
Cystic fibrosis
Assistant prof. Dr.Ahmed Hussein Jasim F.I.B.M.S (resp)

Cystic fibrosis (CF) is the most common fatal genetic disease in
Caucasians, with autosomal recessive inheritance, a carrier rate of 1 in
25, and an incidence of about 1 in 2500 live births
CF is the result of mutations affecting a gene on the long arm of
chromosome 7, which codes for a chloride channel known as cystic
fibrosis transmembrane conductance regulator (CFTR); this influences salt
and water movement across epithelial cell membranes.
The genetic defect causes increased sodium and chloride content in
sweat and increased resorption of sodium and water from respiratory
epithelium. Relative dehydration of the airway epithelium is thought to
predispose to chronic bacterial infection and ciliary dysfunction, leading
to bronchiectasis.
The gene defect also causes disorders in the gut epithelium, pancreas,
liver and reproductive tract
the diagnosis was most commonly made from
the clinical picture
(bowel obstruction, failure to thrive, steatorrhoea and/or chest
symptoms in a young child), supported by sweat electrolyte testing and
genotyping.
Neonatal screening for CF using immunoreactive trypsin and genetic
testing of newborn blood samples is now routine in the UK, and should
reduce delayed diagnosis and improve outcomes.
Clinical features
The lungs are macroscopically normal at birth, but bronchiolar
inflammation and infections usually lead to bronchiectasis in childhood.
At this stage, the lungs are most commonly infected with Staphylococcus
aureus; however, in adulthood, many patients become colonised with
Pseudomonas aeruginosa.

Recurrent exacerbations of bronchiectasis, initially in the upper lobes but
subsequently throughout both lungs, cause progressive lung damage,
resulting ultimately in death from respiratory failure.
Most men with CF are infertile due to failure of development of the
vas deferens, but microsurgical sperm aspiration and in vitro fertilisation
are possible.
Genotype is a poor predictor of disease severity in individuals


Management
Treatment of CF lung disease
All patients with CF who produce sputum should perform chest
physiotherapy regularly, and more frequently during exacerbations.
While infections with Staph. aureus can often be managed with oral
antibiotics
intravenous treatment (frequently self-administered at home through an
implanted subcutaneous vascular port) is usually needed for Pseudomonas
infections.
Regular nebulised antibiotic therapy (colistin or tobramycin) is used between
exacerbations in an attempt to suppress chronic Pseudomonas infection.

