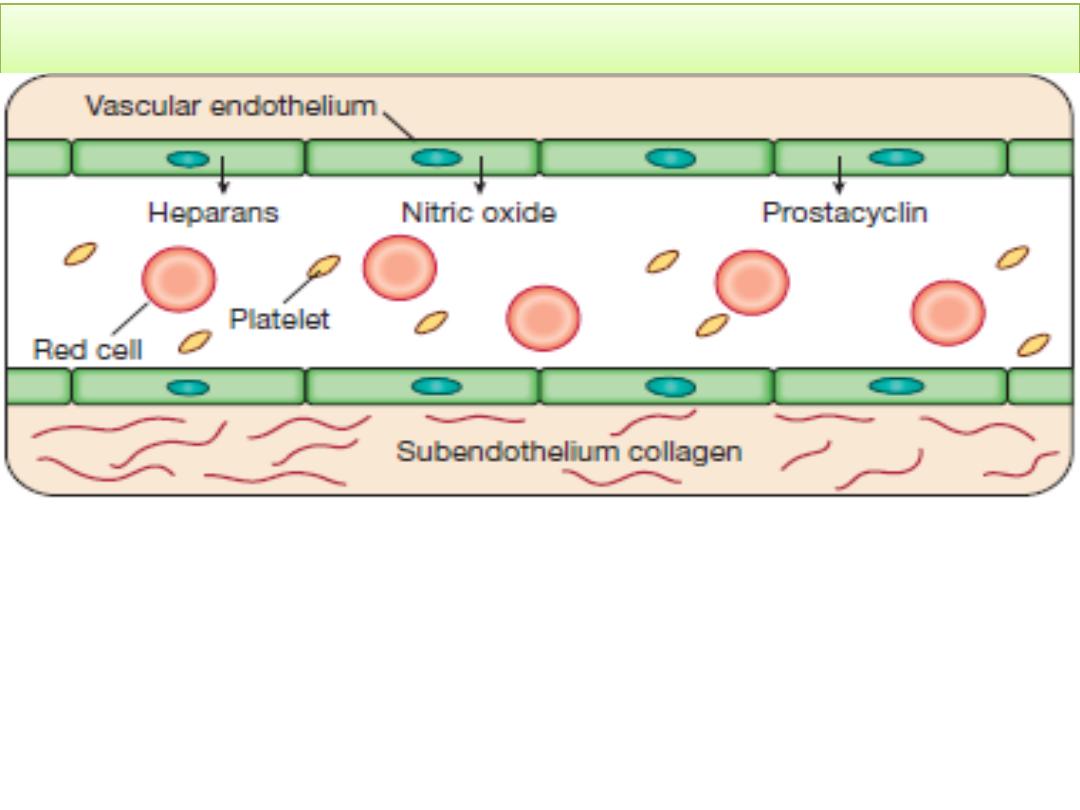
The stages of normal haemostasis
Stage 1
Pre-injury conditions encourage flow. The vascular
endothelium produces substances (including nitric oxide,
prostacyclin and heparans) to prevent adhesion of platelets
and white cells to the vessel wall. Platelets and coagulation
factors circulate in a non-activated state.
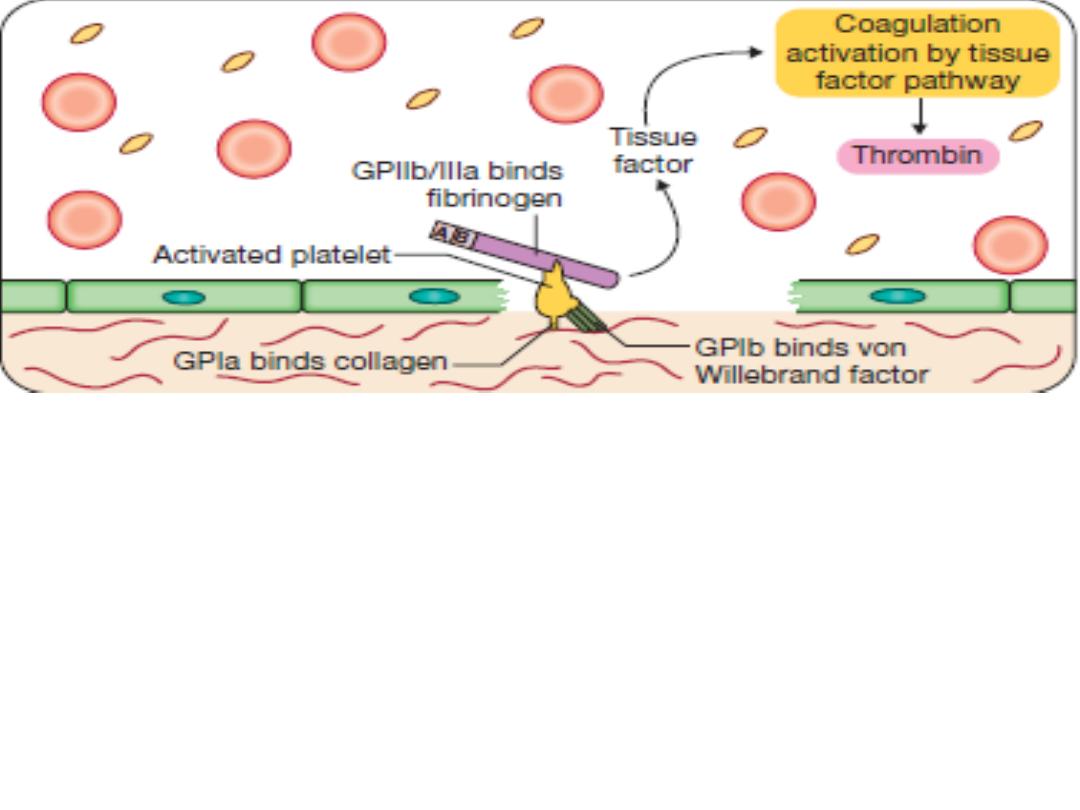
Stage 2
Early haemostatic response: platelets adhere,
coagulation is activated. At the site of injury, the
endothelium is breached, exposing subendothelial collagen.
Small amounts of tissue factor (TF) are released. Platelets
bind by specific receptors (GPIb and GPIIb/IIIa) to von
Willebrand factor and fibrinogen, respectively. Coagulation
is activated by the tissue factor (extrinsic) pathway,
generating small amounts of thrombin.
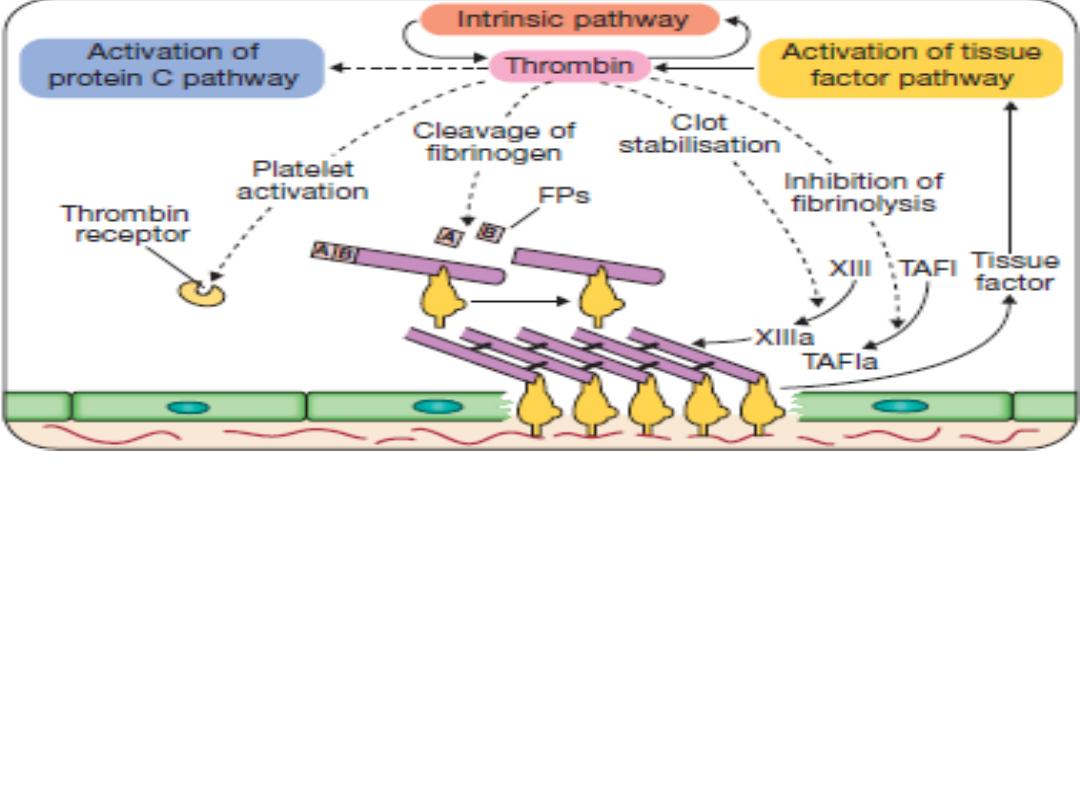
Stage 3
Fibrin clot formation: platelets become activated and
aggregate results in release of the platelet granule contents, enhancing
coagulation further. Thrombin plays a key role in the control of
coagulation: the small amount generated via the TF pathway, the
‘intrinsic’ pathway becomes activated and large amounts of thrombin
are generated. Thrombin directly causes clot formation. Fibrin
monomers are cross-linked by factor XIII, which is also activated by
thrombin. Having had a key role in clot formation and stabilisation.
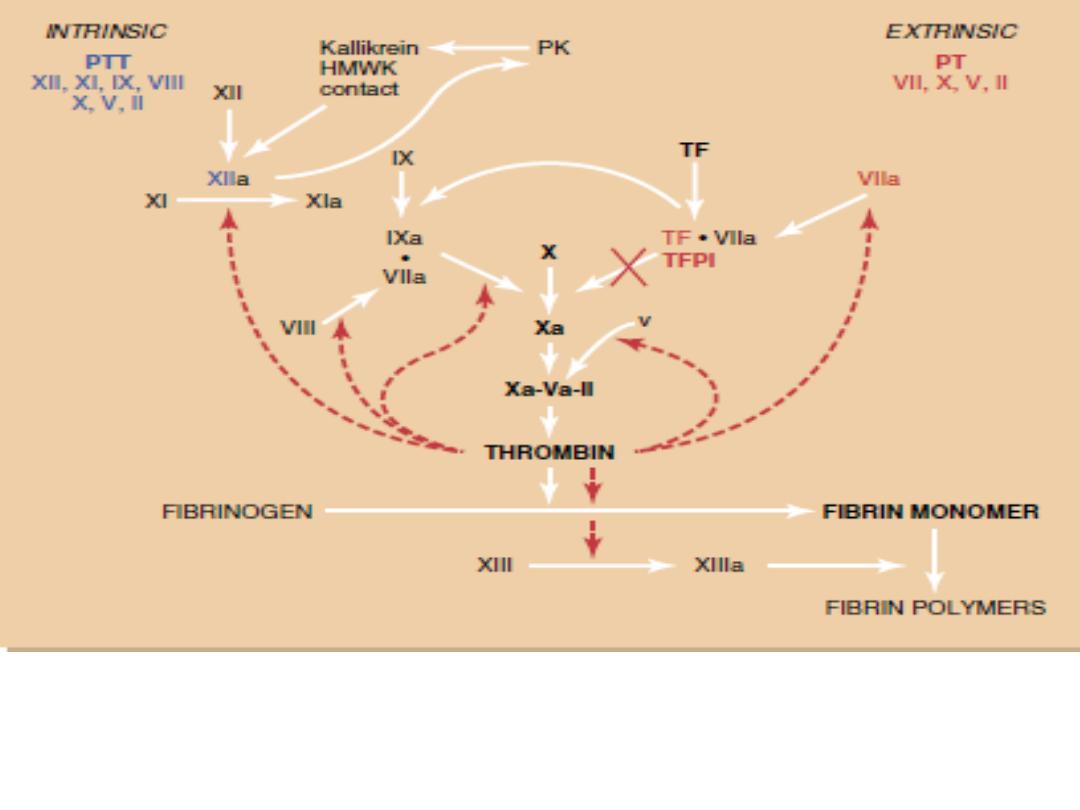
The coagulation cascade. The extrinsic and intrinsic pathways allow monitoring of
anticoagulation by the prothrombin time (PT) and partial thromboplastin time (PTT),
respectively. HMWK, highmolecular- weight kininogen; PK, prekallikrein; TF, tissue
factor; TFPI, tissue factor pathway inhibitor.

