Myocardial Diseases

Introduction Of Myocardial Diseases:
Although the myocardium is involved in most types
of heart disease, the term myocarditis and
cardiomyopathy are usually reserved for conditions
that primerly affect the heart muscle.
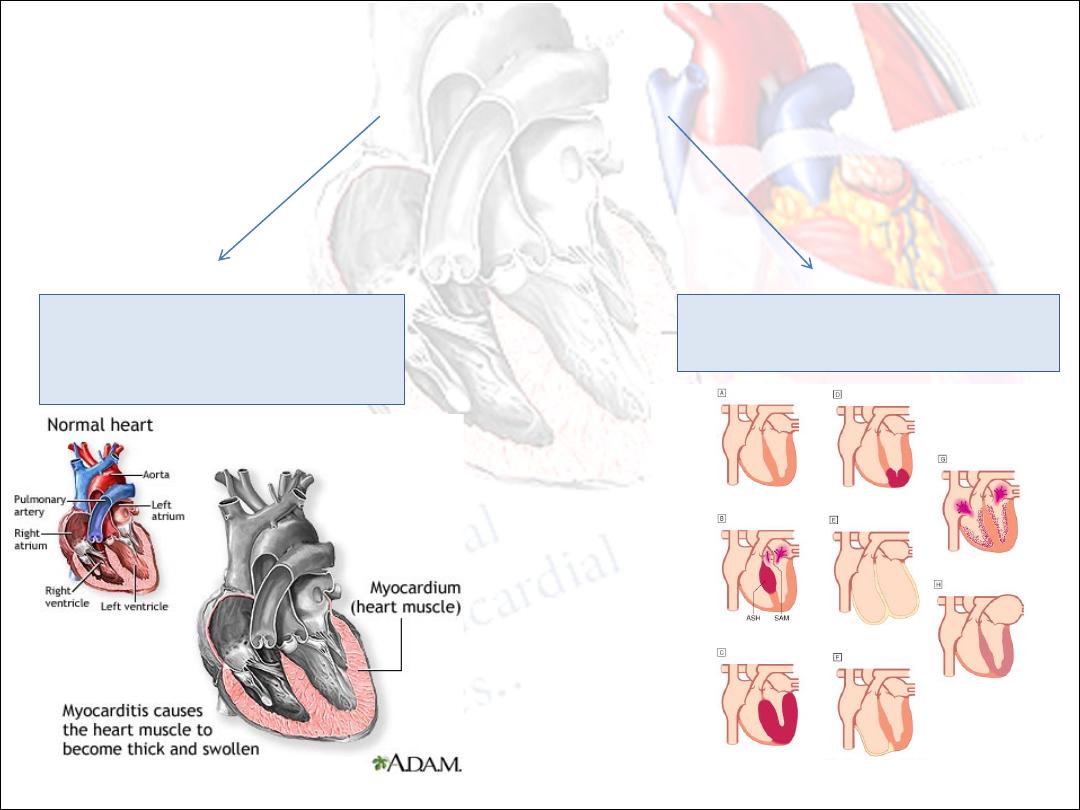
Myocardial diseases may be caused by:
1. an acute or chronic
inflammatory pathology
(myocarditis)
2. idiopathic myocardial disease
(cardiomyopathy).

Myocarditis
•
Acute inflammation of the myocardium.
•
It has many causes:
1.
Idiopathic
2.
Infective
Ø
Viral: Coxsackievirus, adenovirus, CMV, echovirus, influenza, polio, hepatitis, HIV.
Ø
Parasitic: Trypanosoma cruzi, Toxoplasma gondii (a cause of myocarditis in the newborn or
immunocompromised)
Ø
Bacterial: Streptococcus (most commonly rheumatic carditis), diphtheria (toxin-mediated
heart block common)
Ø
Spirochaetal: Lyme disease (heart block common),leptospirosis.
Ø
Fungal.
Ø
Rickettsial.
3- Toxins and Drugs: Causing hypersensitivity reactions, e.g. methyldopa, penicillin,
sulphonamides, antituberculous,modafinil.
4- Radiation: May cause myocarditis but pericarditis more common.
5- Autoimmune: An autoimmune form with autoactivated T cells and organ specific antibodies
may occur.
Infectious
Noninfectious
Viruses –
1.
Coxsackie B
2.
Influenza A & B
3.
Adeno
4.
Hepatitis
5.
HIV
Systemic Diseases:
1.
SLE
2.
Sarcoidosis
3.
Vasculitides(Wegener’s)
4.
Celiac disease
Bacterial –
1.
Corynebacterium diphtheriae
2.
streptococcus
Neoplastic infiltration
Protozoan –
1. Trypanosoma cruzi (Chagas
disease)
Drugs & toxins:
1.
Ethanol
2.
Cocaine
3.
Radiation
4.
Chemotherapeutic agents - Doxorubicin
Spirochete
1.
Borrelia burgdorferi
(Lyme disease)

Pathology:
•
In the acute phase myocarditic hearts are flabby with
focal haemorrhages; in chronic cases they are enlarged
and hypertrophied.
•
Histologically an inflammatory infiltrate is present.
•
lymphocytes predominating in viral causes.
•
Polymorphonuclear cells in bacterial causes.
•
eosinophils in allergic and hypersensitivity causes.