the bronchi of many CF patients eventually become colonised with pathogens
that are resistant to most antibiotics.
Resistant strains of P. aeruginosa,
Stenotrophomonas maltophilia and Burkholderia cepacia are the main
culprits, and may require prolonged treatment with unusual combinations
of antibiotics.
Aspergillus and non-tuberculous mycobacteria are also frequently found in
the sputum of CF patients but, in most cases, these behave as benign
‘colonisers’ of the bronchiectatic airways and do not require specific
therapy.
Some patients have coexistent asthma, which is treated with inhaled
bronchodilators and corticosteroids;
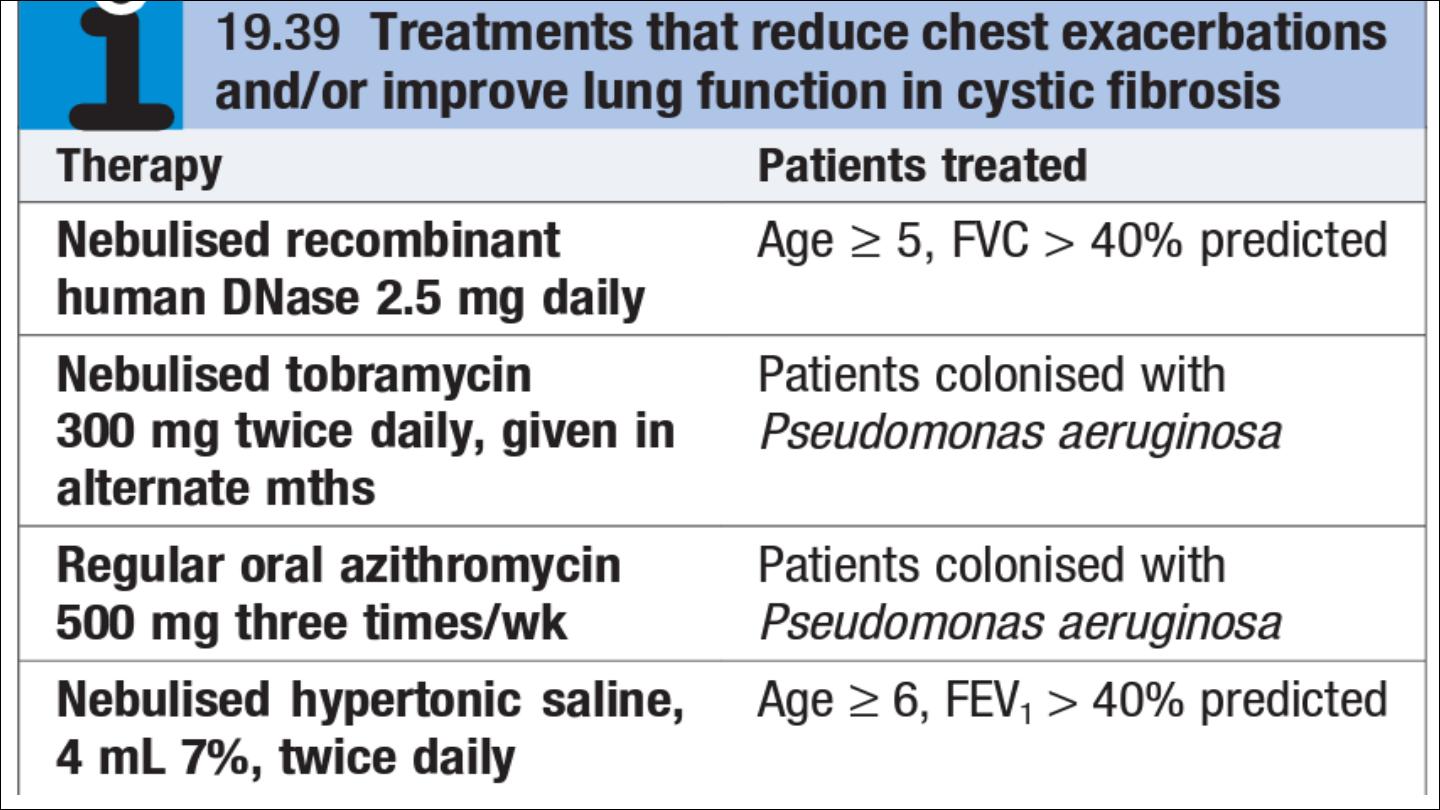
Four maintenance treatments have been shown to cause modest rises in
lung function and/or to reduce the frequency of chest exacerbations in CF
patients

Treatment of non-respiratory manifestations of CF
Malabsorption due to exocrine pancreatic failure is treated with oral
pancreatic enzymes
and vitamin supplements. The increased calori
requirements of CF patients are met by supplemental feeding, including
nasogastric or gastrostomy tube feeding if required.
Diabetes eventually develops in over 25% of patients and often requires
insulin therapy.
Osteoporosis secondary to malabsorption and chronic ill health should be
sought and treated.
Novel therapies for cystic fibrosis
Small molecules designed to correct the function of particular CFTR defects
are being developed, and one (ivacaftor) gives significant clinical benefits in
patients with the G551D mutation.

Bronchiolitis
Definition and epidemiology
Bronchioles are small airways of <2mm diameter, lined by bronchial epithelium and
with no cartilage in their walls
. Terminal bronchioles lead to alveoli. Many bronchioles
need to be affected by disease before a patient becomes symptomatic, when there will
be increased airway resistance unresponsive to β2 stimulants. Bronchiolitis is poorly
understood and is a mixture of conditions.
There are two main pathological patterns of bronchiolitis.
Both can exist in the same
patient.
1.
Proliferative bronchiolitis
More common of the two patterns. non-specific reaction to
bronchiolar injury, with organizing exudate within the bronchiolar lumen. Proliferation of
intraluminal fibrotic buds, called Masson bodies, seen in bronchioles, alveoli, and alveolar
ducts. Associated alveolar wall inflammation and foamy macrophages in alveolar spaces.
May completely or partially resolve. Tends to be more responsive to steroids.

2.
Constrictive bronchiolitis
Less common. Concentric narrowing of the bronchiolar wall
due to cellular infiltrates ± smooth muscle hyperplasia, which may cause extrinsic
compression, obliteration, distortion, mucus collection, peribronchiolar fibrosis, and
scarring. Patchy in distribution.
Typically progressive and unresponsive to steroid
therapy. Usually leads to respiratory failure and death.