The coagulation factors:
• I: Fibrinogen
• II: Thrombin
• III: Tissue factor
• IV: Ca++
• V: Proaccerin / Labile factor
• VI: No longer used
• VII: Proconvertin
• VIII: Antihemophilic factor (AHF)
• IX: Christmas F/ Antihemophilic factor B
• X: Stuart factor
• XI: Plasma thromboplastin antecedent (PTA)/ antihemophilic factor
C
• XII: Hageman factor
• XIII: Fibrin stabilisig factor
• All produce by the liver. Fact. V
and VIII produce also by
platelets and endothelial Cells.
• VIT. K dependent factors: II, VII,
IX and X (inhibited by
warfarin)
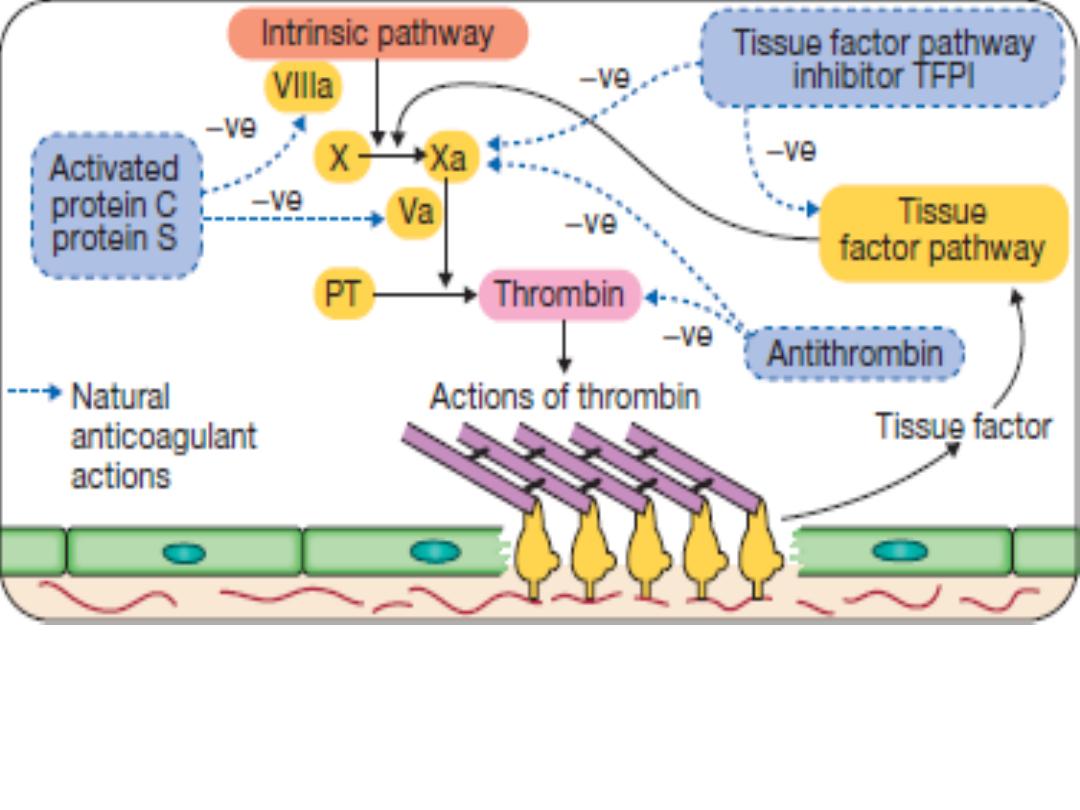
Stage 4
Limiting clot formation: natural anticoagulants reverse
activation of coagulation factors. Once haemostasis has been secured,
the propagation of clot is curtailed by anticoagulants: Antithrombin,
protein C (PC) and protein S (PS).
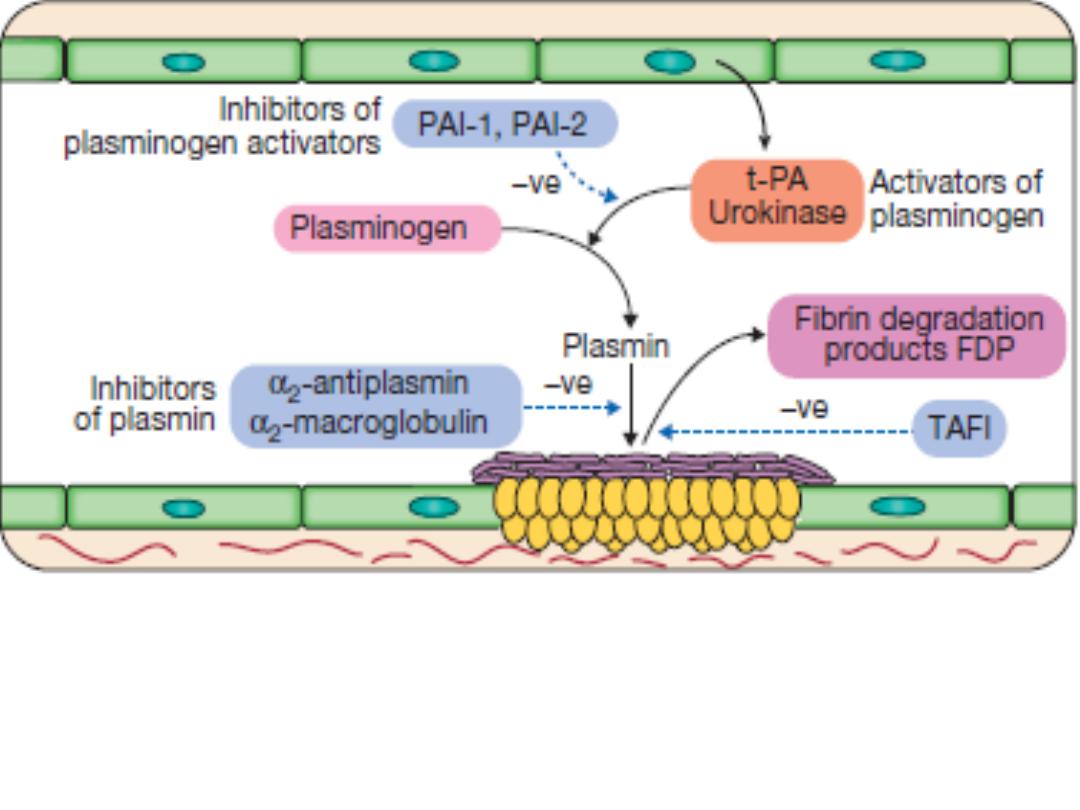
Stage 5
Fibrinolysis: plasmin degrades fibrin to allow vessel
recanalisation and tissue repair. Plasmin, the main fibrinolytic enzyme,
is produced when plasminogen is activated, e.g. by tissue plasminogen
activator (t-PA) or urokinase in the clot. Plasmin hydrolyses the fibrin
clot, producing fibrin degradation products, including the D-dimer.