Clinical features:
•
Myocarditis may be an acute or chronic
process (days to weeks after febrile illness ).
•
its clinical presentations range from:
Ø
an asymptomatic state associated with limited
and focal inflammation.
Ø
fatigue, palpitations, chest pain, dyspnoea
and fulminant congestive cardiac failure due
to diffuse myocardial involvement.

Physical examination:
•
includes soft heart sounds.
•
a prominent third sound.
•
often a tachycardia.
•
A pericardial friction rub may be heard.

Investigations:
q
Chest X-ray may show some cardiac enlargement.
q
ECG demonstrates ST- and T wave abnormalities and arrhythmias.
q
Cardiac enzymes are elevated.
q
Echocardiogram helps evaluate cardiac function & exclude other causes
q
Cardiac MRI improving in ability to see abnormalities in myocardium
q
Endomyocardial biopsy -
:
The Dallas criteria require an inflammatory
infiltrate and associated myocyte necrosis or damage not characteristic of
an ischemic event.
q
Viral RNA can be measured from biopsy material using polymerase chain
reaction (PCR). Specific diagnosis requires demonstration of active viral
replication within myocardial tissue.


Treatment:
•
The underlying cause must be identified, treated,
eliminated or avoided.
•
Bed rest is recommended in the acute phase of the illness
and athletic activities should be avoided for 6 months.
•
Heart failure should be treated conventionally with the use
of diuretics, ACE inhibitors/AII receptor antagonists, beta-
blockers, spironolactone ± digoxin.
•
Specific antimicrobial therapy may be used if a causative
organism has been identified.
•
NSAIDs are contraindicated in the acute phase of the illness
but may be used in the late phase.

•
The use of corticosteroids is controversial and
There is no evidence for any benefit from
treatment with corticosteroids and
immunosuppressive agents.
•
Novel and effective
antiviral,immunomodulating agents (e.g.
gamma-interferon and IL-10) may become
available in the future to treat viral myocarditis.

Other types:
•
Giant cell myocarditis:
•
This is a severe form of myocarditis characterized by the presence
of multinucleated giant cells within the myocardium.
•
The cause is unknown but it may be associated with sarcoidosis,
thymomas and autoimmune disease.
•
It has a rapidly progressive course and a poor prognosis.
•
Immunosuppression is recommended.
•
Chagas’ disease
•
Chagas’ disease is caused by the protozoan Trypanosoma cruzi and
is endemic in South America where upwards of 20 million people
are infected.
•
Acutely, features of myocarditis are present with fever and
congestive heart failure.
•
Chronically, there is progression to a dilated cardiomyopathy with a
propensity towards heart block and ventricular arrhythmias.

CARDIOMYOPATHY
•
Cardiomyopathies are a group of diseases of the myocardium
that affect the mechanical or electrical function of the heart.
•
They are not secondary to coronary artery diseases,
hypertension, or congenital, valvular or pericardial
abnormalities.
•
They are frequently genetic and may produce inappropriate
ventricular hypertrophy or dilatation and can be primarily a
cardiac disorder or part of a multi-system disease.

Four main types:
1. Dilated.
2. Hypertrophic.
3. Restrictive.
4. Arrhythmogenic right venticular.
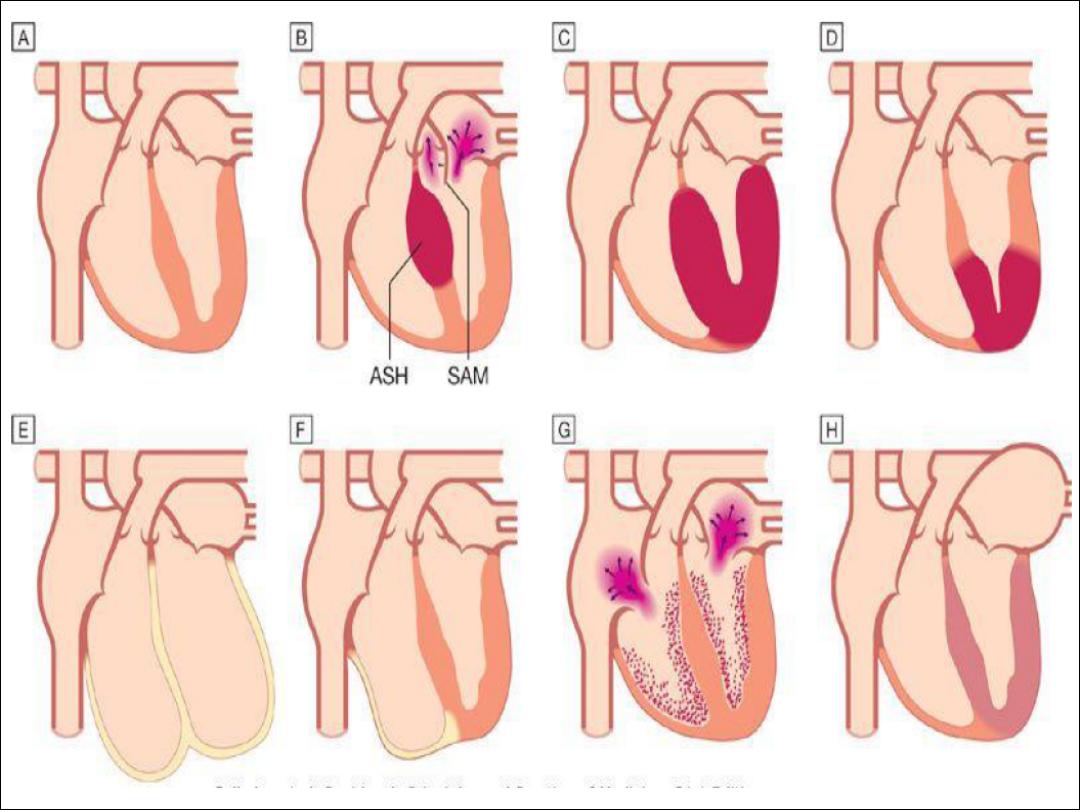

