
INTERSTITIAL AND INFILTRATIVE
PULMONARY DISEASES
DR. AHMED HUSSEIN JASIM F.I.B.M.S (RESP)
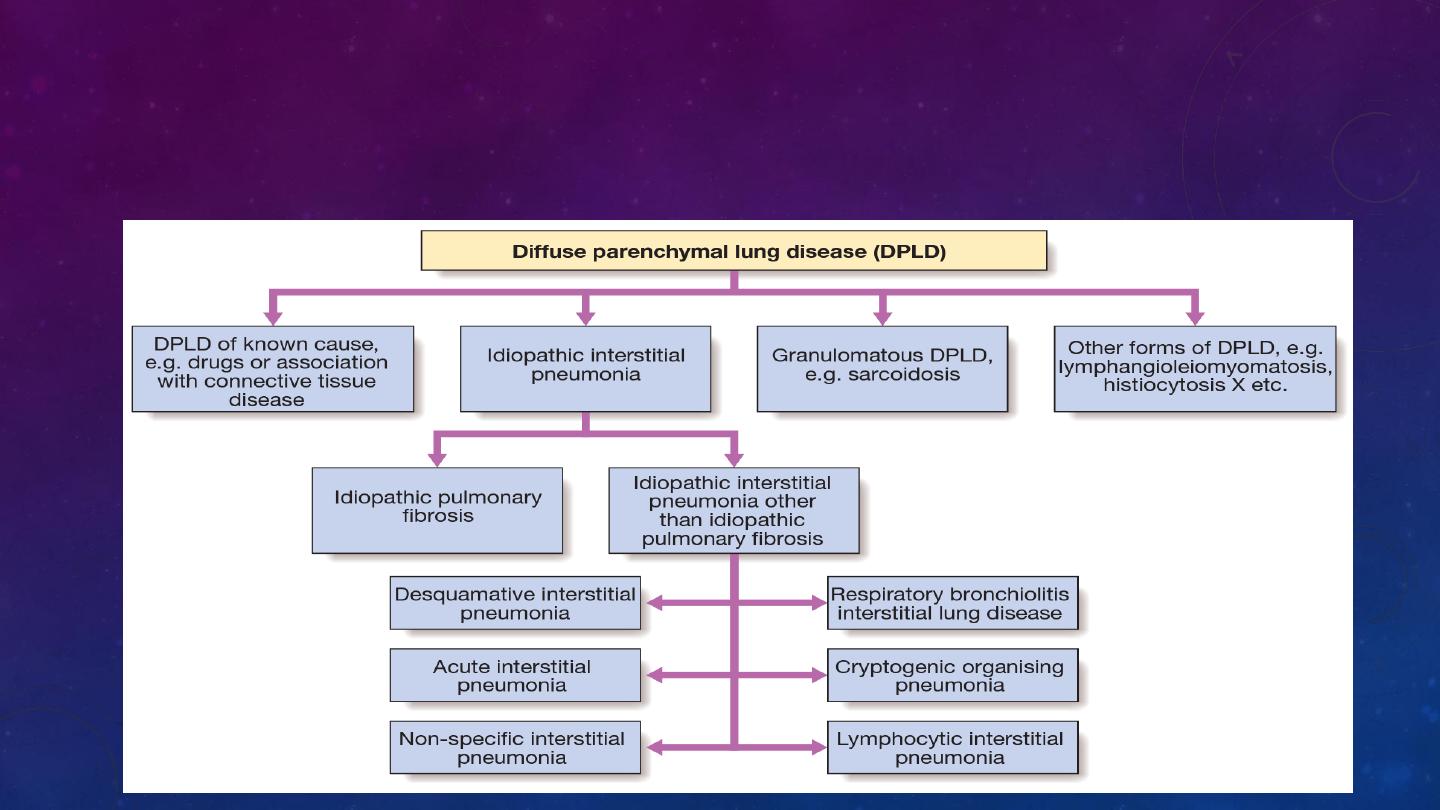
The diffuse parenchymal lung diseases (DPLDs) are a heterogeneous group of
conditions affecting the pulmonary parenchyma (interstitium) and/or alveolar lumen,
which are frequently considered collectively as they share a sufficient number of
clinical physiological and radiographic similarities

Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is defined as a progressive fibrosing interstitial
pneumonia of unknown cause, occurring in adults and associated with the histological
or radiological pattern of usual interstitial pneumonia (UIP).
The histological features of the condition are suggestive of repeated episodes of
focal damage to the alveolar epithelium consistent with an autoimmune process but
the aetiology remains elusive; speculation has included exposure to viruses (e.g.
Epstein–Barr virus), occupational dusts (metal or wood), drugs (antidepressants) or
chronic gastro-oesophageal reflux. Familial cases are rare but genetic factors that
control the inflammatory and fibrotic response are likely to be important.
There is a
strong association with cigarette smoking.
Clinical features
IPF usually presents in the older adult and is uncommon before the age of 50 years.
typically presents with progressive breathlessness (which may have been insidious) and
a non-productive cough. Constitutional symptoms are unusual. Clinical findings include
finger clubbing and the presence of bi-basal fine late inspiratory crackles likened to the
unfastening of Velcro.
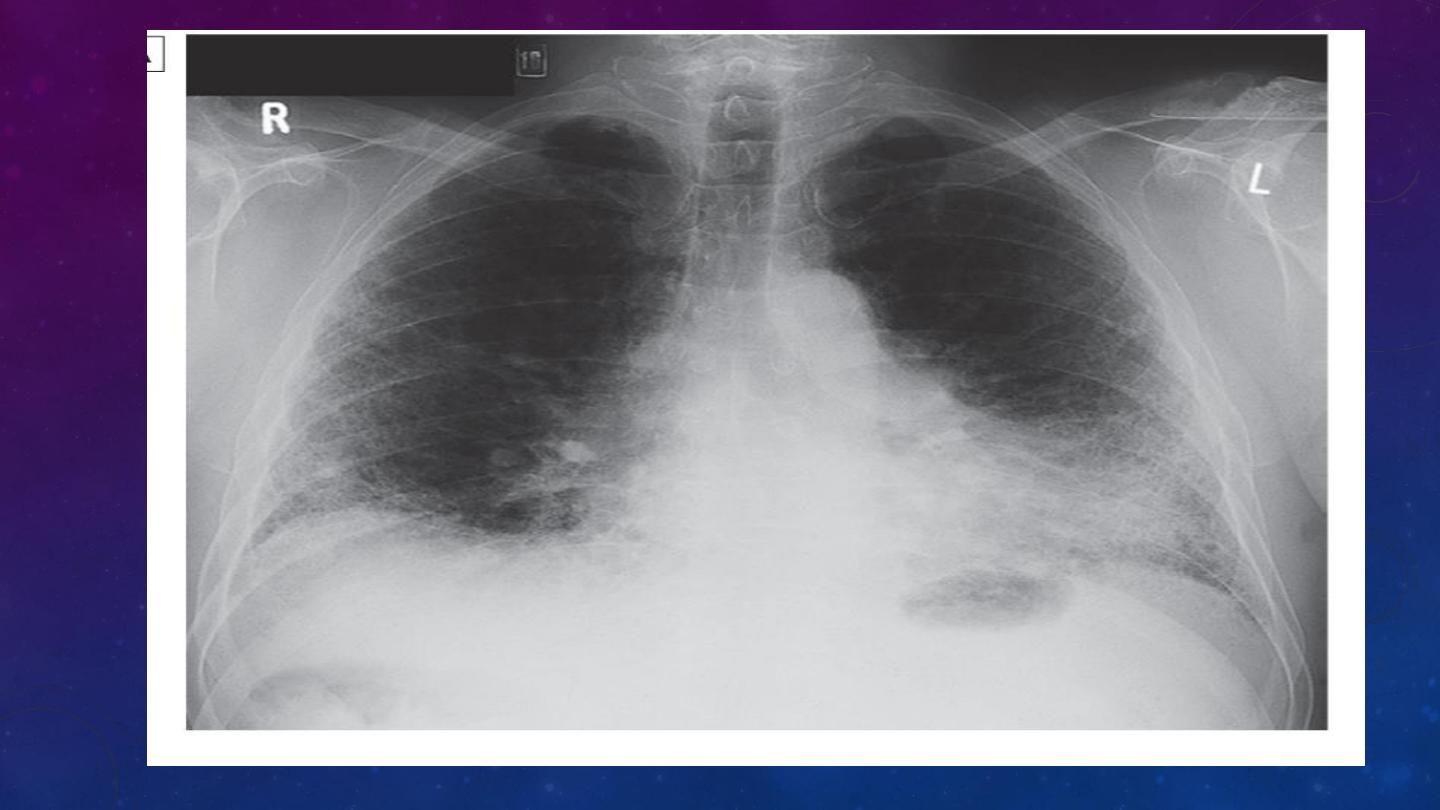