Bleeding disorders
Normal bleeding is seen following surgery and trauma.
Pathological bleeding occurs when structurally abnormal
vessels rupture or a defect in haemostasis.
Causes:
1- Vascular Causes:
vasculitis, senile purpura, scurvy or
vitamin C deficiency, hereditary hemorrhagic telangiectasia
(Osler-Weber-Rendu syndrome), pseudoxanthoma elasticum
and Ehlers Danlos syndrome.
2- Deficiency or dysfunction of platelets
3- Coagulation factors disorders
4- Von Willebrand disease
5- Drugs:
excessive antiplatelets, anticoagulant and
fibrinolytic drugs.

Clinical assessment
It is important to consider the following:
1- Site of bleeding:
• Bleeding into muscle and joints, along with
retroperitoneal and intracranial haemorrhage, indicates a
likely defect in coagulation factors.
• Petechiae, purpura, ecchymoses, prolonged bleeding from
superficial cuts, epistaxis and gastrointestinal
haemorrhage indicates thrombocytopenia, a platelet
function disorder or von Willebrand disease.
• Recurrent bleeds at a single site suggest a local structural
abnormality.
2- Duration of history:
to assess whether the disorder is
congenital or acquired.

3- Precipitating causes:
Bleeding arising spontaneously
indicates a more severe defect than bleeding that occurs
only after trauma.
4- Surgery:
Ask about operations.
• Dental extractions, tonsillectomy and circumcision are
stressful tests of the haemostatic system.
• Immediate post-surgical bleeding suggests defective
platelet plug formation and primary haemostasis
• Delayed haemorrhage is more suggestive of a coagulation
defect.
• In postsurgical patients, persistent bleeding from a single
site is more likely to indicate surgical bleeding than a
bleeding disorder.
5- Drugs:
Use of antiplatelets, anticoagulant and fibrinolytic
drugs must be elicited.

6- Family history:
a positive family history may indicates
inherited disorders, like haemophilias or deficiencies of factor
VII, X and XIII, which are recessively inherited.
Examination
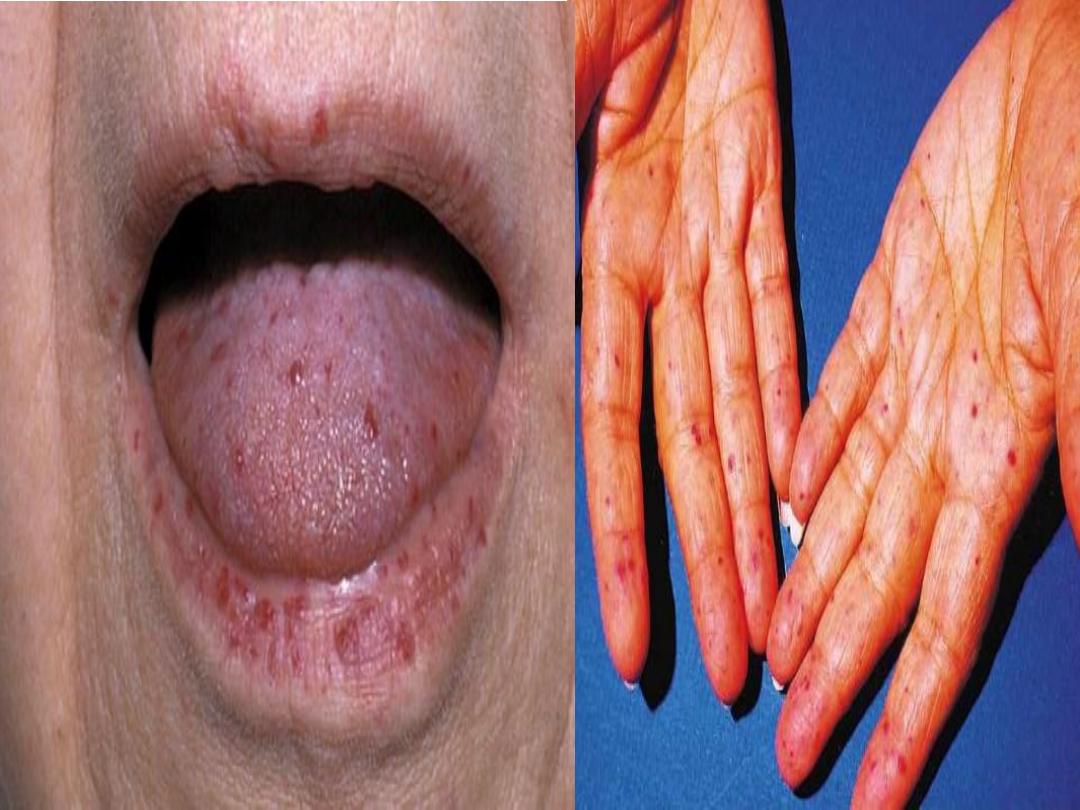
• Petechiae, purpura and ecchymosis, in the skin, mucous
membranes, or in the gastrointestinal tract tends to occur
more often in patients with thrombocytopenia, qualitative
platelet defects, vascular abnormalities, and von Willebrand
disease (vWD). Palpable purpura occurs in vasculitis.
• Bleeding in deep organs; joints, or muscles or retroperitoneal
space is more commonly associated with factor deficiencies,
such as hemophilia.
• Telangiectasia of lips and tongue points to hereditary
haemorrhagic telangiectasia.
• Underlying associated systemic illness such as a
haematological or other malignancy, liver disease, renal
failure, connective tissue disease.

Vessel wall abnormalities
1. Congenital, such as hereditary haemorrhagic
telangiectasia
2. Acquired, as in a vasculitis or scurvy
Hereditary haemorrhagic telangiectasia (HHT) or Osler-
Weber- Rendu syndrome
It is an autosomal dominant inherited condition
Telangiectasia and small aneurysms are found on the
fingertips, face and tongue, and in the nasal passages, lung
and gastrointestinal tract.
large pulmonary arteriovenous malformations (PAVMs) that
cause arterial hypoxaemia due to a right-to-left shunt. These
predispose to paradoxical embolism, resulting in stroke or
cerebral abscess.

Patients present either with recurrent bleeds, particularly
epistaxis, or with iron deficiency due to occult GI bleeding.
Treatment:
• Can be difficult because of the multiple bleeding points
• Regular iron therapy often allows the marrow to
compensate for blood loss.
• Local cautery or laser therapy may prevent single lesions
from bleeding.
Platelet disorders (previous lecture)

Coagulation disorders
• Congenital: X-linked like Haemophilia A and B. Autosomal
like Von Willebrand disease
• Acquired: may be due to under-production (e.g. in liver
failure), increased consumption (e.g. in disseminated
intravascular coagulation) or inhibition of function such as
heparin therapy or inhibition of synthesis such as warfarin
therapy.