1. Dilated cardiomyopathy (DCM):
•
is characterized by dilatation and impaired contraction of the LV
(sometimes RV ) ; The LV mass increased but the wall thickness normal or
dicreased.
•
Histological changes include myofibrillary loss, interstitial fibrosis and T-
cell infiltrates
•
incidence of 20 per 100 000 and a prevalence of 38 per 100 000
•
Men are affected more than twice as often as women
•
Black > white
•
25% it is a familial disease(autosomal dominant) .
•
Sporadic DCM can be caused by multiple conditions:
■
myocarditis – late autoimmune reaction to viral myocarditis
■
toxins – alcohol, chemotherapy, metals (cobalt, lead, mercury, arsenic)
■
autoimmune.
■
endocrine.
■
neuromuscular.

Clinical features:
DCM can present with:
•
heart failure.
•
cardiac arrhythmias.
•
conduction defects.
•
thromboembolism.
•
sudden death.
•
sporadic chest pain is a surprisingly frequent symptom
•
evaluation of relatives of DCM patients is allowing
identification of early asymptomatic disease, prior to
the onset of these complications.

Investigations:
• ■
Chest X-ray demonstrates generalized cardiac enlargement.
• ■
ECG may demonstrate diffuse non-specific ST segment and T
wave changes. Sinus tachycardia, conduction abnormalities and
arrhythmias are also seen.
• ■
Echocardiogram reveals dilatation of the left and/or right
ventricle with poor global contraction function.
• ■
Cardiac MR may demonstrate other aetiologies of left
ventricular dysfunction (e.g. previous myocardial infarction) or
demonstrate abnormal myocardial fibrosis. Cardiac MR is also
useful for identifying myocardial thrombus .
• ■
Coronary angiography should be performed to exclude coronary
artery disease in all individuals at risk (generally patients > 40 years
or younger if symptoms or risk factors are present).

Treatment:
•
Treatment consists of the conventional management of
heart failure.
•
Disease progression is slowed by ACE-inhibitar, angiotensin
II receptor antagonists and spironolactone, which along
with B-blockers are indicated in most cases.
•
Ventricular tachycardia is best treated with an ICD
•
Anticoagulation : In AF or history of emobolization
•
Severe cardiomyopathy is ttt with cardiac transplantation.

2. Hypertrophic cardiomyopathy
(HCM)
•
the most common with a prevalence of approximately 100 per 100000
•
It is characterized by marked inappropriat ventricular hypertrophy in the
absence of an alternate cause (e.g. aortic stenosis or hypertension).
•
May be generalized or confined to the IVS (ASH ), or apical region (commom
in far east).
•
The hypertrophic non-compliant ventricles impair diastolic filling, so that
stroke volume is reduced.
•
Septal hypertrophy may also cause LVOTO(HOCM) and mitral regurgitation
due to abnormal SAM
•
Most cases are familial, autosomal dominant and caused by mutation in
genes coding for proteins that regulate conraction, e.g troponin and B-
myosin.

Clinical features:
Symptoms:
• ■
many are asymptomatic and are detected through family
screening of an affected individual or following a routine ECG
examination.
• ■
chest pain, dyspnoea, syncope or pre-syncope (typically with
exertion), cardiac arrhythmias and sudden death are seen.
• ■
sudden death occurs at any age but the highest rates (up to 6%
per annum) occur in adolescents or young adults.
■
If a patient develops atrial fibrillation there is often a rapid
deterioration in clinical status due to the loss of atrial contraction
and the tachycardia – resulting in elevated left atrial pressure and
acute pulmonary oedema.