Investigations
chest X-ray as bilateral lower lobe and subpleural reticular shadowing. However, the
chest X-ray may be normal in individuals with early or limited disease.
HRCT typically demonstrates a patchy, predominantly peripheral, subpleural and basal
reticular pattern and, in more advanced disease, the presence of honeycombing cysts
and traction bronchiectasis
Pulmonary function tests classically show a restrictive defect with reduced lung
volumes and gas transfer.
IPF advances, arterial hypoxaemia and hypocapnia are present at rest.
Lung biopsy should be considered in cases of diagnostic uncertainty or with atypical
features.
UIP is the histological pattern predominantly

Management and prognosis
pirfenidone may slow the rate of decline of lung function and, by inference, improve
mortality
Oxygen may help breathlessness but opiates may be required to relieve severe
dyspnoea.
It remains sensible to treat gastro-oesophageal reflux
lung transplantation should be considered.
The natural history is usually one of steady decline but some patients are prone to
exacerbations, which are accompanied by an acute deterioration in breathlessness,
disturbed gas exchange, and new ground glass changes or consolidation on HRCT.
A median survival of 3 years
IPF is associated with an increased risk of carcinoma of the lung.

Sarcoidosis
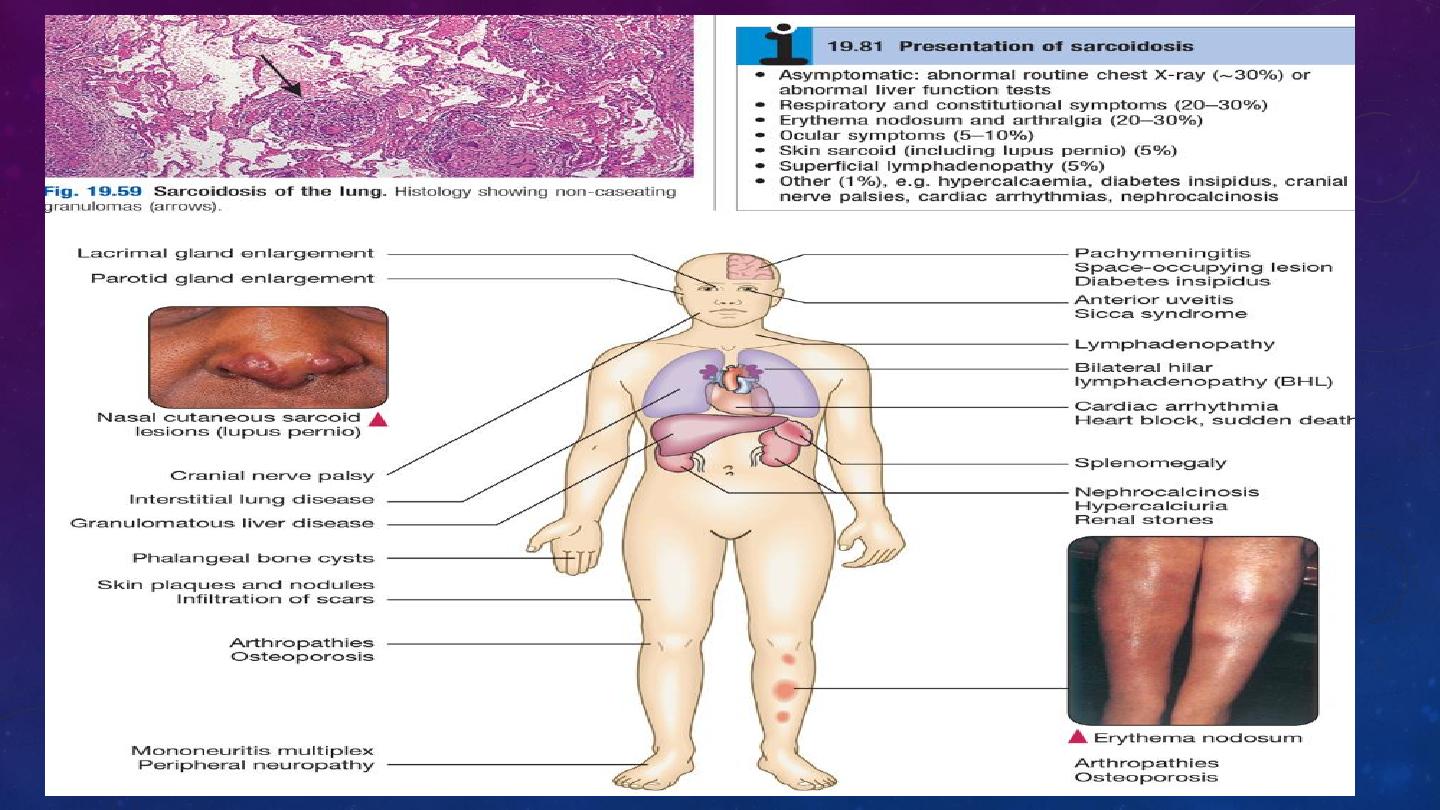
Sarcoidosis is a multisystem granulomatous disorder of unknown aetiology characterised
by the presence of non-caseating granulomas
It also appears to be more common and more severe in those from a West Indian or
Asian background, while Eskimos, Arabs and Chinese are rarely affected.
The tendency for sarcoid to present in spring and summer has led to speculation
about the role of infective agents, including mycobacteria, propionibacteria and viruses,
but the cause remains elusive.
Genetic susceptibility is supported by familial clustering; a range of class II HLA alleles
confer protection from or susceptibility to the condition.
Sarcoidosis occurs less
frequently in smokers.
Clinical features
Sarcoidosis is considered with other DPLDs, as over 90% of cases affect the lungs, but
the condition can involve almost any organ
Löfgren’s syndrome – an acute illness characterised by erythema nodosum, peripheral
arthropathy, uveitis, bilateral hilar lymphadenopathy (BHL), lethargy and occasionally fever
– is often seen in young women.