Haemophilia A
Factor VIII deficiency resulting in haemophilia A affects
1/10 000 individuals. It is the most common congenital
coagulation factor deficiency. Factor VIII is primarily
synthesised by the liver and endothelial cells, and has a half-
life of about 12 hours. It is protected from proteolysis in the
circulation by binding to von Willebrand factor (vWF).
Genetics
It is X-linked recessive disease. Thus all daughters of
haemophiliacs are obligate carriers and they, in turn, have a
1 in 4 chance of each pregnancy resulting in the birth of an
affected male.
All family members have the same factor VIII gene mutation
and a similarly severe or mild phenotype.
Inherited bleeding disorders
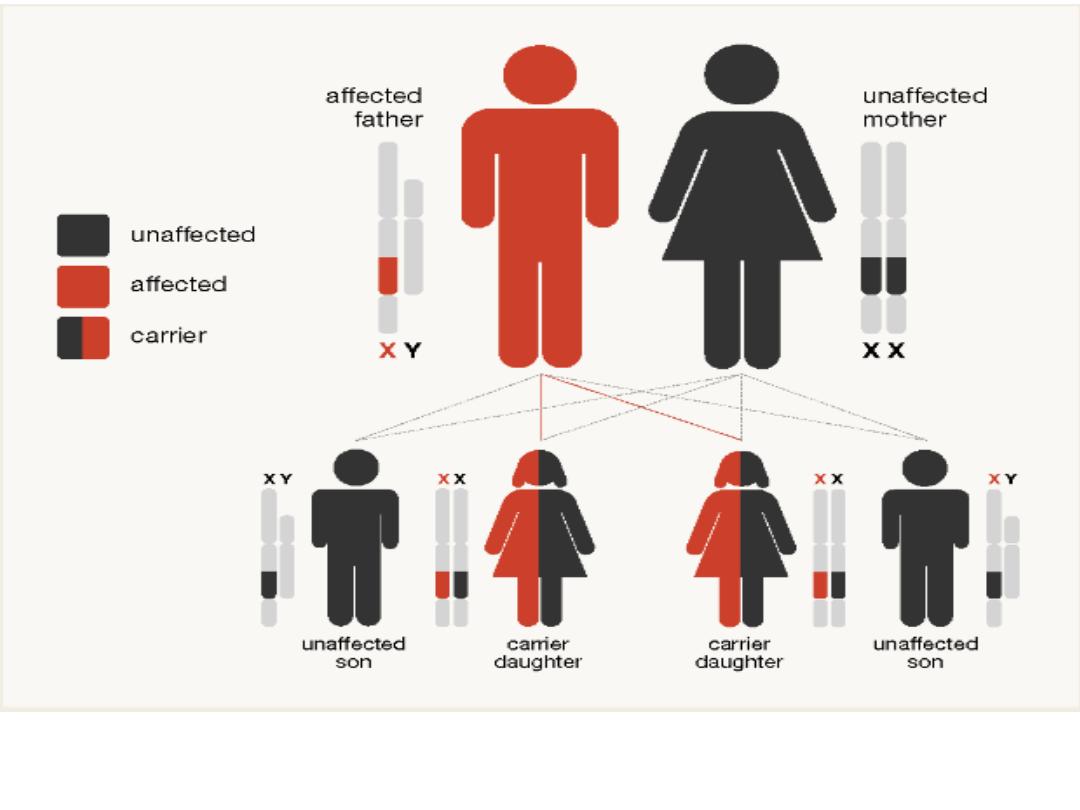
Inheritance of haemophilia in the case of an affected father and a
mother who is not a haemophilia carrier.
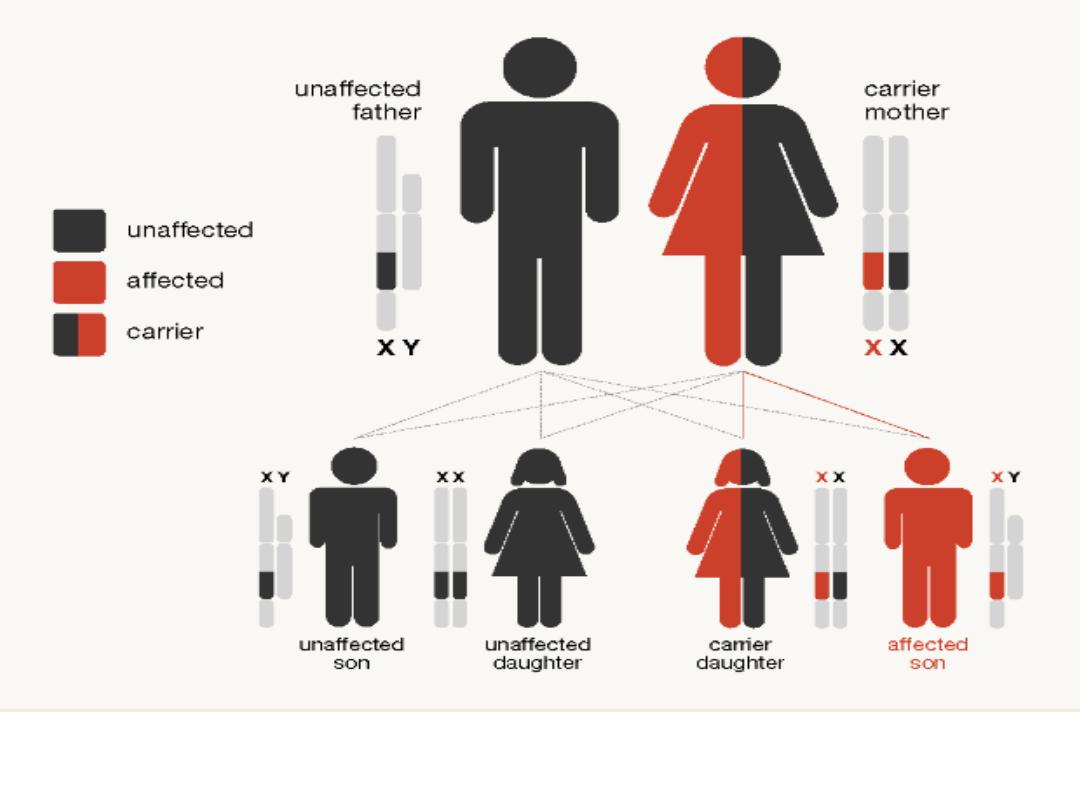
Inheritance of haemophilia in the case of a 'carrier' mother (with one
copy of the haemophilia gene) and a father without haemophilia