Signs:
• ■
double apical pulsation (forceful atrial
contraction producing a fourth heart sound).
•
jerky carotid pulse because of rapid ejection
and sudden obstruction to left ventricular
outflow during systole
• ■
ejection systolic murmur due to left
ventricular outflow obstruction late in systole.
• ■
pan-systolic murmur due to mitra
regurgitation.
• ■
fourth heart sound (if not in AF).

Investigations:
• ■
ECG abnormalities of HCM include left ventricular hypertrophy,
ST and T wave changes, and abnormal Q waves especially in the
infero-lateral leads.
• ■
Echocardiography is usually diagnostic and in classical HCM
there is asymmetric left ventricular hypertrophy (involving the
septum more than the posterior wall), systolic anterior motion of
the mitral valve, and a vigorously contracting ventricle.
• ■
Cardiac MR can detect both the hypertrophy but also abnormal
myocardial fibrosis.
• ■
Genetic analysis, where available, may confirm the diagnosis and
provide prognostic information for the patient and relatives.

Treatment:
•
The management of HCM includes treatment of symptoms and the prevention of
sudden cardiac death in the patient and relatives.
•
Beta-blockers, rate-limiting calcium antagonists (e.g. verapamil) and disopyramide
can help to relieve symptoms and sometimes prevent syncopal attacks.
•
LVOTO can be improved by myectomy or by alcohol septal ablation using a
catheter-delivered alcohol solution
•
Digoxin and vasodilators may increase outflow tract obstruction and should be
avoided.
•
ICD should be considered in patients with clinical risk factors for sudden death
•
Risk factors for sudden death:
•
■
massive left ventricular hypertrophy (> 30 mm on echocardiography).
•
■
family history of sudden cardiac death (< 50 years old).
•
■
non-sustained ventricular tachycardia on 24-hour Holter monitoring.
•
■
prior unexplained syncope.
•
■
abnormal blood pressure response on exercise (flat or hypotensive response).
•
Family members should be screened

Restrictive cardiomyopathy
•
In this rare condition, ventricular filling is
impaired because the ventricles are 'stiff' .
•
This leads to è high atrial pressures with
atrial hypertrophy è dilatation è later atrial
fibrillation.

Restrictive cardiomyopathy
Aetiology:
1. Amyloidosis is the most common cause of
restrictive cardiomyopathy in the UK.
2. other forms of infiltration (e.g. glycogen
storage diseases), idiopathic perimyocyte
fibrosis .

Restrictive cardiomyopathy
Clinical features :
1. Dyspnoea.
2. fatigue .
3. embolic symptoms are the presenting features.
NB : Restriction to ventricular filling (especially
right) results in persistently elevated venous
pressures, consequent hepatic enlargement,
ascites, and dependent oedema.

Restrictive cardiomyopathy
Physical signs :
are similar to those of constrictive pericarditis :
- a high jugular venous pressure with diastolic collapse
(Friedreich's sign)
- and elevation of venous pressure with inspiration
(Kussmaul's sign).
- S4 is common in early disease and cardiac enlargement,.
- S3 may be present in advanced disease.
- In idiopathic RCM , however, cardiac size may remain
normal.

Restrictive cardiomyopathy
Investigations:
1.
Chest X-ray: may show pulmonary venous
congestion. The heart can be normal or show
cardiomegaly and/or atrial enlargement.
2.
ECG usually has low-voltage and ST segment and T
wave abnormalities.
3.
Echocardiogram shows symmetrical myocardial
thickening and often a normal systolic ejection
fraction, but impaired ventricular filling.
4.
Cardiac catheterization and haemodynamic studies
help distinction from constrictive pericarditis.
5.
Endomyocardial biopsy in contrast with other
cardiomyopathies is often useful in this condition and
may permit a specific diagnosis such as amyloidosis to
be made.

Restrictive cardiomyopathy
Treatment:
-
Treatment is symptomatic but the prognosis is usually poor and transplantation
may be indicated in the severe cases especially the idiopathic variety.
- In primary amyloidosis, combination therapy :
with MELPHALAN plus PREDNISOLONE with or without COLCHICINE may improve
survival.
-
However, patients with cardiac amyloidosis have a worse prognosis than those
with other forms of the disease, and the disease often recurs after
transplantation.
-
Liver transplantation may be effective in familial amyloidosis (due to production
of mutant prealbumin) and may lead to reversal of the cardiac abnormalities.