Investigations
Lymphopenia is characteristic and liver function
tests may be mildly deranged.
Hypercalcaemia may be present (reflecting increased formation of calcitrol – 1,25-
dihydroxyvitamin D3 – by alveolar macrophages), particularly if the patient has been exposed
to strong sunlight.
Hypercalciuria may also be seen and may lead to nephrocalcinosis. Serum ACE may provide a
non-specific marker of disease activity and can assist in moni toring the clinical course.
Chest radiography has been used to stage sarcoid
pulmonary function testing may show a restrictive defect accompanied by impaired gas
exchange. Exercise tests may reveal oxygen desatura-tion.
Bronchoscopy may demonstrate a ‘cobblestone’ appearance of the mucosa, and bronchial and
transbron-chial biopsy usually shows non-caseating granulomas. The BAL fluid typically
contains an increased CD4:CD8 T-cell ratio.
Characteristic HRCT appearances include reticulonodular opacities that follow a perilymphatic
distribution, centred on bronchovascular bundles and the subpleural areas.

the diagnosis should be confirmed by histological examination of the involved organ.

Management
Patients who present with acute illness and erythema nodosum are treated with NSAIDs
systemic corticosteroid therapy can be withheld for 6 months. However, pred-nisolone (at a
starting dose of 20–40 mg/day) should be commenced immediately in the presence of
hypercalcae-mia, pulmonary impairment, renal impairment and uveitis
Features suggesting a less favourable outlook include age over 40 years, Afro-Caribbean
ethnicity, persistent symptoms for more than 6 months, the involvement of more than
three organs, lupus pernio and a stage III/IV chest X-ray.
Selected patients may be referred for con-sideration of single lung transplantation.
Chloroquine, hydroxychloroquine and low-dose thalidomide may be useful in cutaneous
sarcoid with limited pulmonary involvement.
The overall mortality is low (1–5%) and usually reflects cardiac involvement or pulmonary
fibrosis.