Clinical features
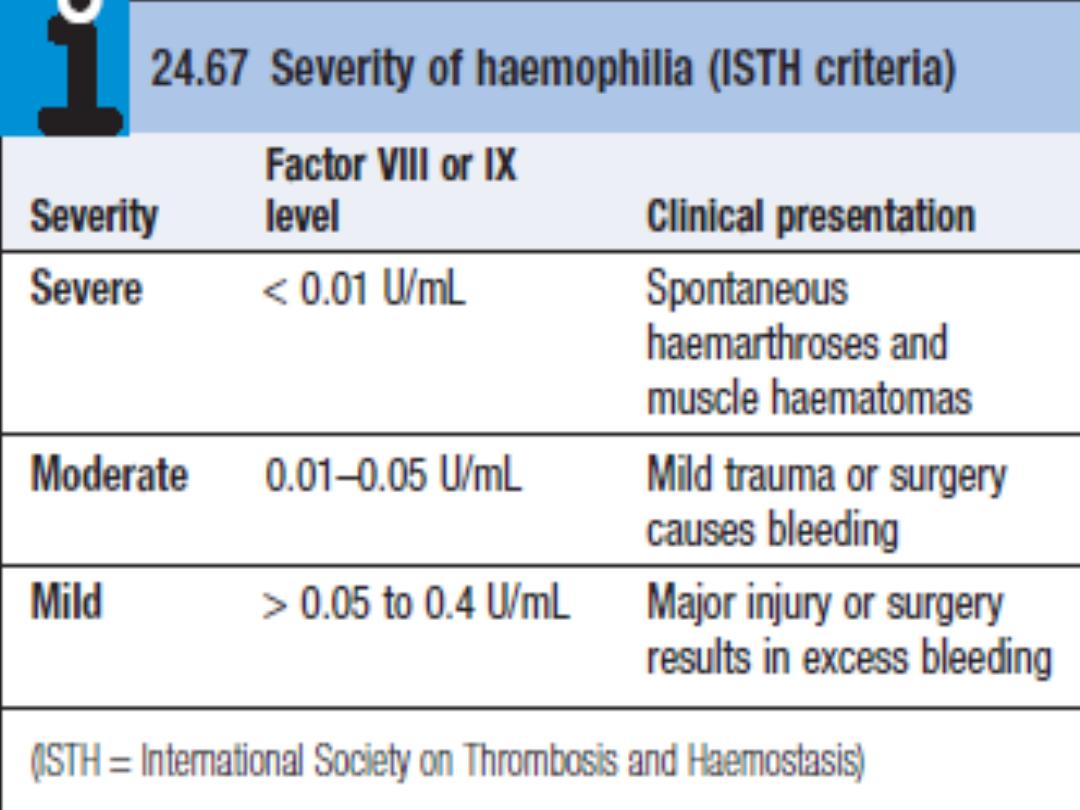
C/F closely related to residual factor VIII levels:
• Severe haemophilia (< 1% of normal factor VIII levels)
present with spontaneous bleeding into skin, muscle and
joints. Retroperitoneal and intracranial bleeding is also a
feature. Babies with severe haemophilia have an
increased risk of intracranial haemorrhage.
• Moderate and mild haemophilia (factor VIII levels 1–40%)
present with the same pattern of bleeding, but usually
after trauma or surgery.
Bleeding is typically into large joints, especially knees,
elbows, ankles and hips. Muscle haematomas are also
characteristic, most commonly in the calf and psoas
muscles.
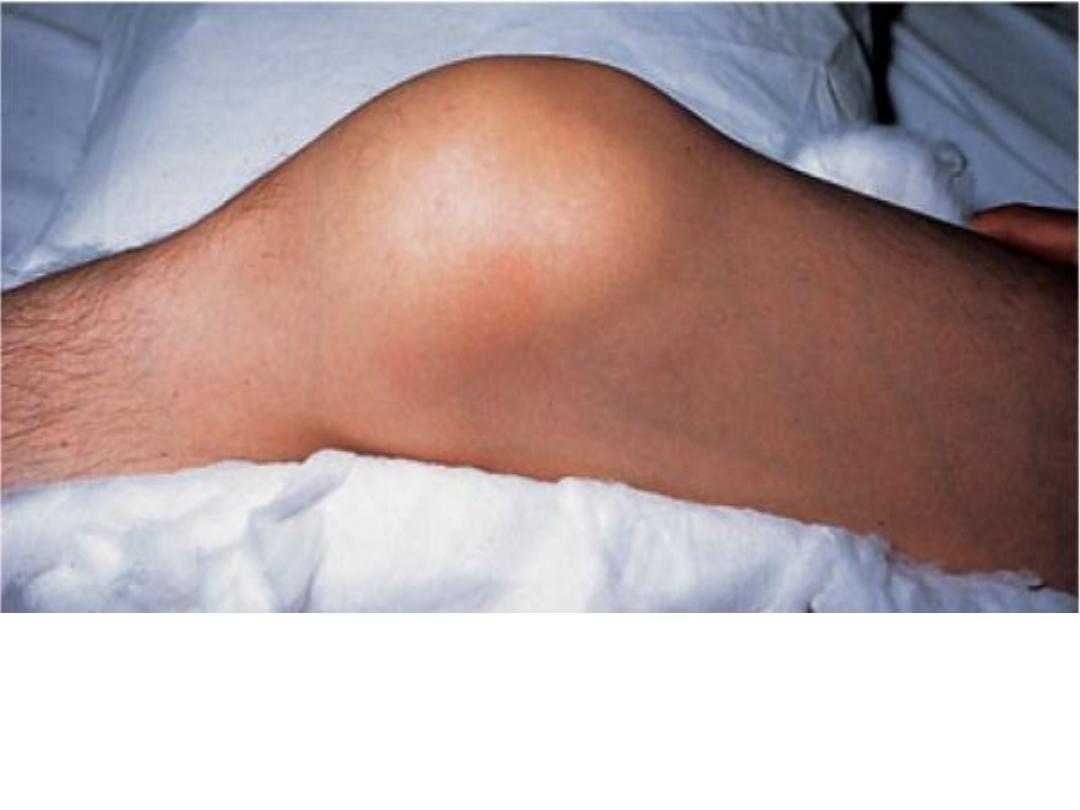
Acute hemarthrosis of the knee is a common complication of
hemophilia. It may be confused with acute infection unless the
patient's coagulation disorder is known, because the knee is hot, red,
swollen, and painful
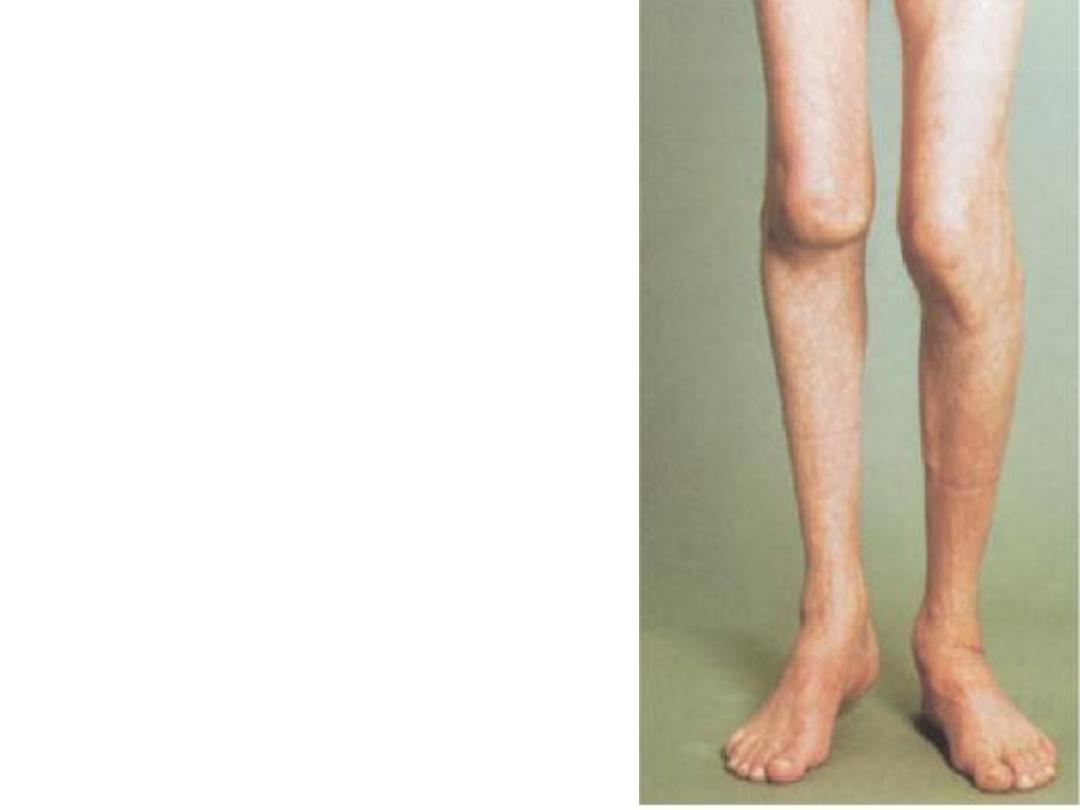
Severe chronic arthritis in
hemophilia. The knee is the most
commonly affected joint. Both
knees are severely deranged in
this patient. Note that he is
unable to stand with both feet flat
on the floor.
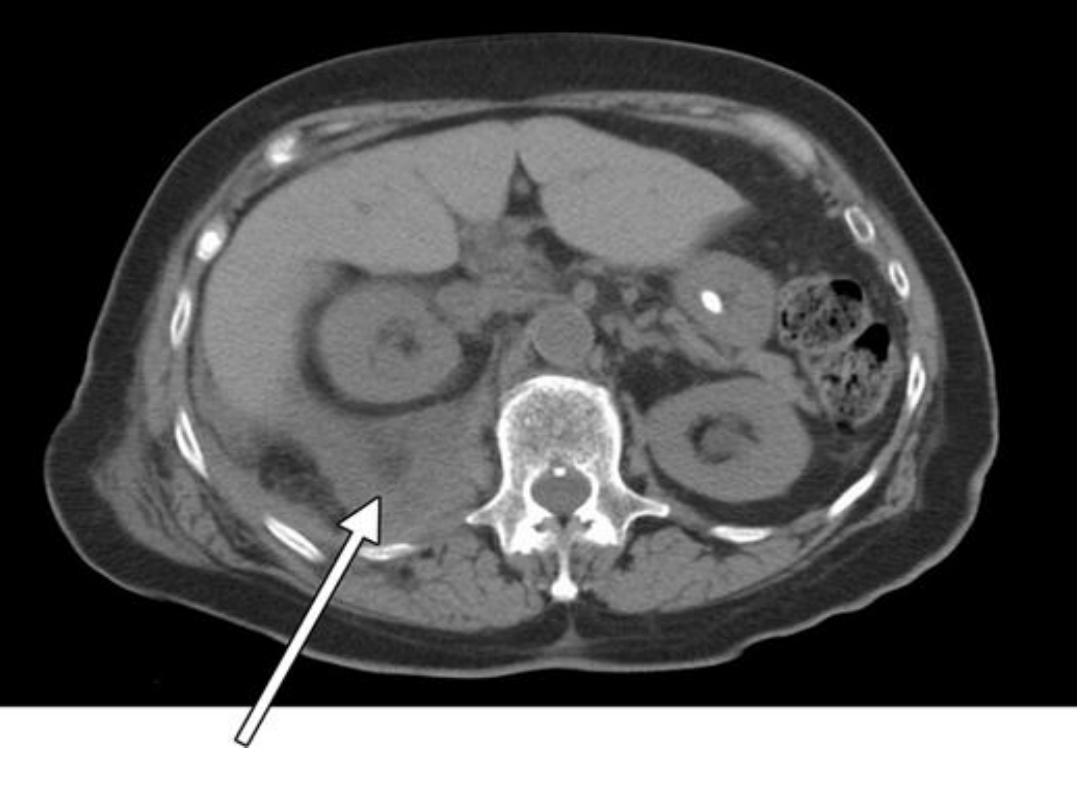
Right retroperitoneal hematoma displacing the kidney anteriorly

Diagnosis
Is confirmed by detection of significantly reduced factor
VIII (hemophilia A) or IX (hemophilia B) activities in the
plasma of:
• Male babies born into families known to be affected by
hemophilia or
• Male children who present with excessive bruising or
bleeding at the time of circumcision
• When intramuscular injections of immunizing vaccinations
are administered, or after trauma during the toddler
years.
prolongation of aPTT.