Arrhythmogenic right ventricular cardiomyopathy
In this condition, patches of the right ventricular
myocardium are replaced with fibrous and fatty tissue.
Arrhythmogenic right ventricular cardiomyopathy
•
This leads to ventricular arrhythmia and risk of sudden death in
its early stages and right ventricular or biventricular failure in its
later stages.
•
The disease is inherited as an autosomal dominant trait.
•
A rare form of ARVC which is associated with dermatological
abnormalities (Naxos disease)
Arrhythmogenic right ventricular cardiomyopathy
Clinical features :
1-commonly with severe symptomatic ventricular arrhythmias or
syncope.
2- Occasionally presentation is with right heart failure.
Heart failure, however, is more commonly associated with a later
stage of disease, in which left ventricular dilatation may also
occur, and severity of arrhythmia may paradoxically diminish.
3- The condition is often asymptomatic and the first presentation
may be with sudden death or alternatively it may be
diagnosed as a result of routine medical evaluation or family
screening.
Arrhythmogenic right ventricular cardiomyopathy
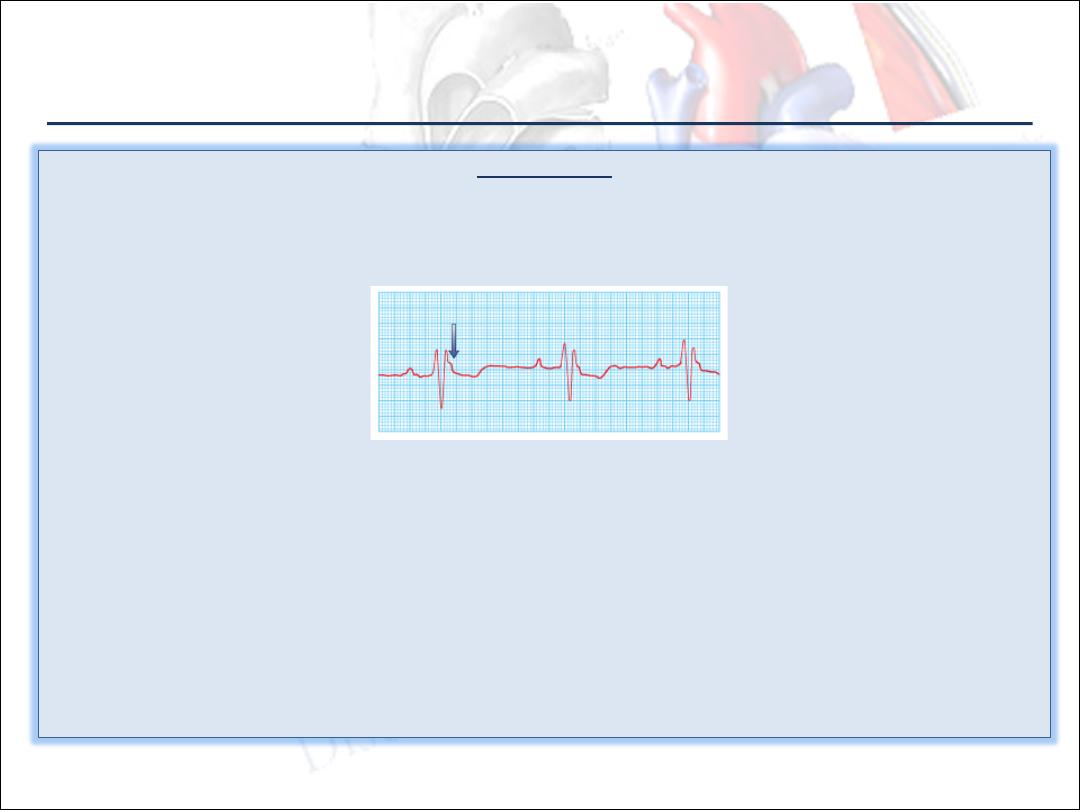
Investigations:
1.
Chest X-ray is usually unremarkable except in advanced disease.
2.
ECG typically shows inverted T waves in the right precordial leads related to the right ventricle (V
1
-V
3
).
Small-amplitude potentials occurring at the end of the QRS complex (epsilon waves) may be present .
Incomplete or complete right bundle branch block RBBB is seen.
3.
Echocardiogram. In early cases this is often normal and in more advanced cases may demonstrate
right ventricular dilatation and aneurysm formation, associated in some cases with concomitant left
ventricular dilatation.
4.
MRI demonstrates morphological abnormalities of the RV and is capable of demonstrating fatty
infiltration.
5.
RV angiography demonstrates enlargement and abnormal motion of right ventricular myocardium.
6.
RV biopsy may demonstrate fibrofatty replacement but is often falsely negative.
7.
Genetic testing, although currently in its infancy, may be a vital diagnostic tool, particularly in variably
penetrant disease.
Arrhythmogenic right ventricular cardiomyopathy
Treatment:
-Beta-blockers are first-line treatment for patients with non-
life-threatening arrhythmias.
-Amiodarone or sotalol may be used for symptomatic
arrhythmias, and for refractory or life-threatening
arrhythmias an ICD may be required.
-Occasionally cardiac transplantation is indicated, either for
intractable arrhythmia or cardiac failure.