Management
In severe haemophilia A, bleeding episodes should be
treated by raising the factor VIII level, usually by intravenous
infusion of factor VIII concentrate. Factor VIII concentrates
are freeze-dried and stable at 4°C and can therefore be
stored in domestic refrigerators, allowing patients to treat
themselves at home at the earliest indication of bleeding.
Resting of the bleeding site by either bed rest or a splint
reduces continuing haemorrhage. Once bleeding has settled,
the patient should be mobilised with physiotherapy.
The vasopressin receptor agonist DDAVP raises the vWF and
factor VIII levels by 3–4-fold, which is useful in arresting
bleeding in patients with mild or moderate haemophilia A.
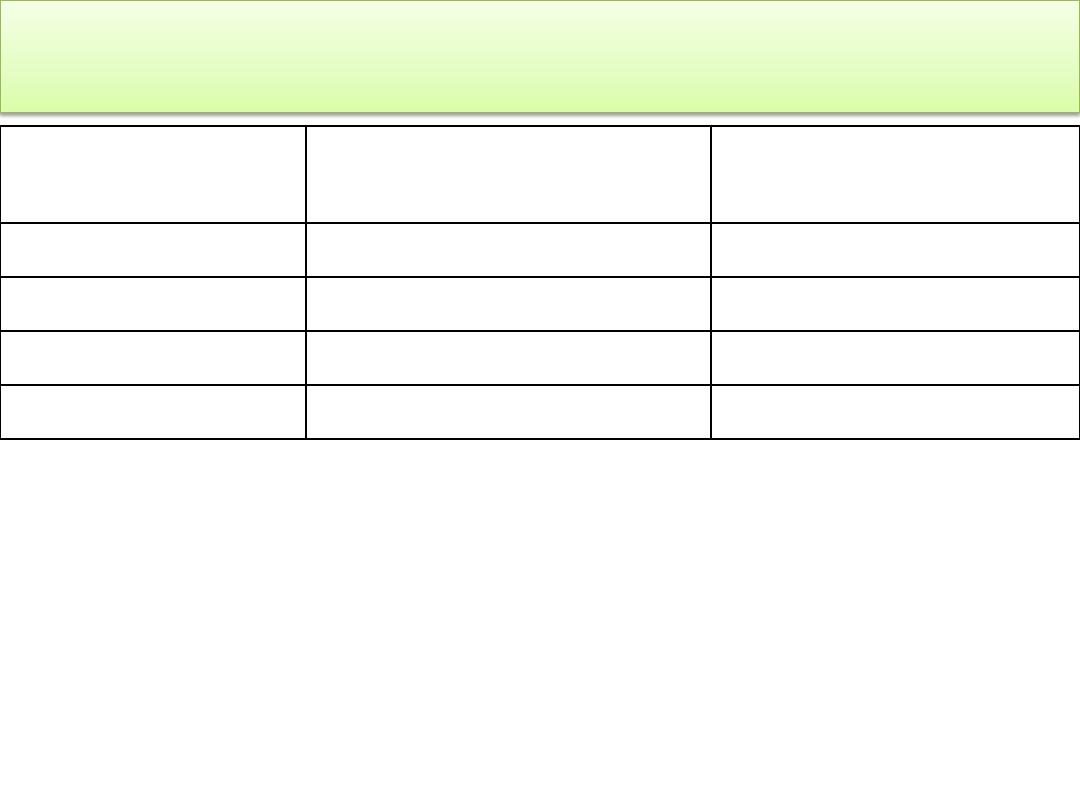
INJURY
FACTOR VIII INITIAL DOSE
(U/kg)*
FACTOR IX INITIAL
DOSE (U/kg)†
Dental prophylaxis
15-25
20-30
Hemarthrosis
15-25
30-50
Muscle hematoma
15-25
30-50
Trauma or surgery
50
100
*
Dosing intervals should be based on a Factor VIII half-life of about 12 hours.
Maintenance doses of one half the listed dose may be additionally provided
at these intervals.
†Dosing intervals should be based on a Factor IX half-life of about 18 to 24
hours. Maintenance doses of one half the listed dose may be additionally
provided at these intervals.
FACTOR REPLACEMENT GUIDELINES FOR HEMOPHILIA A
AND B

Complications
Complications of hemophilia:
Osteoarthrosis. A large psoas bleed may extend to compress
the femoral nerve. Calf haematomas causing a compartment
syndrome with subsequent contraction and shortening of
the Achilles tendon.
Complications of coagulation factor therapy:
• Infection with HIV and hepatitis viruses HBV and HCV.
• Development of anti-factor VIII antibodies ( 20% of severe
haemophiliacs). Such antibodies rapidly neutralise
therapeutic infusions, making treatment relatively
ineffective. Infusions of activated clotting factors, e.g. VIIa
or factor VIII inhibitor bypass activity (FEIBA), may stop
bleeding.

Haemophilia B (Christmas disease)
• It is X-linked recessive disease result in a reduction of the
plasma factor IX level
• It is clinically indistinguishable from hemophilia A but is
less common.
• The frequency of bleeding episodes is related to the
severity of the deficiency of the plasma factor IX level.
• Treatment is with a factor IX concentrate, used in much
the same way as factor VIII for hemophilia A. Although
they do not commonly induce inhibitor antibodies (< 1%
patients).
Acquired Hemophilias:
Autoantibody inhibitors can occur
spontaneously in individuals with previously normal
hemostasis (nonhemophiliacs).

Von Willebrand disease:
• Von Willebrand disease is a common but usually mild
bleeding disorder caused by a quantitative (types 1 and 3)
or qualitative (type 2) deficiency of von Willebrand factor
(vWF).
• The gene for vWF is located on chromosome 12 and the
disease is usually inherited as an autosomal dominant,
except in cases of type 2N and type 3, when it is recessive.
• vWF a protein synthesised by endothelial cells and
megakaryocytes, which is involved in both platelet
function and coagulation. It normally forms a multimeric
structure which is essential for its interaction with
subendothelial collagen and platelets.
• vWF acts as a carrier protein for factor VIII; deficiency of
vWF lowers the plasma factor VIII level.
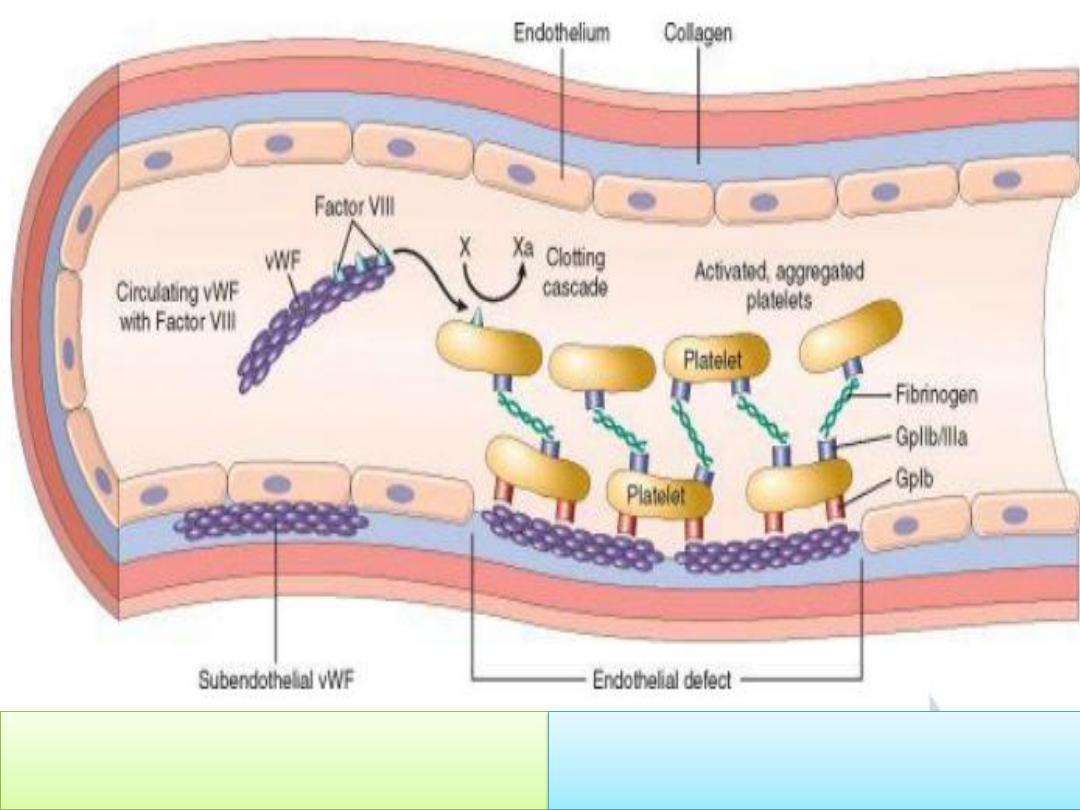
vWF tethers the platelet to
exposed collagen
vWF serves as a carrier protein for
factor VIII
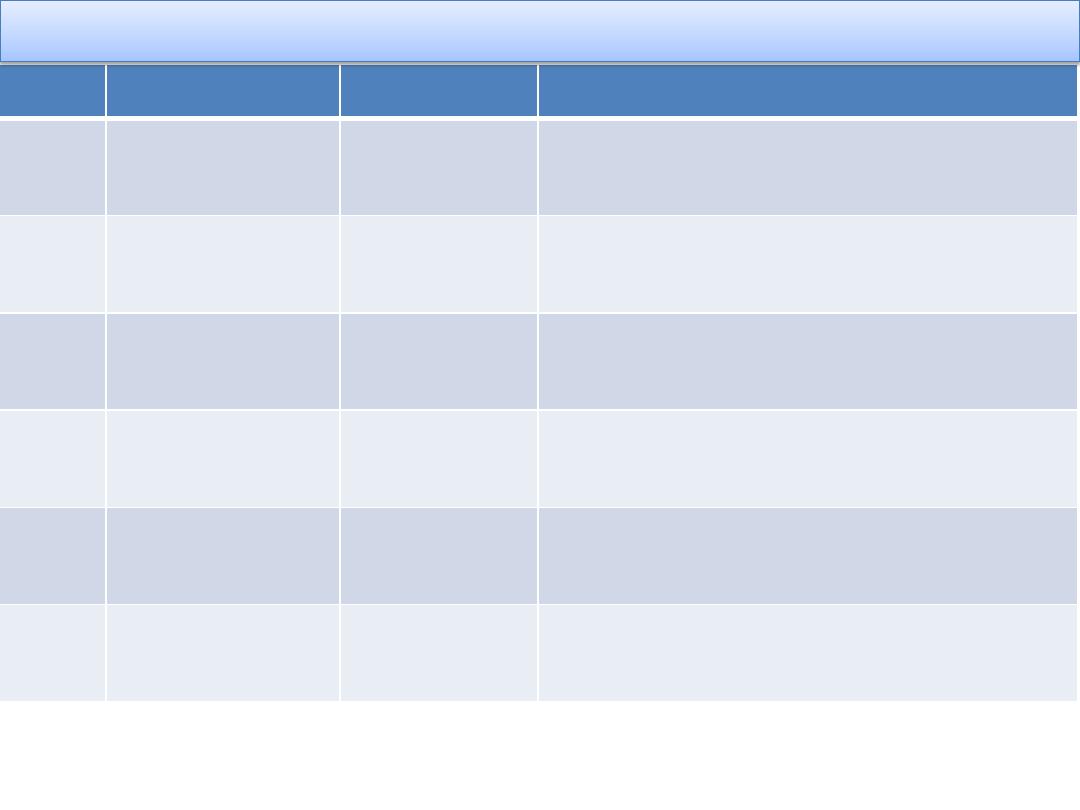
Type
Defect
Inheritance Investigations
1
Partial
quantitative
AD
Parallel decrease in vWF: Ag and
VIII:c
2A
Qualitative
AD
Absent HWM of vWF Ratio of vWF
activity to antigen < 0.7
2B
Qualitative
AD
Reduced HWM of vWF Enhanced
platelet agglutination (RIPA)
2M
Qualitative
AD
Normal multimers of vWF
Abnormal platelets Interactions
2N
Qualitative
AR
Defective binding of vWF to VIII
Low VIII
3
Severe
quantitative
AR or CH
Very low vWF activity and VIII:c
Absent multimers
Classification of von Willebrand disease
CH = compound heterozygote. HWM = high-weight multimers

Most patients with von Willebrand disease have a type 1
disorder, characterised by a quantitative decrease in a
normal functional protein.
Clinical features
Patients present with haemorrhagic manifestations similar
to those in individuals with reduced platelet function.
Superficial bruising, epistaxis, menorrhagia and
gastrointestinal haemorrhage are common.
Within a single family, the disease has variable penetrance,
so that some members may have quite severe and frequent
bleeds, whereas others are relatively asymptomatic.
Investigations
Reduced vWF level, activity of vWF and factor VIII.
Prolonged bleeding time and aPTT time, but normal PT time.

In addition, analysis for mutations in the vWF gene is
informative in most cases.
Treatment:
• Mild haemorrhage can be successfully treated by local
means or with DDAVP and tranexamic acid.
• Serious or persistent bleeds, haemostasis can be achieved
with selected factor VIII concentrates which contain
considerable quantities of vWF in addition to factor VIII.
• Cryoprecipitate (contain fibrinogen, factor VIII and VWF)
is used for prophylaxis or treatment of vWD-related
bleeding complications.

A pool of cryoprecipitate: it containing only fibrinogen, fibronectin,
von Willebrand factor, factor VIII, and factor XIII. Individual units of
cryoprecipitate typically contain 10 to 20 mL. Cryoprecipitate is
stored in the frozen state at less than −18° C and must be thawed
before issuing and administration. The shelf life while frozen is 1
year.

Acquired bleeding disorders
• DIC (previous lecture)
• Renal failure (previous lecture)
• Liver failure:
There is reduced hepatic synthesis of factors V,
VII, VIII, IX, X, XI, prothrombin and fibrinogen.
Thrombocytopenia may occur secondary to hypersplenism in
portal hypertension. In cholestatic jaundice, there is reduced
vitamin K absorption, leading to deficiency of factors II, VII, IX
and X.
The PT is a sensitive measure of liver function and becomes
elevated in liver disorders.
Treatment with plasma products or platelet transfusion should
be reserved for acute bleeds or to cover interventional
procedures such as liver biopsy. Vitamin K deficiency can be
readily corrected with parenteral administration of vitamin K.
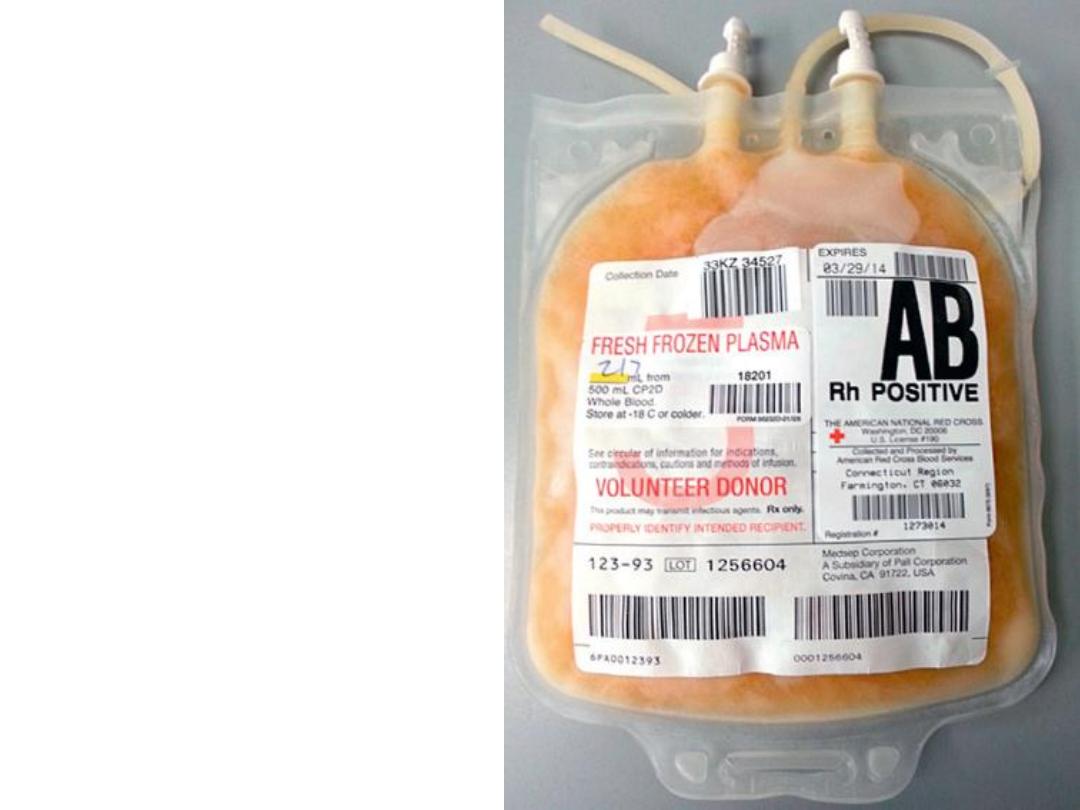
Single unit of fresh-frozen
plasma (FFP). contain a total
volume of about 200 to 250
mL. FFP possesses all
elements found in peripheral
blood plasma, including
coagulation factors, albumin,
complement, and
immunoglobulins, although
they are primarily
administered for coagulation
factor defects. FFP is stored
in the frozen state at less
than −18° C and must be
thawed before issuing and
administration. The shelf life
while frozen is 1 year.