OBLITERATIVE CARDIOMYOPATHY
TAKO-TSUBO CARDIOMYOPATHY
LV NONCOMPACTION
PERIPARTUM CARDIOMYOPATHY
Obliterative cardiomyopathy
Ø
involves the endocardium of one or both ventricles
Ø
characterised by thrombosis and elaborate fibrosis with
gradual obliteration of the ventricular cavities
Ø
The mitral and tricuspid valves become regurgitant. Heart
failure and pulmonary and systemic embolism are prominent
features
Ø
associated with eosinophilia (e.g. eosinophilic leukaemia,
Churg-Strauss syndrome
Ø
Mortality is high at 50% at 2 years
Ø
Anticoagulation and antiplatelet therapy are usually advisable
Ø
Surgery (tricuspid and/or mitral valve replacement with
decortication of the endocardium) may be helpful in selected
cases.
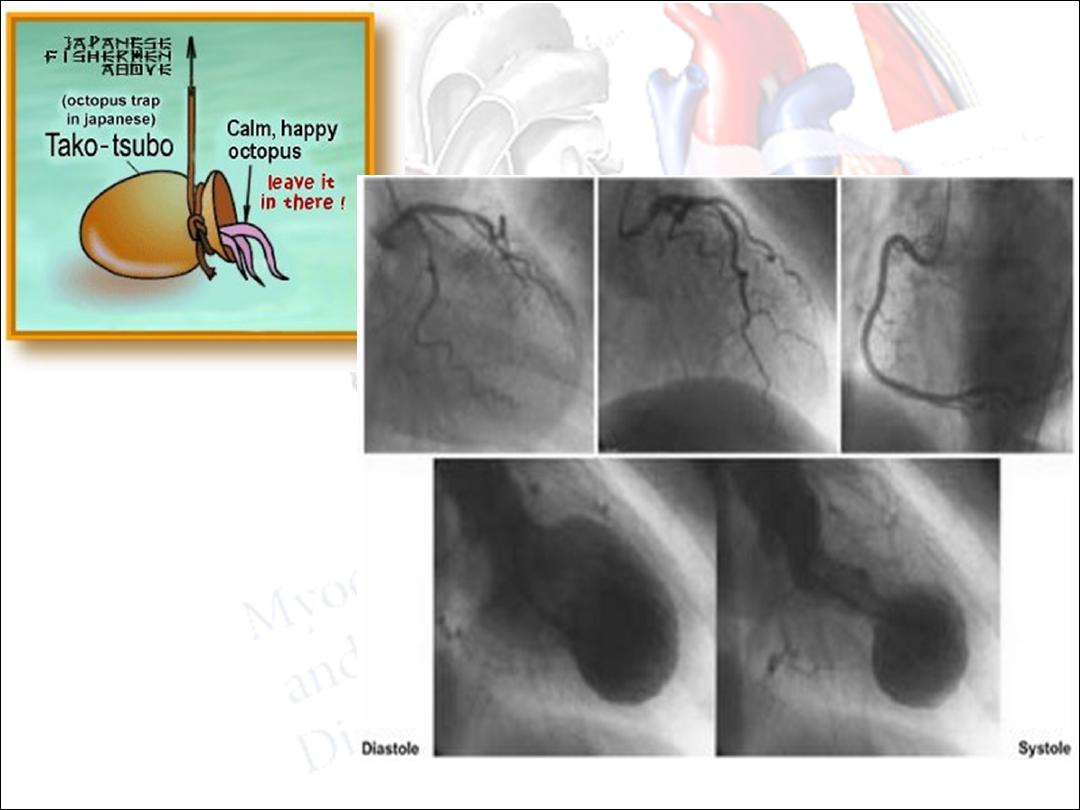
Tako-Tsubo
•
LV apical ballooning after a high catecholamine stress which
results in LV shape similar to octopus pot (takotsubo pot
which is Japanese octopus trap)
•
Has been described following stressful event like
hypoglycemia, earthquakes, following surgery, after emotional
stress
•
Presents as acute anterior MI with chest pain or SOB
•
Usually in post-menopausal women
•
Cardiac catheterization reveals clean coronary arteries
•
Prognosis is good unless there is serious complication (like
MR, ventricular rupture, v-tachycardia
Noncompaction
•
Congenital disorder with hypertrophied LV with deep
trabeculations
•
Decreased systolic fxn
•
Can be isolated or occur with other congenital heart diseases
•
Facial abnormalities and neurologic problems also occur in
high proportion of pts with LVNC
•
Some genetic links, screen 1st degree relatives
Peripartum Cardiomyopathy
•
A form of dilated cardiomyopathy
•
symptoms occur during the last trimester, diagnosed in the
peripartum period
•
1/10,000 in the U.S., 1/100 in parts of Africa
•
greatest in twin preganancies, multiparas, >30 years of age,
African-American
•
50-60% show near complete recovery in 6 months
•
Can recur in subsequent pregnancies