Warfarin therapy bleeding:
warfarin inhibit the vitamin K-dependent carboxylation of factors
II, VII, IX and X in the liver.
Major bleeding is the most common serious side effect of
warfarin and occurs in 1–2% of patients each year. Fatal
haemorrhage, most commonly intracranial, occurs in about
0.25% per annum.
Contraindications to anticoagulation are:
• Recent surgery, especially to eye or CNS
• Pre-existing haemorrhage state, e.g. liver disease,
haemophilia, thrombocytopenia
• Pre-existing structural lesions, e.g. peptic ulcer
• Recent cerebral or gastrointestinal haemorrhage
• Uncontrolled hypertension
• Cognitive impairment
• Frequent falls

The international normalised ratio (INR) is validated only to
assess the therapeutic effect of warfarin. INR is the ratio of
the patient’s PT to that of a normal control, raised to the
power of the international sensitivity index of the
thromboplastin used in the test (ISI, derived by comparison
with an international reference standard material); (patient
PT/mean control PT
ISI
).
warfarin is administered in doses that produce a target INR
of 2.0–3.0 and an increase in bleeding with INR values >4.5.
Treatment of warfarin over-anticoagulation and bleeding:
1- If the INR is above the therapeutic level in asymptomatic
patients whose INR is between 3.5 and 4.5, warfarin should
be withheld until the INR returns to the therapeutic range.

2- If the patient is not bleeding, it may be appropriate to
give a small dose of vitamin K either orally or IV (1–2.5 mg),
especially if the INR is greater than 8.
3- In the event of bleeding, withhold further warfarin.
• Minor bleeding can be treated with 1–2.5 mg of vitamin K
IV.
• Major haemorrhage should be treated as an emergency
with vitamin K 5–10 mg slowly IV, combined with
coagulation factor replacement. This should optimally be
a prothrombin complex concentrate (30–50 U/kg) which
contains factors II, VII, IX and X; if that is not available,
fresh frozen plasma (15–30 mL/kg) should be given.

Heparin:
Heparin is a sulfated polysaccharide acts as an anticoagulant
by activating antithrombin (previously known as
antithrombin III) and accelerating the rate at which
antithrombin inhibits clotting enzymes, particularly
thrombin and factor Xa.
Heparin therapy can be monitored using the activated
partial thromboplastin time (aPTT) or anti-factor Xa level.
Therapeutic heparin levels are achieved with a two- to
threefold prolongation of the aPTT.
Heparin-treated patients with serious bleeding can be given
protamine sulfate to neutralize the heparin and result in
protamine-heparin complexes are then cleared. Typically, 1
mg of protamine sulfate neutralizes 100 units of heparin.
Protamine sulfate is given by slow IV infusion.
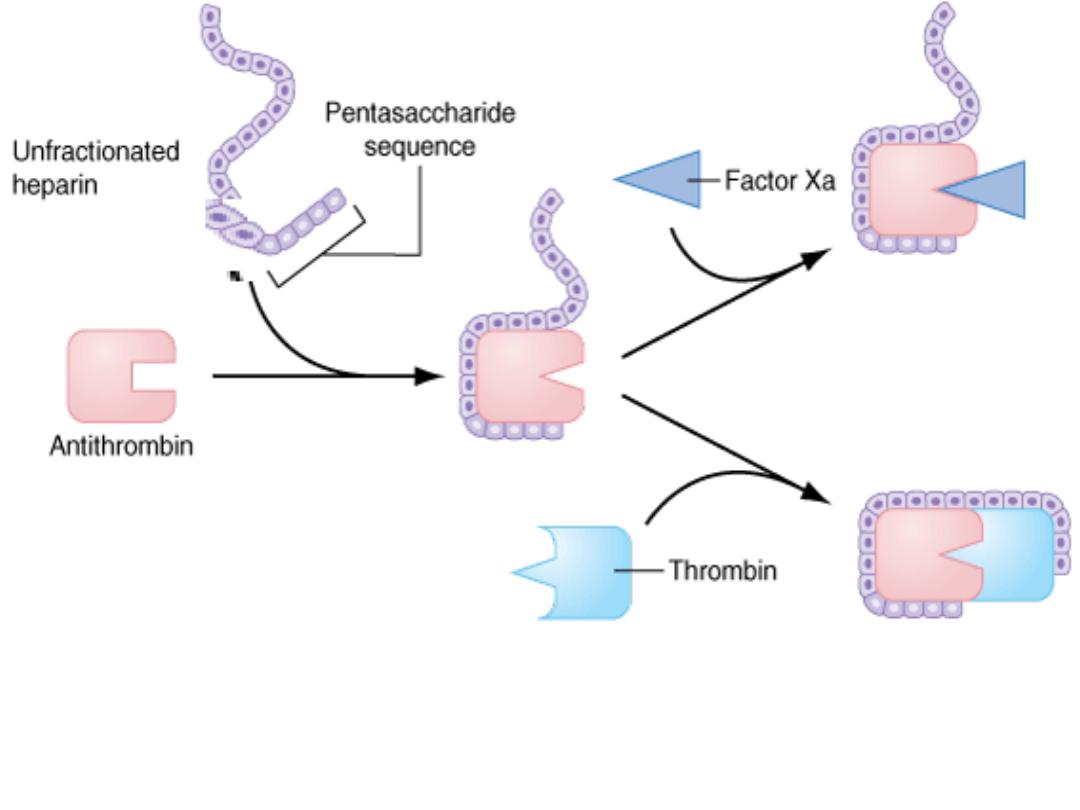
Heparin binds to antithrombin via its pentasaccharide sequence. This induces a
conformational change in the reactive center loop of antithrombin that accelerates
its interaction with factor Xa. To potentiate thrombin inhibition, heparin must
simultaneously bind to antithrombin and thrombin.
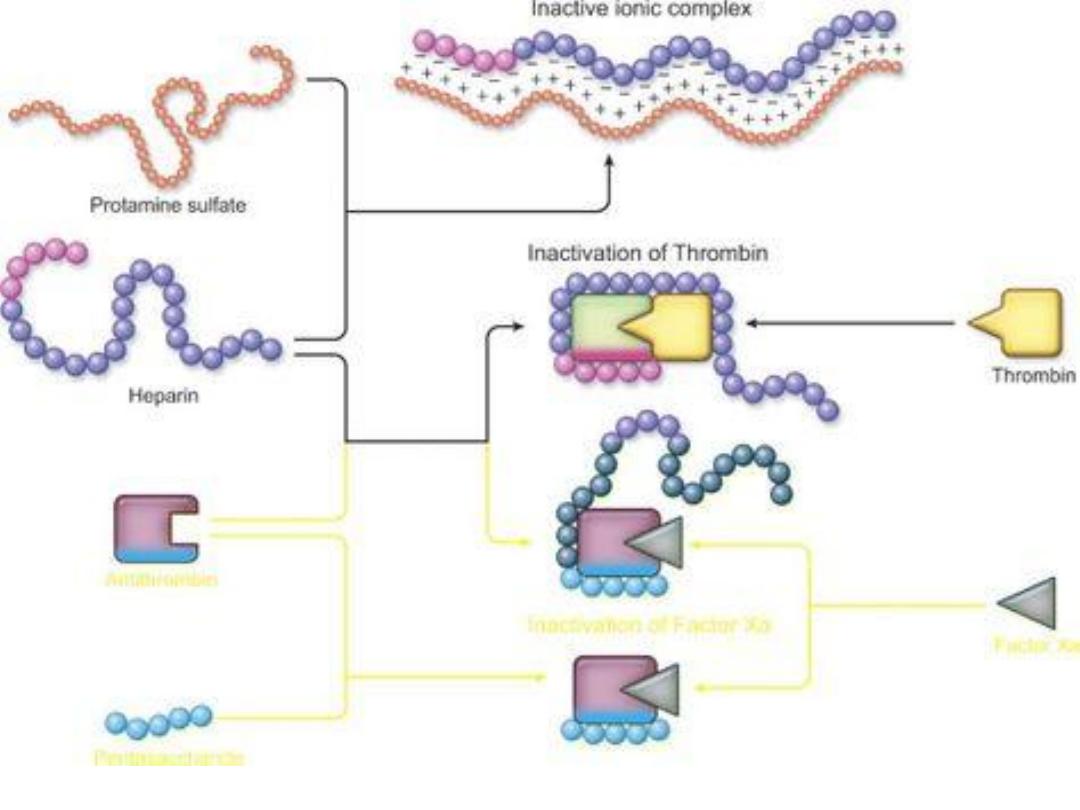
Protamine-heparin complexe

Thanks