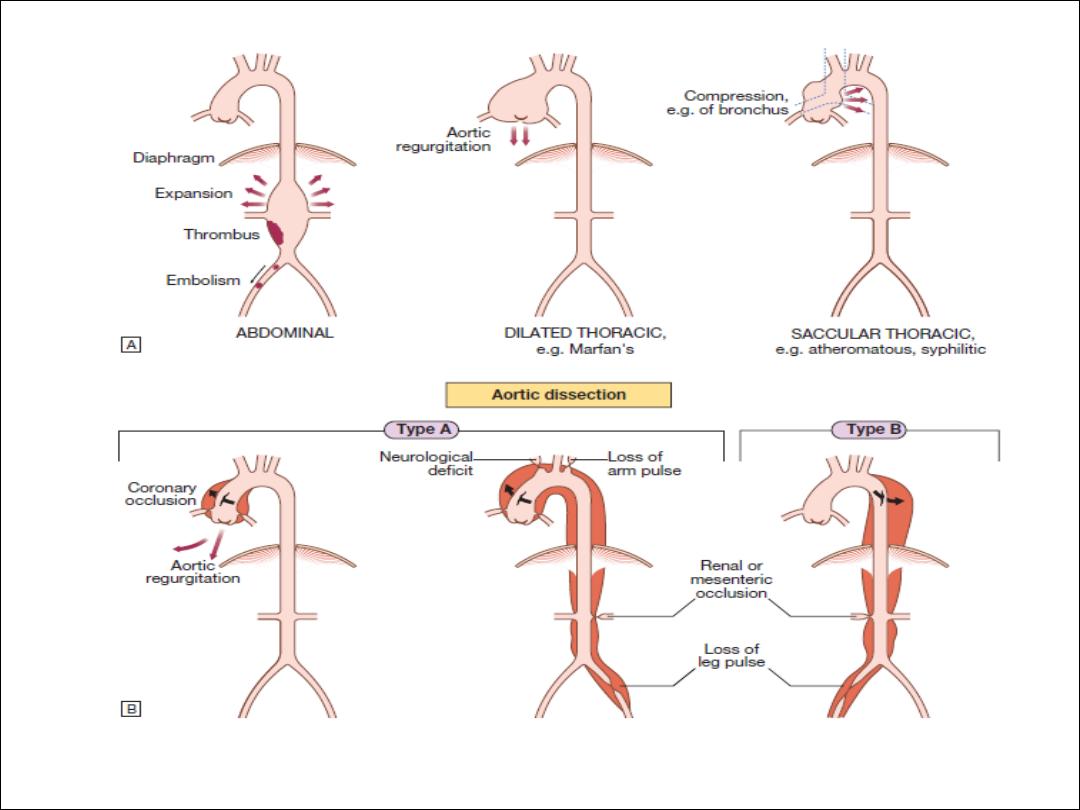
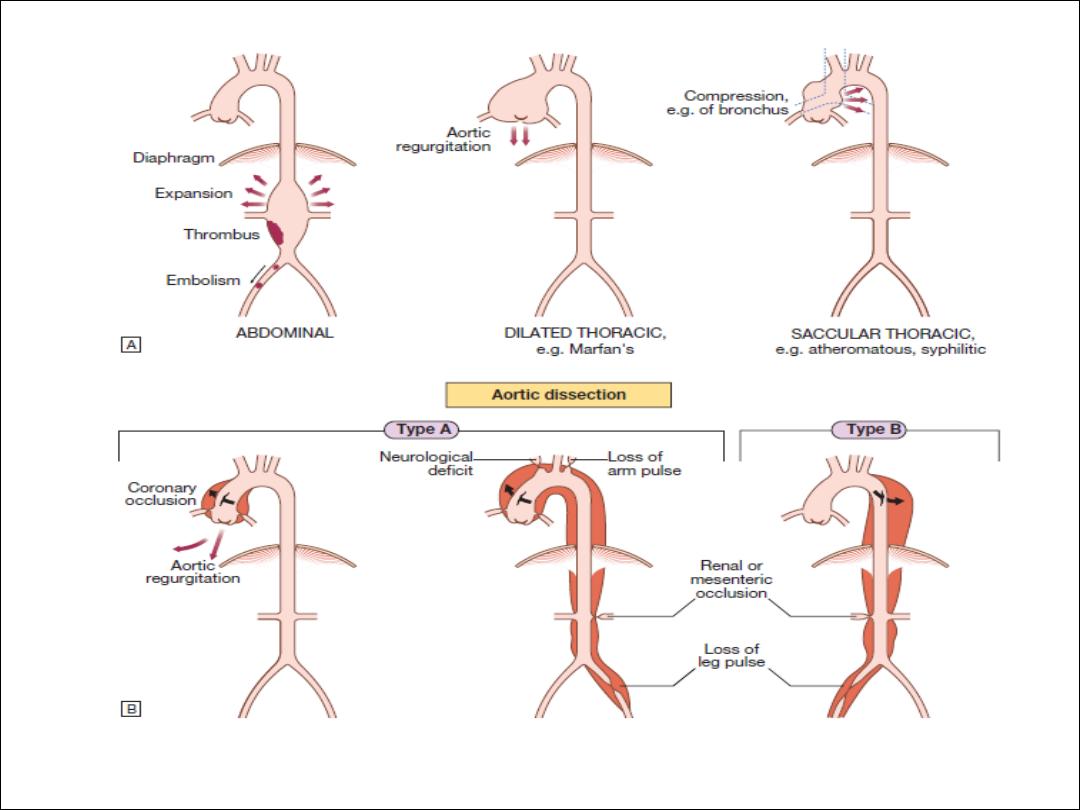
Diseases of the aorta
Aortic aneurysm
Aortic dissection
Aortitis

Aortic aneurysm
•
This is an abnormal dilatation of the aortic
lumen
•
True aneurysm involves all the layers of the
wall, whereas a
•
false aneurysm does not.

Aetiology and types of aneurysm
Non-specific aneurysms
Marfan’s syndrome
Aortitis
Thoracic aortic aneurysms
Abdominal aortic aneurysms

Abdominal aortic aneurysms
•
5% of men aged over 60 years
•
80% are confined to theinfrarenal segment.
•
Men> women
•
The usual age at presentation is 65–75 years
for elective presentations and 75–85 years for
emergency presentations
•
Dx : Ultrasound and CT scan
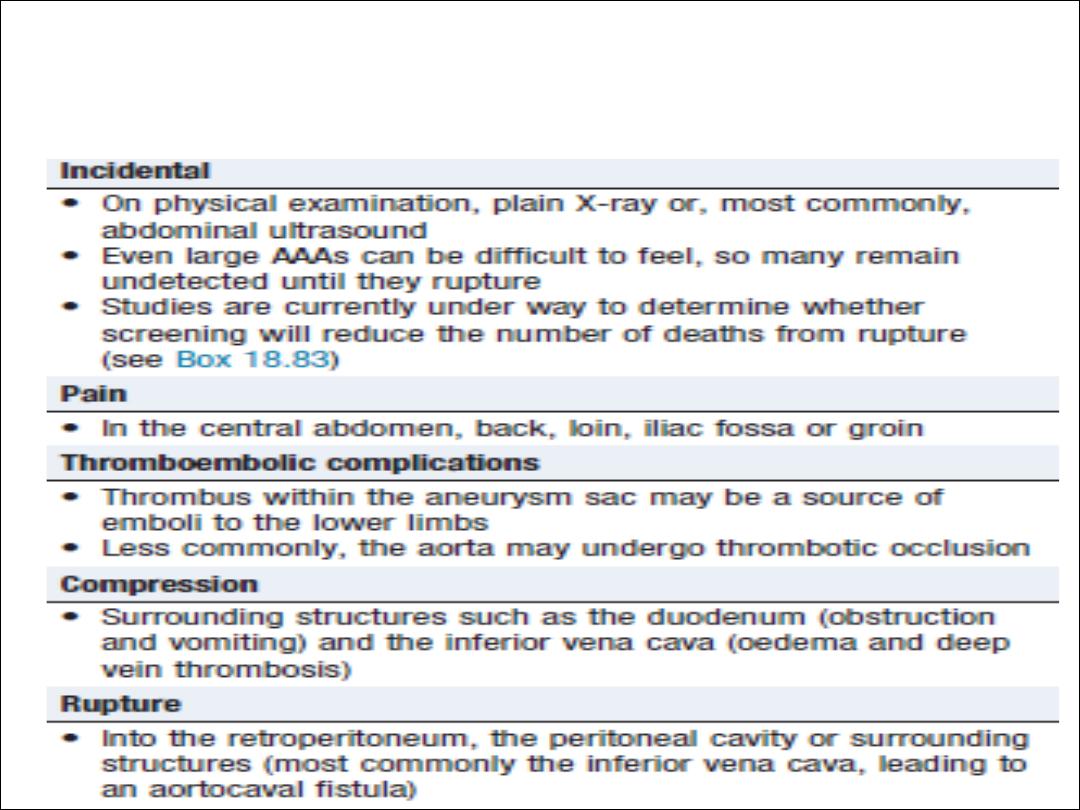
Abdominal aortic aneurysm: common
presentations

Management.
B-blockers
Surgical repair
EVAR

Aortic dissection
Risk Factors
•
Hypertension (in 80%)
•
• Aortic atherosclerosis
•
• Non-specific aortic aneurysm
•
• Aortic coarctation
•
• Collagen disorders (e.g. Marfan’s syndrome, Ehlers–Danlos syndrome)
•
• Fibromuscular dysplasia
•
• Previous aortic surgery (e.g. CABG, aortic valve replacement)
•
• Pregnancy (usually third trimester)
•
• Trauma
•
• Iatrogenic (e.g. cardiac catheterisation, intra-aortic balloon pumping)